
Metal Chelation Enables High-Performance Tea Polyphenol Electrodes for Lithium-Ion Batteries

 ,
, 

Abstract
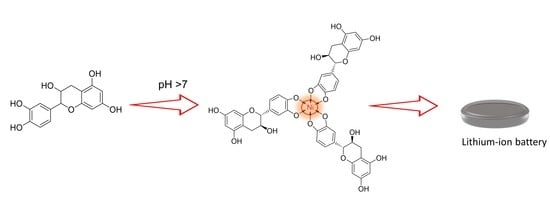
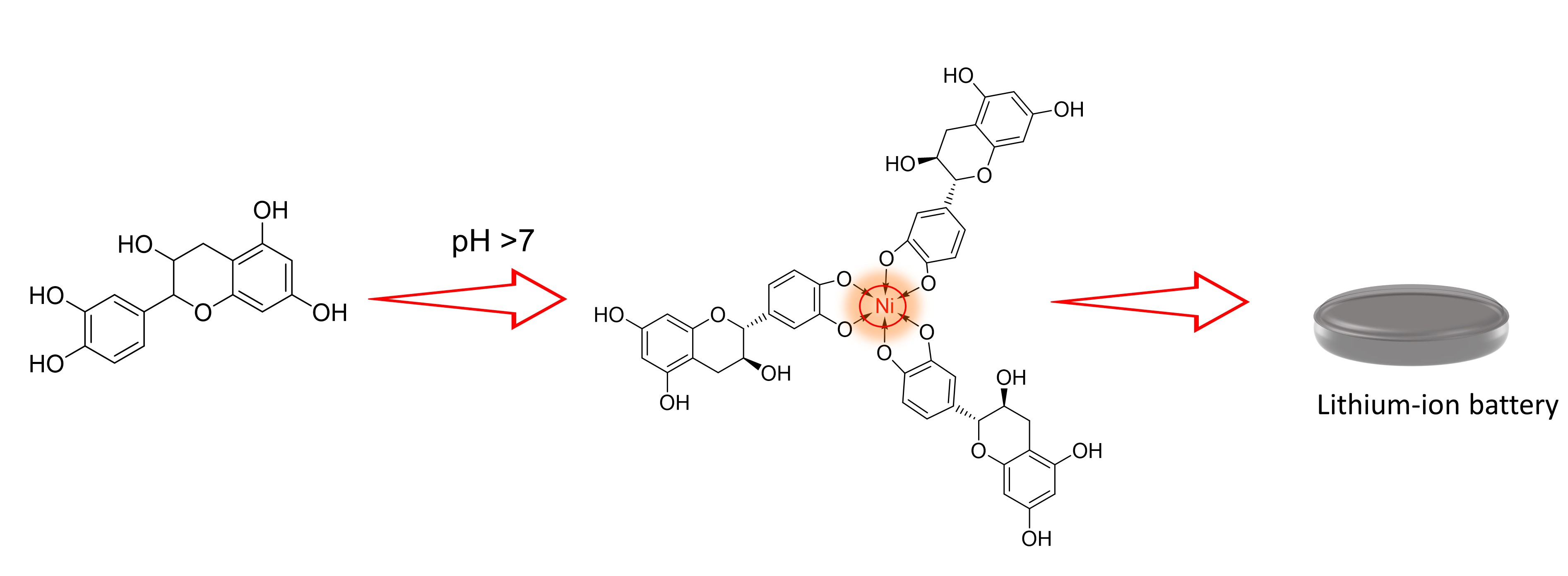{kind=link}
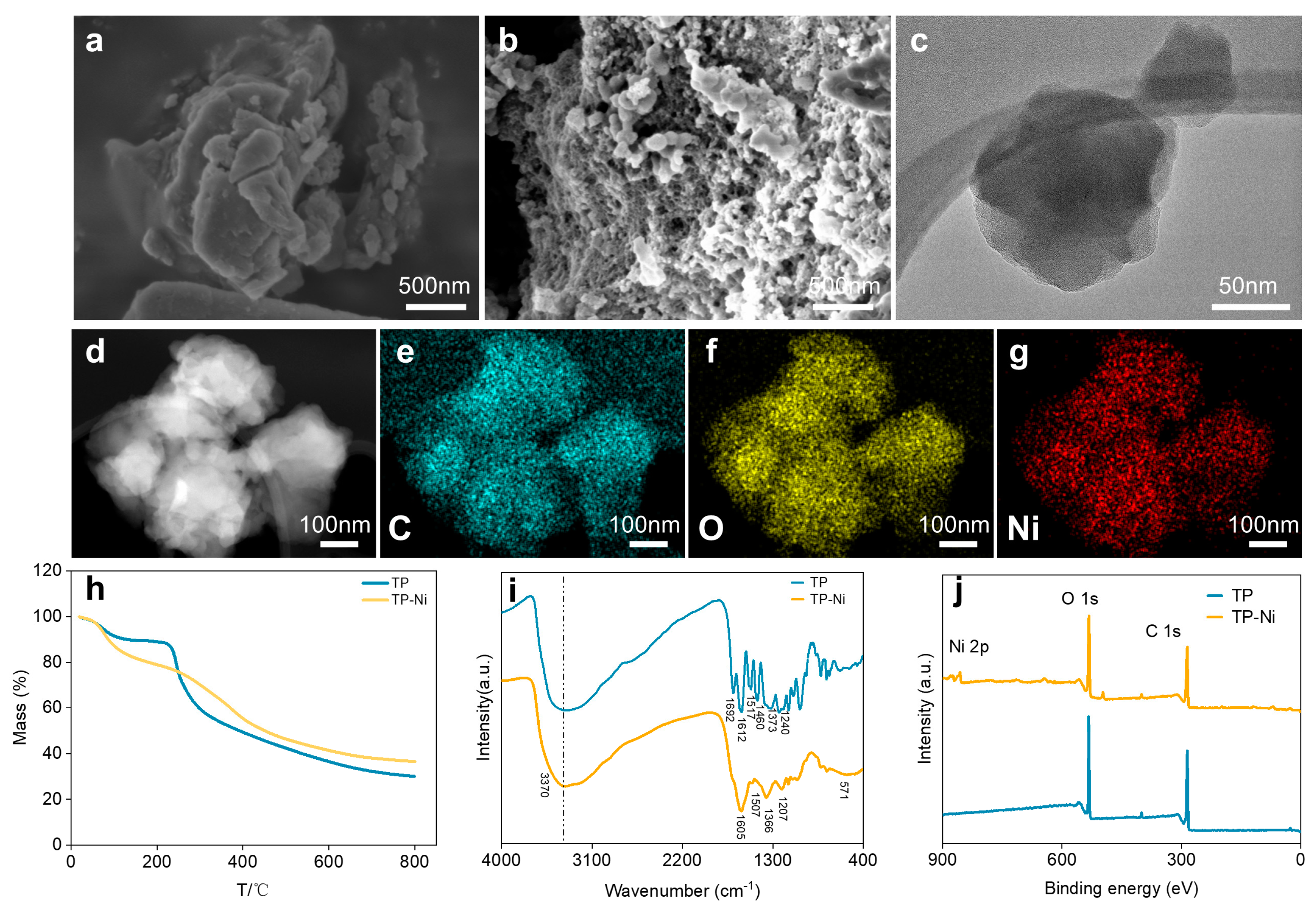{kind=link}
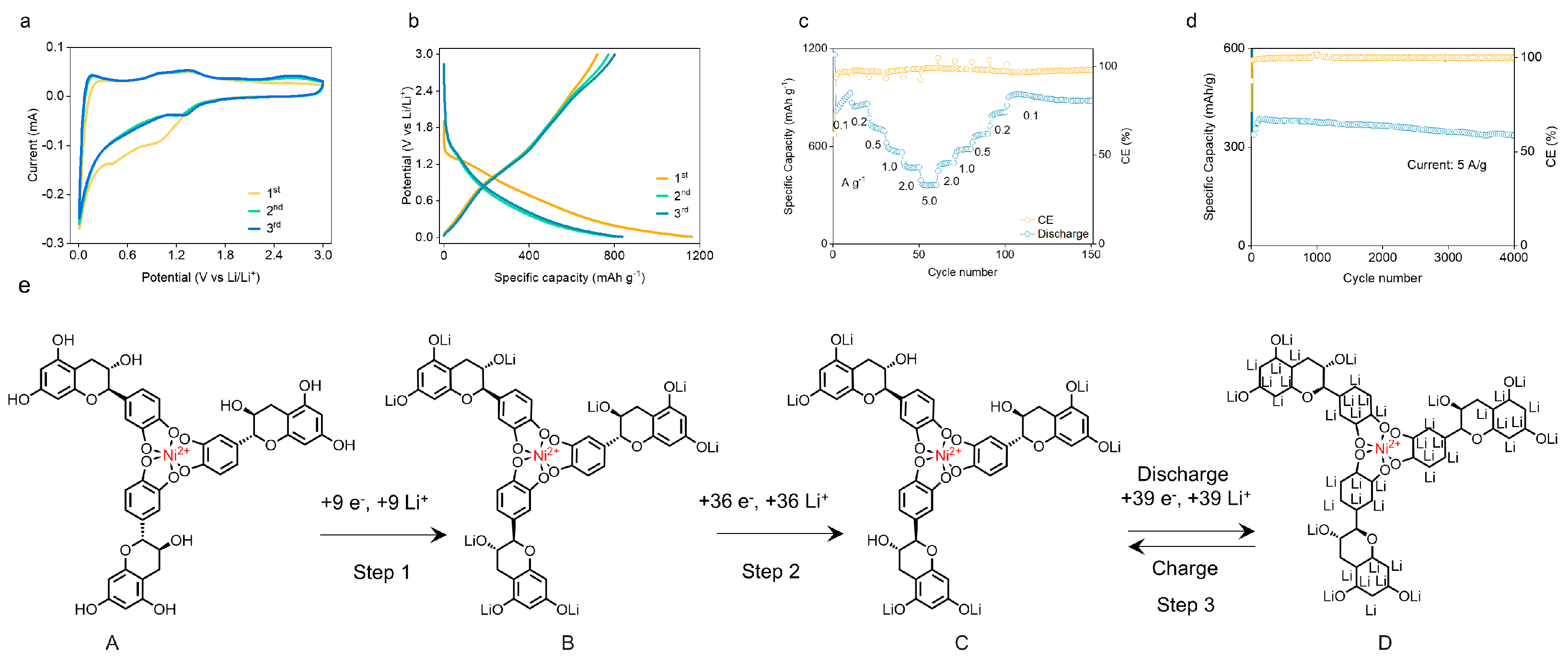{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
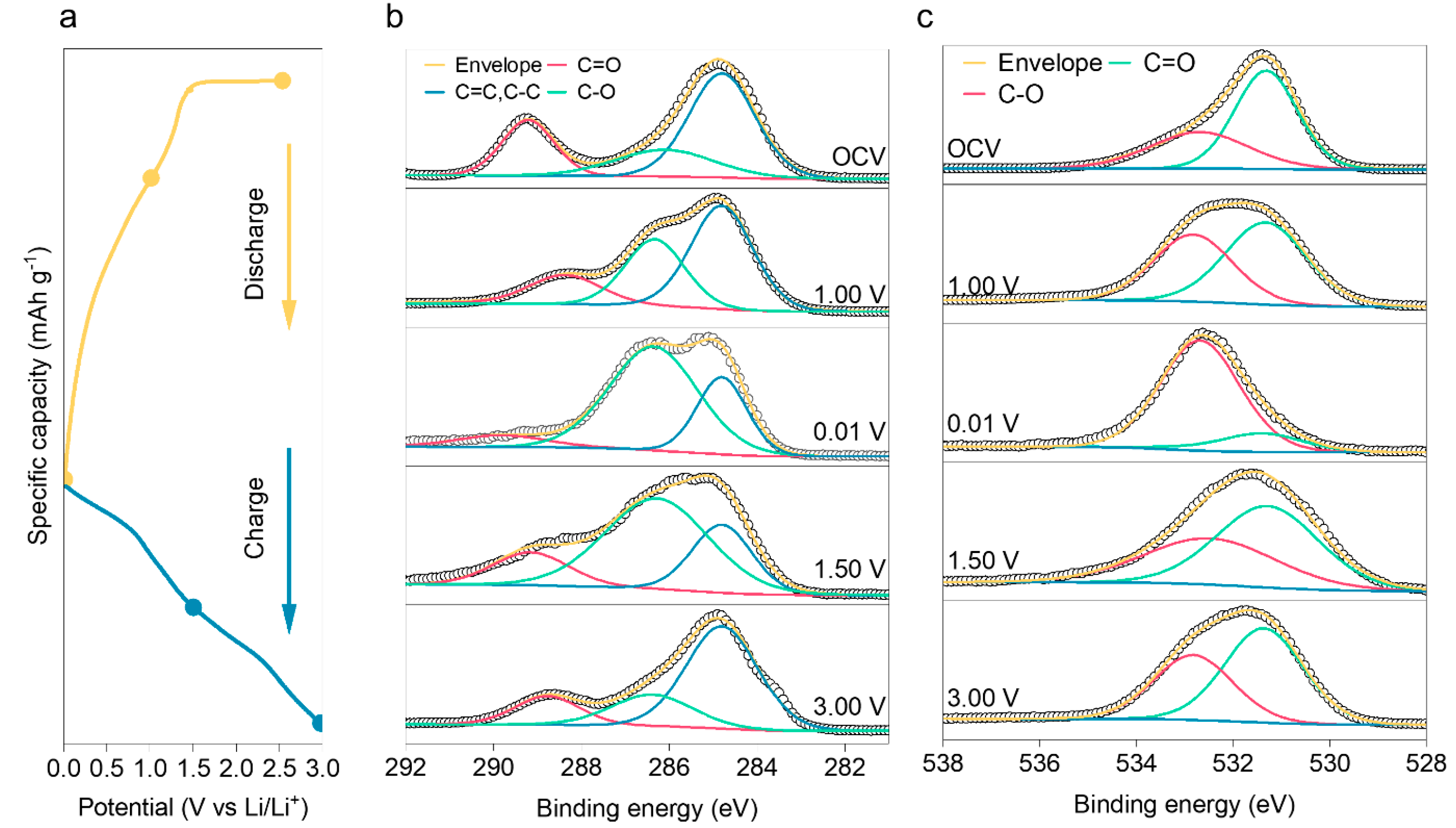
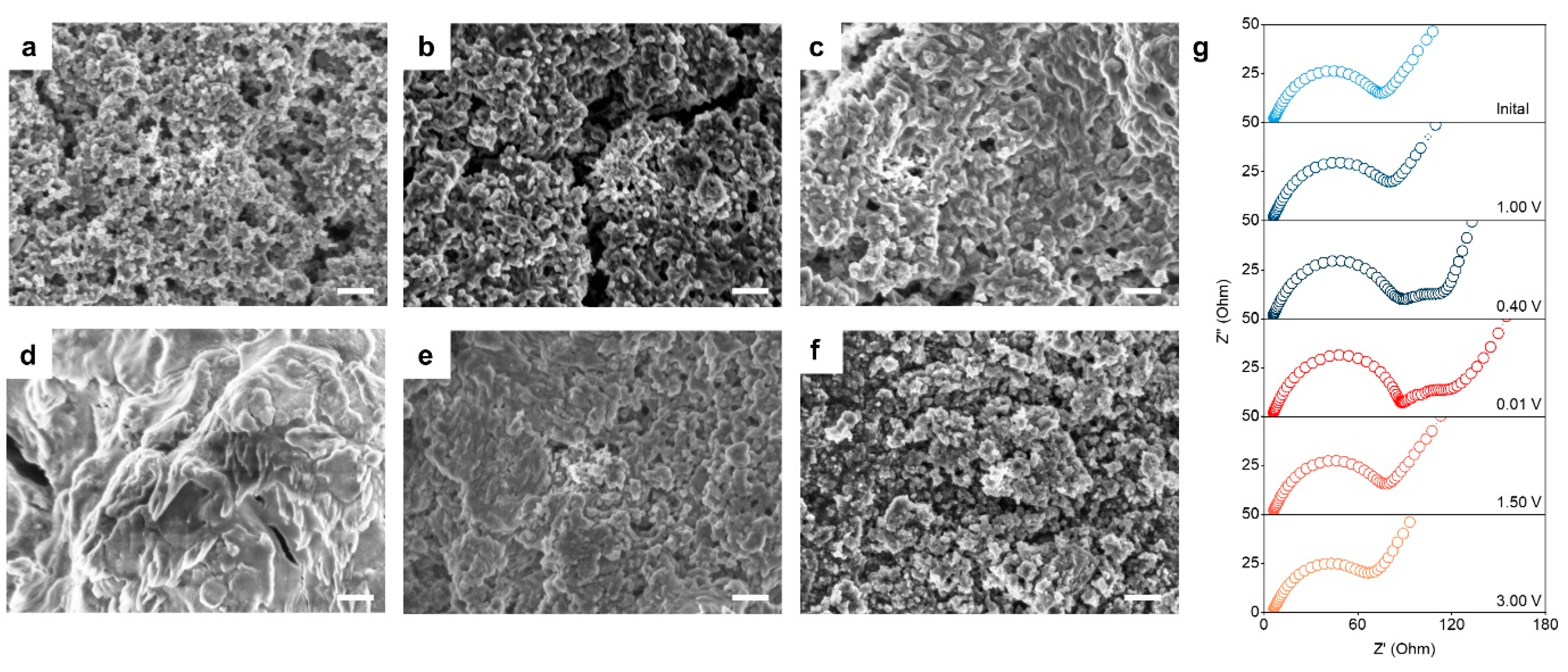
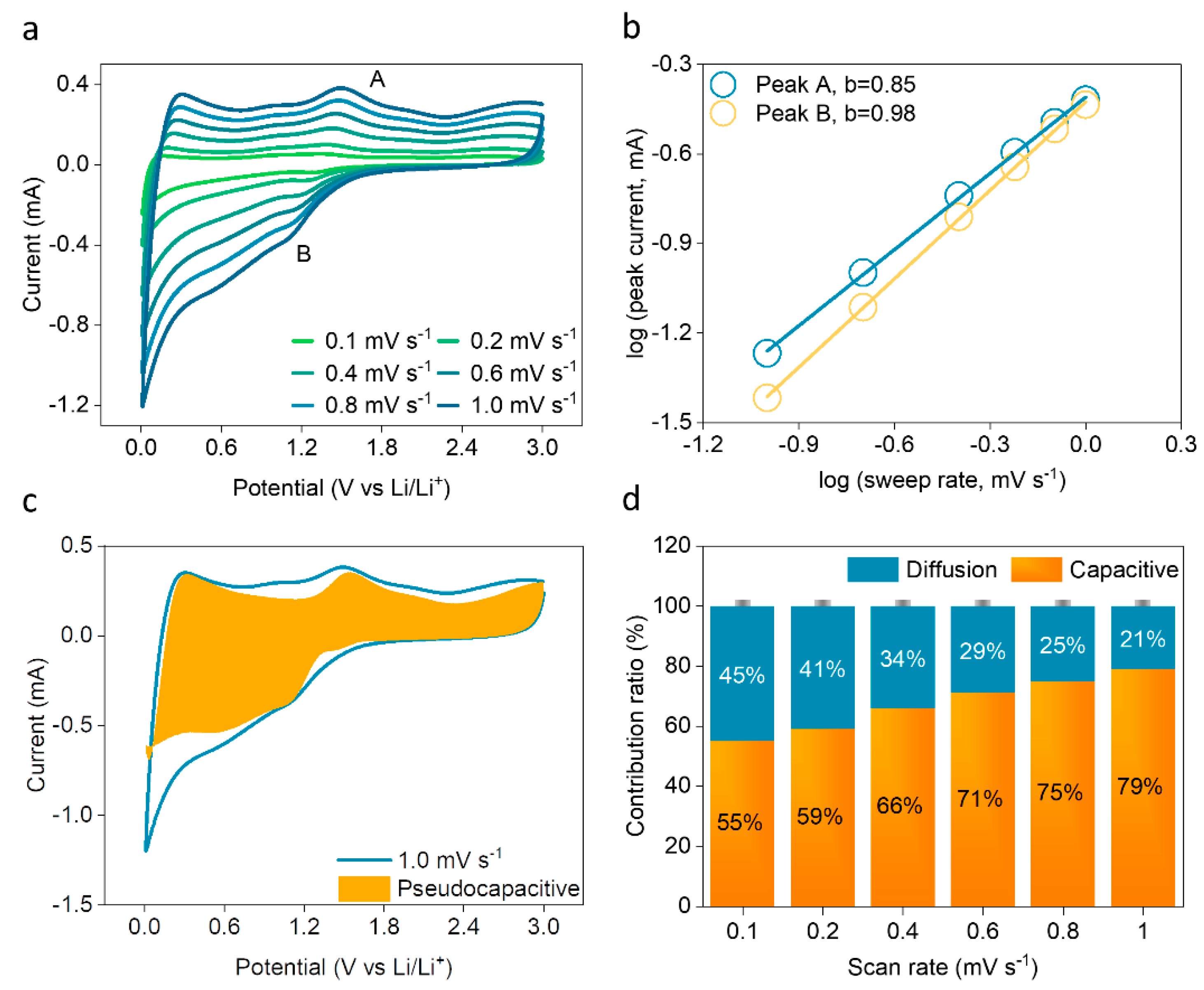
2. Results and Discussion
3. Materials and Methods
3.1. Preparation of TP-Ni
3.2. Electrode Preparation
3.3. Material Characterization
3.4. Electrochemical Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Etacheri, V.; Marom, R.; Elazari, R.; Salitra, G.; Aurbach, D. Challenges in the Development of Advanced Li-Ion Batteries: A Review. Energy Environ. Sci. 2011, 4, 3243–3262. [Google Scholar] [CrossRef]
- Nitta, N.; Wu, F.; Lee, J.T.; Yushin, G. Li-Ion Battery Materials: Present and Future. Mater. Today 2015, 18, 252–264. [Google Scholar] [CrossRef]
- Li, M.; Lu, J.; Chen, Z.; Amine, K. 30 Years of Lithium-Ion Batteries. Adv. Mater. 2018, 30, 1800561. [Google Scholar] [CrossRef] [PubMed]
- Fu, K.; Gong, Y.; Liu, B.; Zhu, Y.; Xu, S.; Yao, Y.; Luo, W.; Wang, C.; Lacey, S.D.; Dai, J.; et al. Toward Garnet Electrolyte–Based Li Metal Batteries: An Ultrathin, Highly Effective, Artificial Solid-State Electrolyte/Metallic Li Interface. Sci. Adv. 2017, 3, e1601659. [Google Scholar] [CrossRef]
- Liu, B.; Zhang, J.-G.; Xu, W. Advancing Lithium Metal Batteries. Joule 2018, 2, 833–845. [Google Scholar] [CrossRef]
- Xie, L.; Tang, C.; Bi, Z.; Song, M.; Fan, Y.; Yan, C.; Li, X.; Su, F.; Zhang, Q.; Chen, C. Hard Carbon Anodes for Next-Generation Li-Ion Batteries: Review and Perspective. Adv. Energy Mater. 2021, 11, 2101650. [Google Scholar] [CrossRef]
- Guo, X.; Wang, C.; Wang, W.; Zhou, Q.; Xu, W.; Zhang, P.; Wei, S.; Cao, Y.; Zhu, K.; Liu, Z.; et al. Vacancy Manipulating of Molybdenum Carbide MXenes to Enhance Faraday Reaction for High Performance Lithium-Ion Batteries. Nano Res. Energy 2022, 1, e9120026. [Google Scholar] [CrossRef]
- Xing, C.; Chen, H.; Zhang, S. Powering 10-Ah-Level Li-S Pouch Cell via a Smart “Skin”. Matter 2022, 5, 2523–2525. [Google Scholar] [CrossRef]
- Sun, H.; Xin, G.; Hu, T.; Yu, M.; Shao, D.; Sun, X.; Lian, J. High-Rate Lithiation-Induced Reactivation of Mesoporous Hollow Spheres for Long-Lived Lithium-Ion Batteries. Nat. Commun. 2014, 5, 4526. [Google Scholar] [CrossRef]
- Kundu, D.; Talaie, E.; Duffort, V.; Nazar, L.F. The Emerging Chemistry of Sodium Ion Batteries for Electrochemical Energy Storage. Angew. Chem. Int. Ed. 2015, 54, 3431–3448. [Google Scholar] [CrossRef]
- Peters, J.F.; Baumann, M.; Zimmermann, B.; Braun, J.; Weil, M. The Environmental Impact of Li-Ion Batteries and the Role of Key Parameters—A Review. Renew. Sustain. Energy Rev. 2017, 67, 491–506. [Google Scholar] [CrossRef]
- Shetti, N.P.; Dias, S.; Reddy, K.R. Nanostructured Organic and Inorganic Materials for Li-Ion Batteries: A Review. Mater. Sci. Semicond. Process. 2019, 104, 104684. [Google Scholar] [CrossRef]
- Zhang, K.; Hu, Z.; Tao, Z.; Chen, J. Inorganic & Organic Materials for Rechargeable Li Batteries with Multi-Electron Reaction. Sci. China Mater. 2014, 57, 42–58. [Google Scholar]
- Lee, S.; Hong, J.; Kang, K. Redox-Active Organic Compounds for Future Sustainable Energy Storage System. Adv. Energy Mater. 2020, 10, 2001445. [Google Scholar] [CrossRef]
- Xie, X.; Criddle, C.; Cui, Y. Design and Fabrication of Bioelectrodes for Microbial Bioelectrochemical Systems. Energy Environ. Sci. 2015, 8, 3418–3441. [Google Scholar] [CrossRef]
- Hussain, I.; Lamiel, C.; Sufyan Javed, M.; Sahoo, S.; Ahmed, M.; Chen, X.; Zhang, K. Plant- and Fungi-Inspired Hierarchical Structures as Electrode Materials: A Review. Mater. Chem. Front. 2022, 6, 3460–3488. [Google Scholar] [CrossRef]
- Lee, B.; Ko, Y.; Kwon, G.; Lee, S.; Ku, K.; Kim, J.; Kang, K. Exploiting Biological Systems: Toward Eco-Friendly and High-Efficiency Rechargeable Batteries. Joule 2018, 2, 61–75. [Google Scholar] [CrossRef]
- Wu, Y.; Zeng, R.; Nan, J.; Shu, D.; Qiu, Y.; Chou, S.-L. Quinone Electrode Materials for Rechargeable Lithium/Sodium Ion Batteries. Adv. Energy Mater. 2017, 7, 1700278. [Google Scholar] [CrossRef]
- Williams, D.L.; Byrne, J.J.; Driscoll, J.S. A High Energy Density Lithium/Dichloroisocyanuric Acid Battery System. J. Electrochem. Soc. 1969, 116, 2. [Google Scholar] [CrossRef]
- Shimizu, A.; Tsujii, Y.; Kuramoto, H.; Nokami, T.; Inatomi, Y.; Hojo, N.; Yoshida, J. Nitrogen-Containing Polycyclic Quinones as Cathode Materials for Lithium-Ion Batteries with Increased Voltage. Energy Technol. 2014, 2, 155–158. [Google Scholar] [CrossRef]
- Pringle, J.M.; Efthimiadis, J.; Howlett, P.C.; Efthimiadis, J.; MacFarlane, D.R.; Chaplin, A.B.; Hall, S.B.; Officer, D.L.; Wallace, G.G.; Forsyth, M. Electrochemical Synthesis of Polypyrrole in Ionic Liquids. Polymer 2004, 45, 1447–1453. [Google Scholar] [CrossRef]
- Momma, T.; Nishimura, K.; Osaka, T.; Kondo, N.; Nakamura, S. Electrochemical Properties of a Polypyrrole/Polystyrenesulfonate Composite Film and Its Application to Rechargeable Lithium Battery Cathodes. J. Electrochem. Soc. 1994, 141, 2326. [Google Scholar] [CrossRef]
- Nishide, H.; Suga, T. Organic Radical Battery. Electrochem. Soc. Interface 2005, 14, 32. [Google Scholar] [CrossRef]
- Nishide, H.; Iwasa, S.; Pu, Y.-J.; Suga, T.; Nakahara, K.; Satoh, M. Organic Radical Battery: Nitroxide Polymers as a Cathode-Active Material. Electrochim. Acta 2004, 50, 827–831. [Google Scholar] [CrossRef]
- Renault, S.; Brandell, D.; Gustafsson, T.; Edström, K. Improving the Electrochemical Performance of Organic Li-Ion Battery Electrodes. Chem. Commun. 2013, 49, 1945–1947. [Google Scholar] [CrossRef] [PubMed]
- Novák, P.; Müller, K.; Santhanam, K.S.V.; Haas, O. Electrochemically Active Polymers for Rechargeable Batteries. Chem. Rev. 1997, 97, 207–282. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.-K.; Hwang, I.; Chang, D.; Park, K.-Y.; Kim, S.J.; Seong, W.M.; Eum, D.; Park, J.; Kim, B.; Kim, J.; et al. Nanoscale Phenomena in Lithium-Ion Batteries. Chem. Rev. 2020, 120, 6684–6737. [Google Scholar] [CrossRef] [PubMed]
- Gorzalski, A.S.; Donley, C.; Coronell, O. Elemental Composition of Membrane Foulant Layers Using EDS, XPS, and RBS. J. Membr. Sci. 2017, 522, 31–44. [Google Scholar] [CrossRef]
- Juma, A.O.; Arbab, E.A.A.; Muiva, C.M.; Lepodise, L.M.; Mola, G.T. 571-Synthesis and Characterization of CuO-NiO-ZnO Mixed Metal Oxide Nanocomposite. J. Alloys Compd. 2017, 723, 866–872. [Google Scholar] [CrossRef]
- Varunkumar, K.; Hussain, R.; Hegde, G.; Ethiraj, A.S. Effect of Calcination Temperature on Cu Doped NiO Nanoparticles Prepared via Wet-Chemical Method: Structural, Optical and Morphological Studies. Mater. Sci. Semicond. Process. 2017, 66, 149–156. [Google Scholar] [CrossRef]
- Appu, M.; Wu, H.; Chen, H.; Huang, J. Tea Polyphenols Mediated Biogenic Synthesis of Chitosan-Coated Cerium Oxide (CS/CeO2) Nanocomposites and Their Potent Antimicrobial Capabilities. Env. Sci. Pollut. Res. 2022, 1–2. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Li, Y.; Meng, X.; Liu, G.; Hu, S.; Pan, F.; Wu, H.; Jiang, Z.; Wang, B.; Li, Z.; et al. Embedding Dopamine Nanoaggregates into a Poly(Dimethylsiloxane) Membrane to Confer Controlled Interactions and Free Volume for Enhanced Separation Performance. J. Mater. Chem. A 2013, 1, 3713–3723. [Google Scholar] [CrossRef]
- Catauro, M.; Papale, F.; Bollino, F.; Piccolella, S.; Marciano, S.; Nocera, P.; Pacifico, S. Silica/Quercetin Sol–Gel Hybrids as Antioxidant Dental Implant Materials. Sci. Technol. Adv. Mater. 2015, 16, 035001. [Google Scholar] [CrossRef] [PubMed]
- Kalinowska, M.; Świderski, G.; Matejczyk, M.; Lewandowski, W. Spectroscopic, Thermogravimetric and Biological Studies of Na(I), Ni(II) and Zn(II) Complexes of Quercetin. J. Anal. Calorim. 2016, 126, 141–148. [Google Scholar] [CrossRef]
- Wang, H.; Wang, X.; Kong, R.-M.; Xia, L.; Qu, F. Metal-Organic Framework as a Multi-Component Sensor for Detection of Fe3+, Ascorbic Acid and Acid Phosphatase. Chin. Chem. Lett. 2021, 32, 198–202. [Google Scholar] [CrossRef]
- Jones, C.D.; Lewis, A.R.; Jones, D.R.; Ottley, C.J.; Liu, K.; Steed, J.W. Lilypad Aggregation: Localised Self-Assembly and Metal Sequestration at a Liquid–Vapour Interface. Chem. Sci. 2020, 11, 7501–7510. [Google Scholar] [CrossRef] [PubMed]
- Matienzo, J.; Yin, L.I.; Grim, S.O.; Swartz, W.E., Jr. X-ray Photoelectron Spectroscopy of Nickel Compounds. Inorg. Chem. 1973, 12, 2762–2769. [Google Scholar] [CrossRef]
- Jurasekova, Z.; Domingo, C.; Garcia-Ramos, J.V.; Sanchez-Cortes, S. Effect of PH on the Chemical Modification of Quercetin and Structurally Related Flavonoids Characterized by Optical (UV-Visible and Raman) Spectroscopy. Phys. Chem. Chem. Phys. 2014, 16, 12802–12811. [Google Scholar] [CrossRef]
- Kim, S.; Regitsky, A.U.; Song, J.; Ilavsky, J.; McKinley, G.H.; Holten-Andersen, N. In Situ Mechanical Reinforcement of Polymer Hydrogels via Metal-Coordinated Crosslink Mineralization. Nat. Commun. 2021, 12, 667. [Google Scholar] [CrossRef]
- Liu, X.; Weng, Q.; Liu, T.; Tang, Z.; Tang, H. A Li3PO4 Coating Strategy to Enhance the Li-Ion Transport Properties of Li2ZnTi3O8 Anode Material for Lithium-Ion Battery. Electrochim. Acta 2023, 447, 142151. [Google Scholar] [CrossRef]
- Tang, S.B.; Lai, M.O.; Lu, L. Study on Li+-Ion Diffusion in Nano-Crystalline LiMn2O4 Thin Film Cathode Grown by Pulsed Laser Deposition Using CV, EIS and PITT Techniques. Mater. Chem. Phys. 2008, 111, 149–153. [Google Scholar] [CrossRef]
- Yang, X.; Rogach, A.L. Electrochemical Techniques in Battery Research: A Tutorial for Nonelectrochemists. Adv. Energy Mater. 2019, 9, 1900747. [Google Scholar] [CrossRef]
- Xia, S.-B.; Yao, L.-F.; Guo, H.; Shen, X.; Liu, J.-M.; Cheng, F.-X.; Liu, J.-J. Li+ Intercalcation Pseudocapacitance in Sn-Based Metal-Organic Framework for High Capacity and Ultra-Stable Li Ion Storage. J. Power Sources 2019, 440, 227162. [Google Scholar] [CrossRef]
- Zhang, G.; Yang, Y.; Zhang, T.; Xu, D.; Lei, Z.; Wang, C.; Liu, G.; Deng, Y. FeIII Chelated Organic Anode with Ultrahigh Rate Performance and Ultra-Long Cycling Stability for Lithium-Ion Batteries. Energy Storage Mater. 2020, 24, 432–438. [Google Scholar] [CrossRef]
- Li, B. Surface Modification of Natural Graphite by Phenolic Resin Carbon Coating for Improvement of Cyclic Performances in Lithium Ion Batteries. Energy Sources 2003, 25, 827–836. [Google Scholar] [CrossRef]
- Xu, S.; Zhang, Z.; Wu, T.; Xue, Y. Nanoporous Carbon Microspheres as Anode Material for Enhanced Capacity of Lithium Ion Batteries. Ionics 2018, 24, 99–109. [Google Scholar] [CrossRef]
- Li, C.; Liu, B.; Jiang, N.; Ding, Y. Elucidating the Charge-Transfer and Li-Ion-Migration Mechanisms in Commercial Lithium-Ion Batteries with Advanced Electron Microscopy. Nano Res. Energy 2022, 1, e9120031. [Google Scholar] [CrossRef]
- Zhang, H.; Zhong, L.; Xie, J.; Yang, F.; Liu, X.; Lu, X. A COF-Like N-Rich Conjugated Microporous Polytriphenylamine Cathode with Pseudocapacitive Anion Storage Behavior for High-Energy Aqueous Zinc Dual-Ion Batteries. Adv. Mater. 2021, 33, 2101857. [Google Scholar] [CrossRef]
- Shen, K.; Chen, H.; Qin, H.; Hou, X. Construct Pseudo-Capacitance of a Flexible 3D-Entangled Carbon Nanofiber Film as Freestanding Anode for Dual-Ion Full Batteries. J. Mater. Sci. Mater. Electron. 2020, 31, 10962–10969. [Google Scholar] [CrossRef]
- Patil, D.R.; Koteswararao, B.; Begari, K.; Yogi, A.; Moussa, M.; Dubal, D.P. Cobalt Cyclotetraphosphate (Co2P4O12): A New High-Performance Electrode Material for Supercapacitors. ACS Appl. Energy Mater. 2019, 2, 2972–2981. [Google Scholar] [CrossRef]





Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guo, Y.; Guo, J.; Li, B.; Zheng, Y.; Lei, W.; Jiang, J.; Xu, J.; Shen, J.; Li, J.; Shao, H. Metal Chelation Enables High-Performance Tea Polyphenol Electrodes for Lithium-Ion Batteries. Inorganics 2023, 11, 148. https://doi.org/10.3390/inorganics11040148
Guo Y, Guo J, Li B, Zheng Y, Lei W, Jiang J, Xu J, Shen J, Li J, Shao H. Metal Chelation Enables High-Performance Tea Polyphenol Electrodes for Lithium-Ion Batteries. Inorganics. 2023; 11(4):148. https://doi.org/10.3390/inorganics11040148
Chicago/Turabian StyleGuo, Yan, Junpo Guo, Bo Li, Yun Zheng, Wen Lei, Jiangmin Jiang, Jincheng Xu, Jingjun Shen, Jielei Li, and Huaiyu Shao. 2023. "Metal Chelation Enables High-Performance Tea Polyphenol Electrodes for Lithium-Ion Batteries" Inorganics 11, no. 4: 148. https://doi.org/10.3390/inorganics11040148
APA StyleGuo, Y., Guo, J., Li, B., Zheng, Y., Lei, W., Jiang, J., Xu, J., Shen, J., Li, J., & Shao, H. (2023). Metal Chelation Enables High-Performance Tea Polyphenol Electrodes for Lithium-Ion Batteries. Inorganics, 11(4), 148. https://doi.org/10.3390/inorganics11040148