
Screening of Carbon-Supported Platinum Electrocatalysts Using Frumkin Adsorption Isotherms

 , ,
, ,  ,
, 
Abstract
1. Introduction
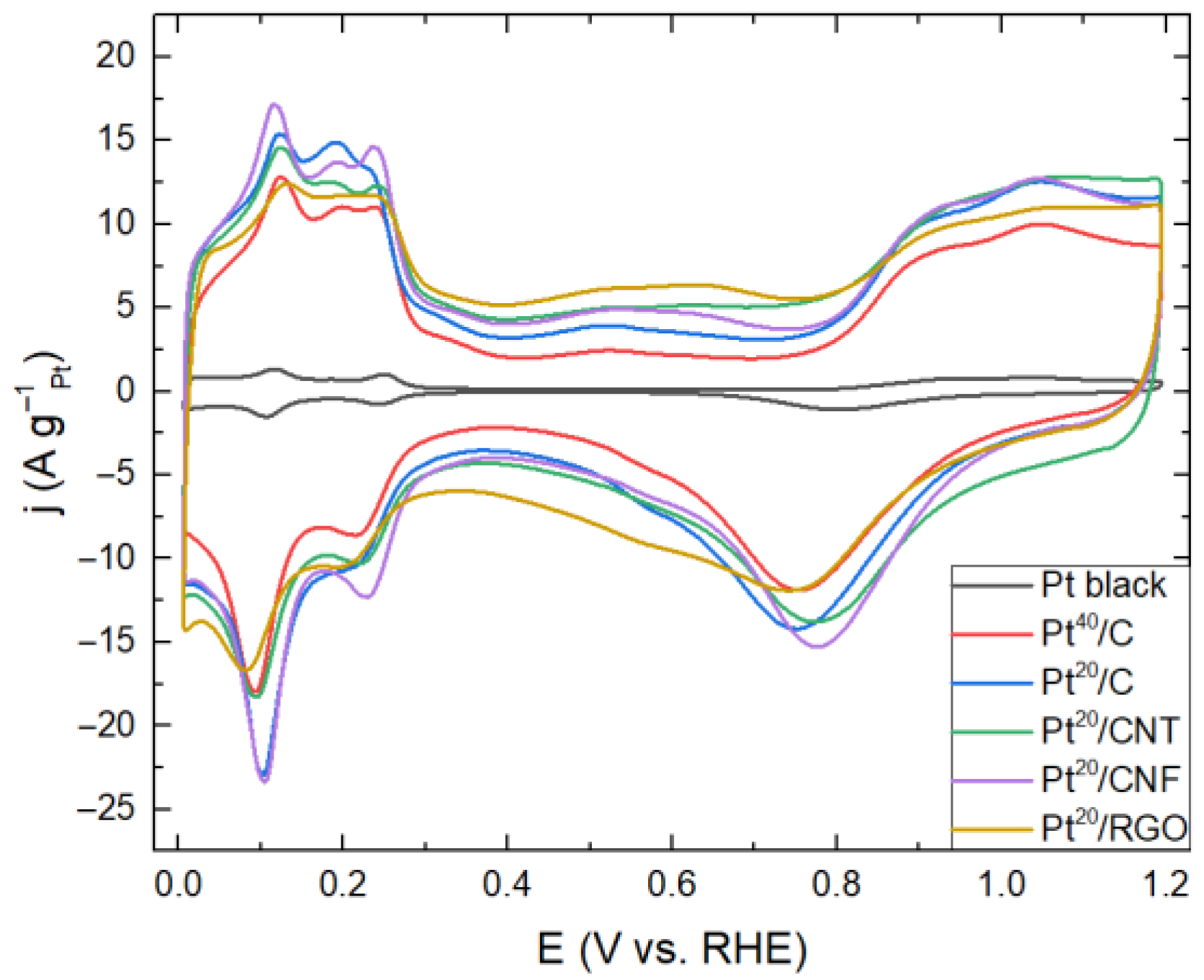
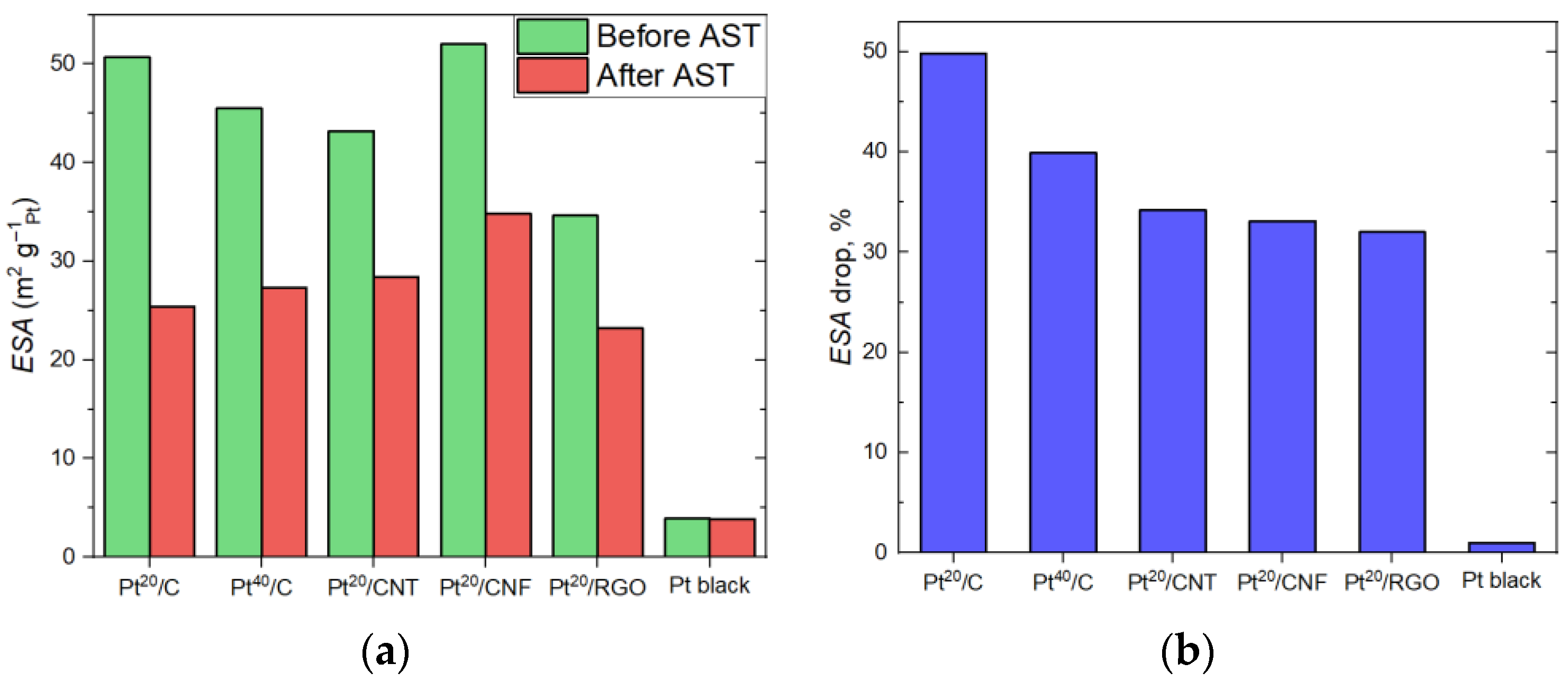
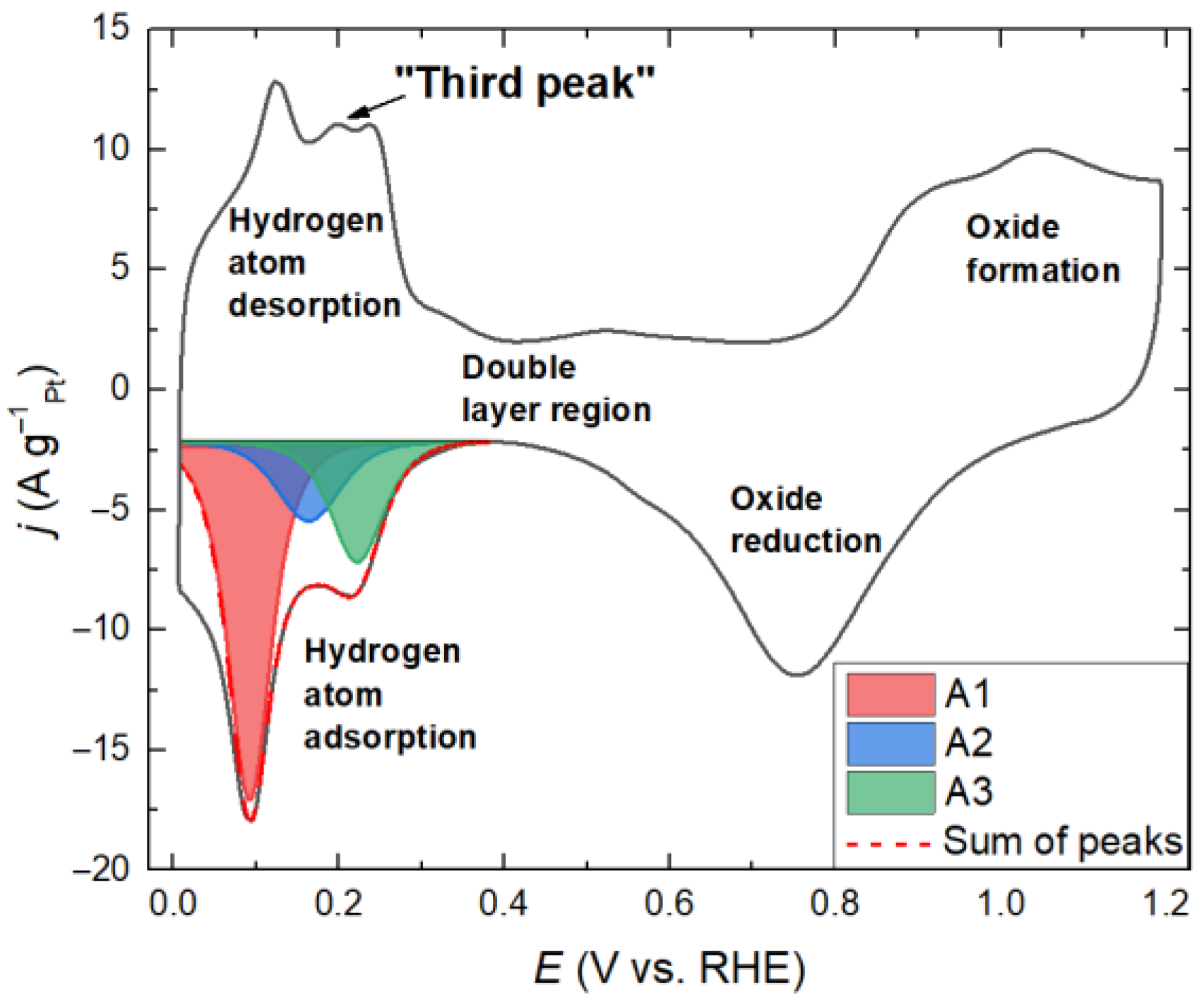
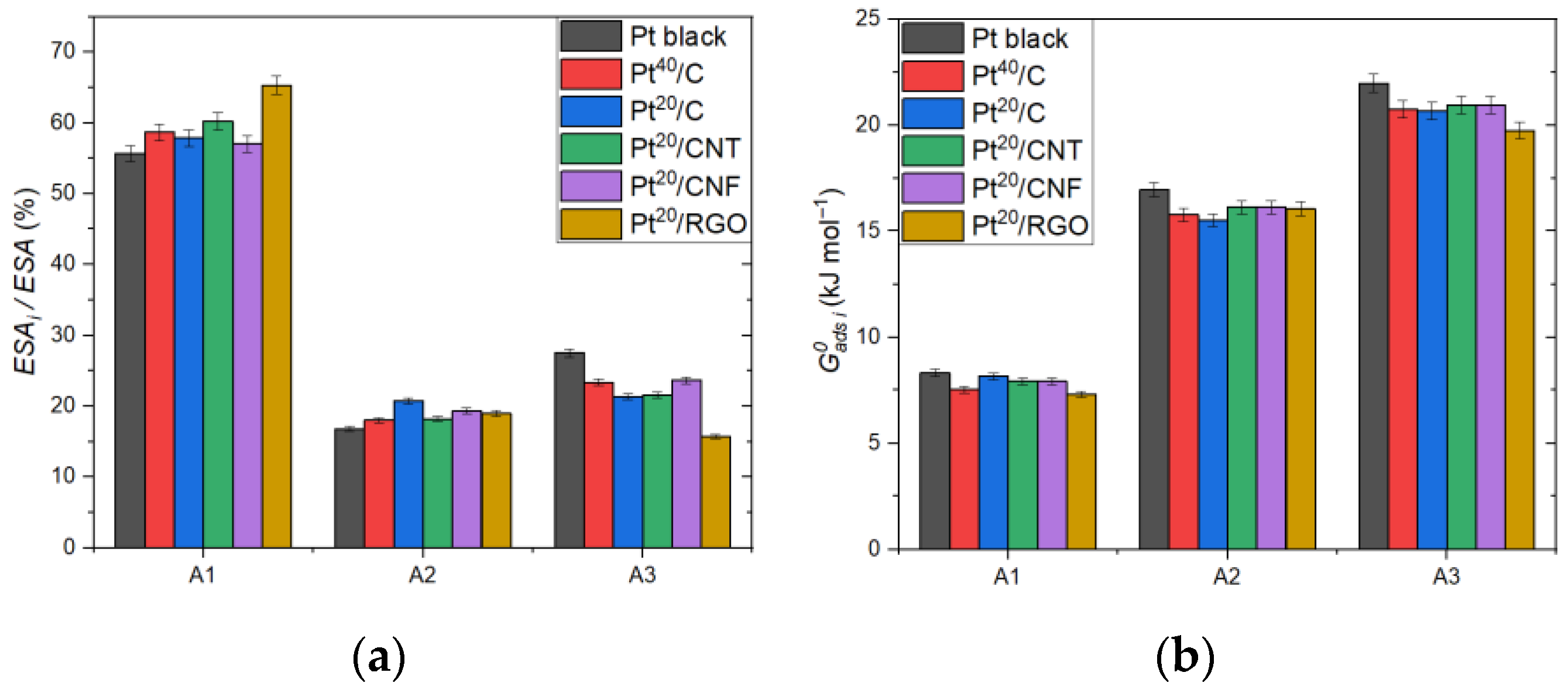
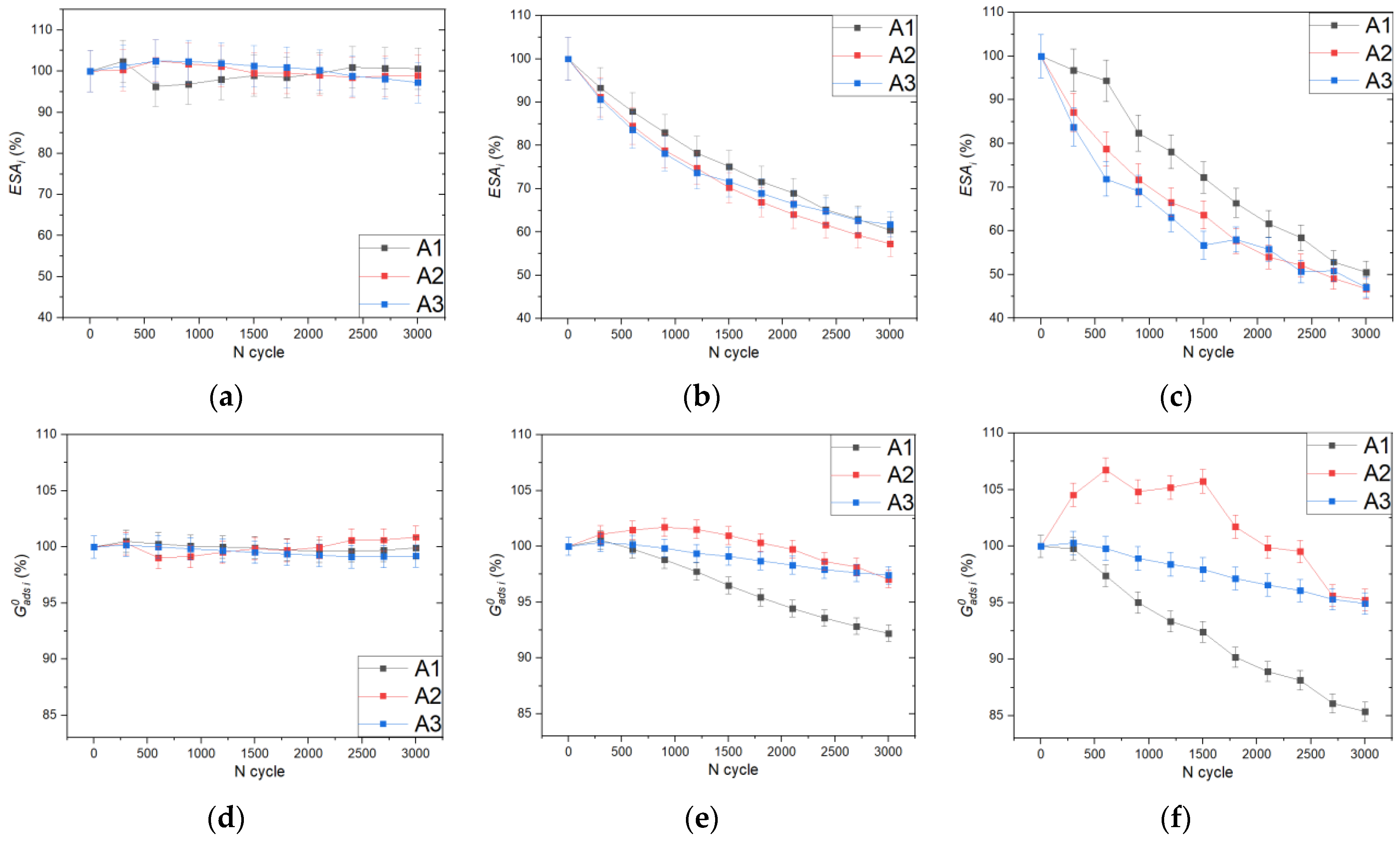
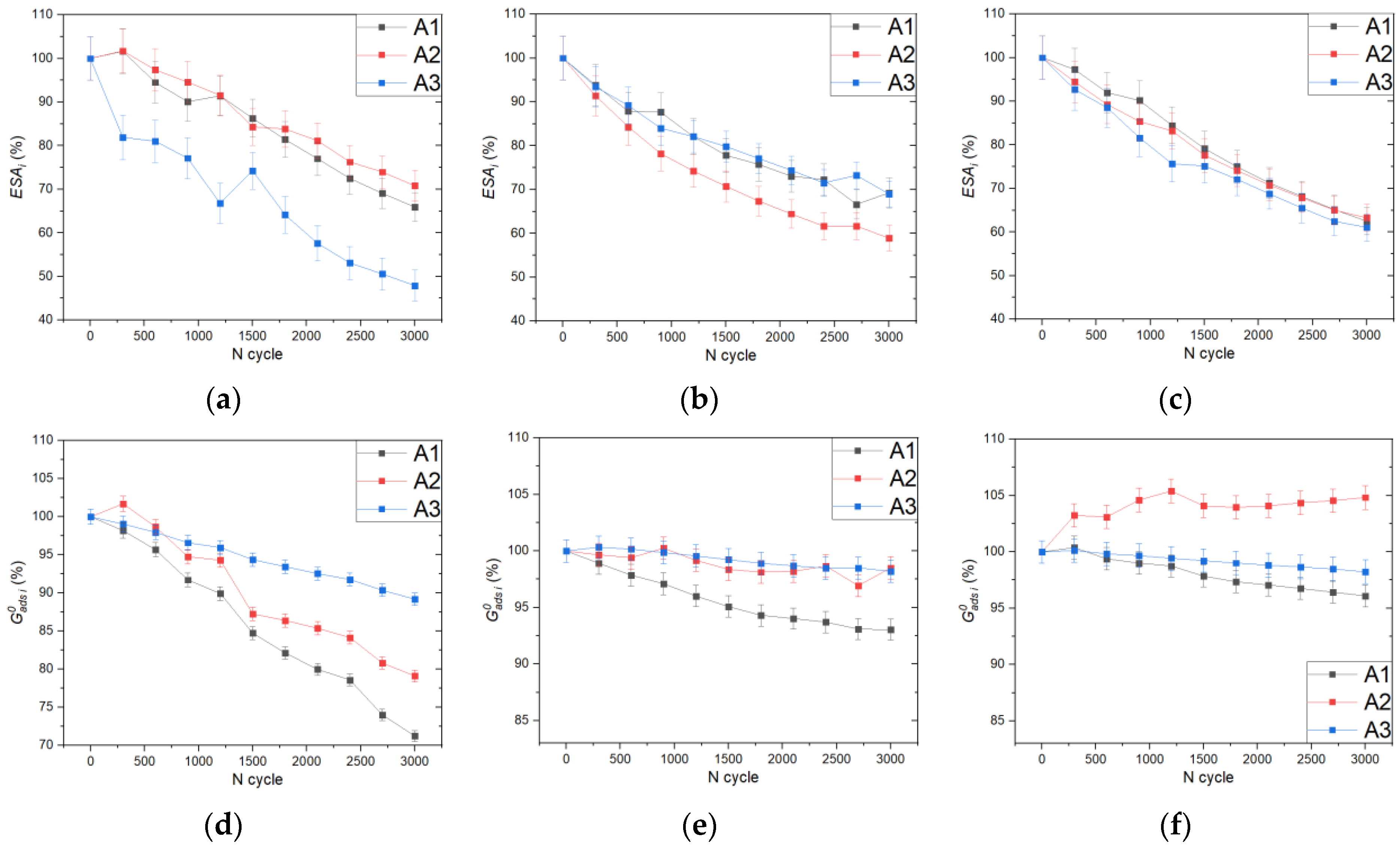
2. Results and Discussion
3. Materials and Methods
3.1. Synthesis of Electrocatalysts
3.2. Electrochemical Studies
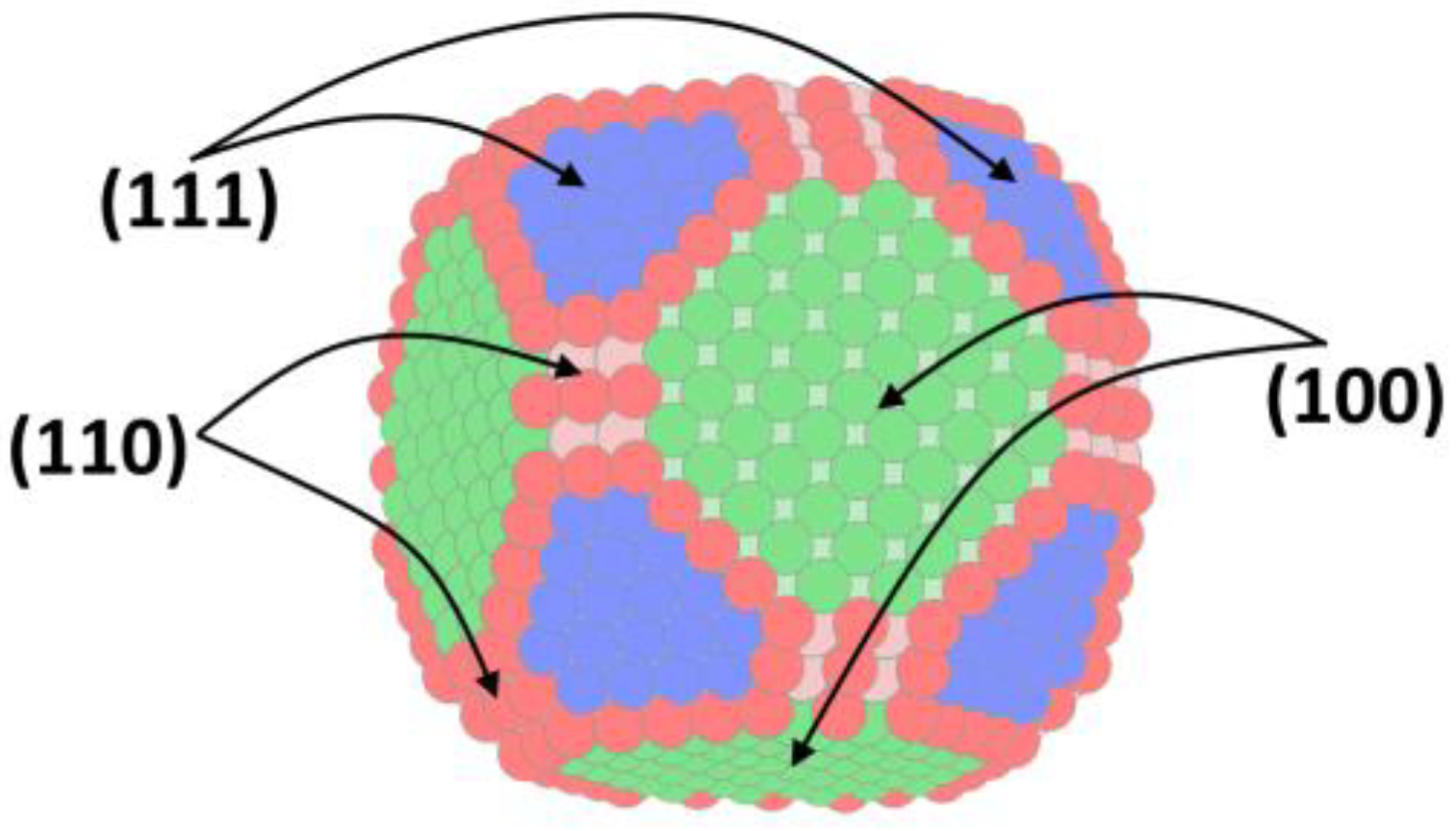
4. Theory
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning |
| PEM | Polymer electrolyte membrane |
| CNTs | Carbon nanotubes |
| CNFs | Carbon nanofibers |
| RGO | Reduced graphene oxide |
| ESA | Electrochemical surface area |
| CV | Cyclic voltammogram |
| HER | Hydrogen evolution reaction |
| HOR | Hydrogen oxidation reaction |
| AST | Accelerated stress testing |
| RHE | Reversible hydrogen electrode |
| FWHM | Full width at half maximum |
References
- Ishaq, H.; Dincer, I.; Crawford, C. A Review on Hydrogen Production and Utilization: Challenges and Opportunities. Int. J. Hydrogen Energy 2022, 47, 26238–26264. [Google Scholar] [CrossRef]
- Grigoriev, A.S.; Skorlygin, V.V.; Grigoriev, S.A.; Melnik, D.A.; Filimonov, M.N. A Hybrid Power Plant Based on Renewables and Electrochemical Energy Storage and Generation Systems for Decentralized Electricity Supply of the Northern Territories. Int. J. Electrochem. Sci 2018, 13, 1822–1830. [Google Scholar] [CrossRef]
- Borup, R.L.; Kusoglu, A.; Neyerlin, K.C.; Mukundan, R.; Ahluwalia, R.K.; Cullen, D.A.; More, K.L.; Weber, A.Z.; Myers, D.J. Recent Developments in Catalyst-Related PEM Fuel Cell Durability. Curr. Opin. Electrochem. 2020, 21, 192–200. [Google Scholar] [CrossRef]
- Mamat, M.S.; Grigoriev, S.A.; Dzhus, K.A.; Grant, D.M.; Walker, G.S. The Performance and Degradation of Pt Electrocatalysts on Novel Carbon Carriers for PEMFC Applications. Int. J. Hydrogen Energy 2010, 35, 7580–7587. [Google Scholar] [CrossRef]
- Mardle, P.; Ji, X.; Wu, J.; Guan, S.; Dong, H.; Du, S. Thin Film Electrodes from Pt Nanorods Supported on Aligned N-CNTs for Proton Exchange Membrane Fuel Cells. Appl. Catal. B Environ. 2020, 260, 118031. [Google Scholar] [CrossRef]
- Baranov, I.E.; Grigoriev, S.A.; Ylitalo, D.; Fateev, V.N.; Nikolaev, I.I. Transfer Processes in PEM Fuel Cell: Influence of Electrode Structure. Int. J. Hydrogen Energy 2006, 31, 203–210. [Google Scholar] [CrossRef]
- Mensharapov, R.M.; Ivanova, N.A.; Zasypkina, A.A.; Spasov, D.D.; Sinyakov, M.V.; Grigoriev, S.A.; Fateev, V.N. Model Study of CNT-Based PEMFCs’ Electrocatalytic Layers. Catalysts 2022, 12, 1227. [Google Scholar] [CrossRef]
- Venkataraman, A.; Amadi, E.V.; Chen, Y.; Papadopoulos, C. Carbon Nanotube Assembly and Integration for Applications. Nanoscale Res. Lett. 2019, 14, 220. [Google Scholar] [CrossRef]
- MoghadamEsfahani, R.A.; Vankova, S.K.; Easton, E.B.; Ebralidze, I.I.; Specchia, S. A Hybrid Pt/NbO/CNTs Catalyst with High Activity and Durability for Oxygen Reduction Reaction in PEMFC. Renew. Energy 2020, 154, 913–924. [Google Scholar] [CrossRef]
- Park, G.-G.; Sohn, Y.-J.; Yim, S.-D.; Yang, T.-H.; Yoon, Y.-G.; Lee, W.-Y.; Eguchi, K.; Kim, C.-S. Adoption of Nano-Materials for the Micro-Layer in Gas Diffusion Layers of PEMFCs. J. Power Sources 2006, 163, 113–118. [Google Scholar] [CrossRef]
- Kannan, A.M.; Munukutla, L. Carbon Nano-Chain and Carbon Nano-Fibers Based Gas Diffusion Layers for Proton Exchange Membrane Fuel Cells. J. Power Sources 2007, 167, 330–335. [Google Scholar] [CrossRef]
- Sharma, S.; Pollet, B.G. Support Materials for PEMFC and DMFC Electrocatalysts—A Review. J. Power Sources 2012, 208, 96–119. [Google Scholar] [CrossRef]
- Grigoriev, S.A.; Fateev, V.N.; Pushkarev, A.S.; Pushkareva, I.V.; Ivanova, N.A.; Kalinichenko, V.N.; Presnyakov, M.Y.; Wei, X. Reduced Graphene Oxide and Its Modifications as Catalyst Supports and Catalyst Layer Modifiers for PEMFC. Materials 2018, 11, 1405. [Google Scholar] [CrossRef] [PubMed]
- Shaari, N.; Kamarudin, S.K. Graphene in Electrocatalyst and Proton Conductiong Membrane in Fuel Cell Applications: An Overview. Renew. Sustain. Energy Rev. 2017, 69, 862–870. [Google Scholar] [CrossRef]
- Hsieh, C.-T.; Wei, J.-M.; Lin, J.-S.; Chen, W.-Y. Pulse Electrodeposition of Pt Nanocatalysts on Graphene-Based Electrodes for Proton Exchange Membrane Fuel Cells. Catal. Commun. 2011, 16, 220–224. [Google Scholar] [CrossRef]
- Hou, J.; Liu, Z.; Zhang, P. A New Method for Fabrication of Graphene/Polyaniline Nanocomplex Modified Microbial Fuel Cell Anodes. J. Power Sources 2013, 224, 139–144. [Google Scholar] [CrossRef]
- Mariani, M.; Latorrata, S.; Patrignani, S.; Stampino, P.G.; Dotelli, G. Characterization of Novel Graphene-Based Microporous Layers for Polymer Electrolyte Membrane Fuel Cells Operating under Low Humidity and High Temperature. Int. J. Hydrogen Energy 2020, 45, 7046–7058. [Google Scholar] [CrossRef]
- Han, T.H.; Mohapatra, D.; Mahato, N.; Parida, S.; Shim, J.H.; Nguyen, A.T.N.; Cho, M.H.; Shim, J.-J. Effect of Nitrogen Doping on the Catalytic Activity of Carbon Nano-Onions for the Oxygen Reduction Reaction in Microbial Fuel Cells. J. Ind. Eng. Chem. 2020, 81, 269–277. [Google Scholar] [CrossRef]
- Yeon, J.H.; Park, S.J.; Choi, I.; Choi, M. Generation of Carbon Nano-Onions by Laser Irradiation of Gaseous Hydrocarbons for High Durability Catalyst Support in Proton Exchange Membrane Fuel Cells. J. Ind. Eng. Chem. 2019, 80, 65–73. [Google Scholar] [CrossRef]
- Fu, X.; Hassan, F.M.; Zamani, P.; Jiang, G.; Higgins, D.C.; Choi, J.-Y.; Wang, X.; Xu, P.; Liu, Y.; Chen, Z. Engineered Architecture of Nitrogenous Graphene Encapsulating Porous Carbon with Nano-Channel Reactors Enhancing the PEM Fuel Cell Performance. Nano Energy 2017, 42, 249–256. [Google Scholar] [CrossRef]
- Zhu, J.; Xiao, M.; Song, P.; Fu, J.; Jin, Z.; Ma, L.; Ge, J.; Liu, C.; Chen, Z.; Xing, W. Highly Polarized Carbon Nano-Architecture as Robust Metal-Free Catalyst for Oxygen Reduction in Polymer Electrolyte Membrane Fuel Cells. Nano Energy 2018, 49, 23–30. [Google Scholar] [CrossRef]
- Morrow, B.H.; Striolo, A. Platinum Nanoparticles on Carbonaceous Materials: The Effect of Support Geometry on Nanoparticle Mobility, Morphology, and Melting. Nanotechnology 2008, 19, 195711. [Google Scholar] [CrossRef] [PubMed]
- Solla-Gullón, J.; Rodríguez, P.; Herrero, E.; Aldaz, A.; Feliu, J.M. Surface Characterization of Platinum Electrodes. Phys. Chem. Chem. Phys. 2008, 10, 1359–1373. [Google Scholar] [CrossRef] [PubMed]
- Hofman, M.S.; Wang, D.Z.; Yang, Y.; Koel, B.E. Interactions of Incident H Atoms with Metal Surfaces. Surf. Sci. Rep. 2018, 73, 153–189. [Google Scholar] [CrossRef]
- Pang, X.-Y.; Xue, L.-Q.; Wang, G.-C. Adsorption of Atoms on Cu Surfaces: A Density Functional Theory Study. Langmuir 2007, 23, 4910–4917. [Google Scholar] [CrossRef]
- Shi, Q.; Sun, R. Adsorption Manners of Hydrogen on Pt (1 0 0), (1 1 0) and (1 1 1) Surfaces at High Coverage. Comput. Theor. Chem. 2017, 1106, 43–49. [Google Scholar] [CrossRef]
- Ferrin, P.; Kandoi, S.; Nilekar, A.U.; Mavrikakis, M. Hydrogen Adsorption, Absorption and Diffusion on and in Transition Metal Surfaces: A DFT Study. Surf. Sci. 2012, 606, 679–689. [Google Scholar] [CrossRef]
- Gremaud, R.; Slaman, M.; Schreuders, H.; Dam, B.; Griessen, R. An Optical Method to Determine the Thermodynamics of Hydrogen Absorption and Desorption in Metals. Appl. Phys. Lett. 2007, 91, 231916. [Google Scholar] [CrossRef]
- Faglioni, F.; Goddard, W.A., III. Energetics of Hydrogen Coverage on Group VIII Transition Metal Surfaces and a Kinetic Model for Adsorption/Desorption. J. Chem. Phys. 2005, 122, 14704. [Google Scholar] [CrossRef]
- Zhai, F.; Li, Y.; Yang, Y.; Jiang, S.; Shen, X. Abnormal Subsurface Hydrogen Diffusion Behaviors in Heterogeneous Hydrogenation Reactions. J. Chem. Phys. 2018, 149, 174704. [Google Scholar] [CrossRef]
- Shen, X.; Chen, J.; Sun, Y.M.; Liang, T. Hydrogen Diffusion on Fe Surface and into Subsurface from First Principles. Surf. Sci. 2016, 654, 48–55. [Google Scholar] [CrossRef]
- Li, Y.; Zhai, F.; Xiao, Z.; Zhang, X.; Shen, X. Enhanced Diffusion and Permeation of Hydrogen Species on the Partially Carbon Covered Iron Surfaces. Appl. Surf. Sci. 2020, 515, 145899. [Google Scholar] [CrossRef]
- Weng, M.H.; Chen, H.-T.; Wang, Y.-C.; Ju, S.-P.; Chang, J.-G.; Lin, M.-C. Kinetics and Mechanisms for the Adsorption, Dissociation, and Diffusion of Hydrogen in Ni and Ni/YSZ Slabs: A DFT Study. Langmuir 2012, 28, 5596–5605. [Google Scholar] [CrossRef] [PubMed]
- Clavilier, J.; El Achi, K.; Rodes, A. In Situ Probing of Step and Terrace Sites on Pt (S)-[n (111) × (111)] Electrodes. Chem. Phys. 1990, 141, 1–14. [Google Scholar] [CrossRef]
- Nørskov, J.K.; Bligaard, T.; Logadottir, A.; Kitchin, J.R.; Chen, J.G.; Pandelov, S.; Stimming, U. Trends in the Exchange Current for Hydrogen Evolution. J. Electrochem. Soc. 2005, 152, J23. [Google Scholar] [CrossRef]
- Marković, N.M.; Schmidt, T.J.; Grgur, B.N.; Gasteiger, H.A.; Behm, R.J.; Ross, P.N. Effect of Temperature on Surface Processes at the Pt (111)—Liquid Interface: Hydrogen Adsorption, Oxide Formation, and CO Oxidation. J. Phys. Chem. B 1999, 103, 8568–8577. [Google Scholar] [CrossRef]
- Marković, N.M.; Grgur, B.N.; Ross, P.N. Temperature-Dependent Hydrogen Electrochemistry on Platinum Low-Index Single-Crystal Surfaces in Acid Solutions. J. Phys. Chem. B 1997, 101, 5405–5413. [Google Scholar] [CrossRef]
- Lasia, A. Modeling of Hydrogen Upd Isotherms. J. Electroanal. Chem. 2004, 562, 23–31. [Google Scholar] [CrossRef]
- Rodes, A.; El Achi, K.; Zamakhchari, M.A.; Clavilier, J. Hydrogen Probing of Step and Terrace Sites on Pt (S)-[n (111) × (100)]. J. Electroanal. Chem. Interfacial Electrochem. 1990, 284, 245–253. [Google Scholar] [CrossRef]
- Kowalczyk, P.; Savard, S.; Lasia, A. Determination of the Adsorption Energy Distribution Function of Upd Hydrogen on Monocrystalline Platinum. J. Electroanal. Chem. 2004, 574, 41–47. [Google Scholar] [CrossRef]
- Dwivedi, S. Graphene Based Electrodes for Hydrogen Fuel Cells: A Comprehensive Review. Int. J. Hydrogen Energy 2022, 47, 41848–41877. [Google Scholar] [CrossRef]
- Wu, J.; Yuan, X.Z.; Martin, J.J.; Wang, H.; Zhang, J.; Shen, J.; Wu, S.; Merida, W. A Review of PEM Fuel Cell Durability: Degradation Mechanisms and Mitigation Strategies. J. Power Sources 2008, 184, 104–119. [Google Scholar] [CrossRef]
- Gerasimova, I.; Belenov, S.; Lyanguzov, N.; Pankov, I.; Tolstunov, M.; Pavlets, A. Role of the Potential Range during Stress Testing of Platinum-Containing Electrocatalysts at Elevated Temperature. Catalysts 2022, 12, 1179. [Google Scholar] [CrossRef]
- Pushkareva, I.V.; Pushkarev, A.S.; Kalinichenko, V.N.; Chumakov, R.G.; Soloviev, M.A.; Liang, Y.; Millet, P.; Grigoriev, S.A. Reduced Graphene Oxide-Supported Pt-Based Catalysts for PEM Fuel Cells with Enhanced Activity and Stability. Catalysts 2021, 11, 256. [Google Scholar] [CrossRef]
- Liu, M.; Zhang, R.; Chen, W. Graphene-Supported Nanoelectrocatalysts for Fuel Cells: Synthesis, Properties, and Applications. Chem. Rev. 2014, 114, 5117–5160. [Google Scholar] [CrossRef]
- Rice, C.A.; Urchaga, P.; Pistono, A.O.; McFerrin, B.W.; McComb, B.T.; Hu, J. Platinum Dissolution in Fuel Cell Electrodes: Enhanced Degradation from Surface Area Assessment in Automotive Accelerated Stress Tests. J. Electrochem. Soc. 2015, 162, F1175–F1180. [Google Scholar] [CrossRef]
- Sharma, R.; Gyergyek, S.; Li, Q.; Andersen, S.M. Evolution of the Degradation Mechanisms with the Number of Stress Cycles during an Accelerated Stress Test of Carbon Supported Platinum Nanoparticles. J. Electroanal. Chem. 2019, 838, 82–88. [Google Scholar] [CrossRef]
- Kinoshita, K.; Lundquist, J.; Stonehart, P. Hydrogen Adsorption on High Surface Area Platinum Crystallites. J. Catal. 1973, 31, 325–334. [Google Scholar] [CrossRef]
- McCrum, I.T.; Bondue, C.J.; Koper, M.T. Hydrogen-Induced Step-Edge Roughening of Platinum Electrode Surfaces. J. Phys. Chem. Lett. 2019, 10, 6842–6849. [Google Scholar] [CrossRef]
- Diaz-Morales, O.; Hersbach, T.J.; Badan, C.; Garcia, A.C.; Koper, M.T. Hydrogen Adsorption on Nano-Structured Platinum Electrodes. Faraday Discuss. 2018, 210, 301–315. [Google Scholar] [CrossRef]
- Furuya, N.; Koide, S. Hydrogen Adsorption on Platinum Single-Crystal Surfaces. Surf. Sci. 1989, 220, 18–28. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA 3 for Three-Dimensional Visualization of Crystal, Volumetric and Morphology Data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]
- Santos, V.P.; Camara, G.A. Platinum Single Crystal Electrodes: Prediction of the Surface Structures of Low and High Miller Indexes Faces. Results Surf. Interfaces 2021, 3, 100006. [Google Scholar] [CrossRef]
- Søgaard, M.; Odgaard, M.; Skou, E.M. An Improved Method for the Determination of the Electrochemical Active Area of Porous Composite Platinum Electrodes. Solid State Ion. 2001, 145, 31–35. [Google Scholar] [CrossRef]
- Shao, M.; Peles, A.; Shoemaker, K. Electrocatalysis on Platinum Nanoparticles: Particle Size Effect on Oxygen Reduction Reaction Activity. Nano Lett. 2011, 11, 3714–3719. [Google Scholar] [CrossRef] [PubMed]
- Oudenhuijzen, M.K.; Bitter, J.H.; Koningsberger, D.C. The Nature of the Pt-H Bonding for Strongly and Weakly Bonded Hydrogen on Platinum. A XAFS Spectroscopy Study of the Pt- H Antibonding Shaperesonance and Pt-H EXAFS. J. Phys. Chem. B 2001, 105, 4616–4622. [Google Scholar] [CrossRef]
- Chaparro, A.M.; Martín, A.J.; Folgado, M.A.; Gallardo, B.; Daza, L. Comparative Analysis of the Electroactive Area of Pt/C PEMFC Electrodes in Liquid and Solid Polymer Contact by Underpotential Hydrogen Adsorption/Desorption. Int. J. Hydrogen Energy 2009, 34, 4838–4846. [Google Scholar] [CrossRef]
- Koper, M.T. Structure Sensitivity and Nanoscale Effects in Electrocatalysis. Nanoscale 2011, 3, 2054–2073. [Google Scholar] [CrossRef]
- Spasov, D.D.; Ivanova, N.A.; Pushkarev, A.S.; Pushkareva, I.V.; Presnyakova, N.N.; Chumakov, R.G.; Presnyakov, M.Y.; Grigoriev, S.A.; Fateev, V.N. On the Influence of Composition and Structure of Carbon-Supported Pt-SnO2 Hetero-Clusters onto Their Electrocatalytic Activity and Durability in PEMFC. Catalysts 2019, 9, 803. [Google Scholar] [CrossRef]
- Garsany, Y.; Singer, I.L.; Swider-Lyons, K.E. Impact of Film Drying Procedures on RDE Characterization of Pt/VC Electrocatalysts. J. Electroanal. Chem. 2011, 662, 396–406. [Google Scholar] [CrossRef]
- Garsany, Y.; Baturina, O.A.; Swider-Lyons, K.E.; Kocha, S.S. Experimental Methods for Quantifying the Activity of Platinum Electrocatalysts for the Oxygen Reduction Reaction. Anal. Chem. 2010, 82, 6321–6328. [Google Scholar] [CrossRef] [PubMed]
- Ivanova, N.A.; Alekseeva, O.K.; Fateev, V.N.; Shapir, B.L.; Spasov, D.D.; Nikitin, S.M.; Presnyakov, M.Y.; Kolobylina, N.N.; Soloviev, M.A.; Mikhalev, A.I. Activity and Durability of Electrocatalytic Layers with Low Platinum Loading Prepared by Magnetron Sputtering onto Gas Diffusion Electrodes. Int. J. Hydrogen Energy 2019, 44, 29529–29536. [Google Scholar] [CrossRef]
- Ivanova, N.A.; Spasov, D.D.; Zasypkina, A.A.; Alekseeva, O.K.; Kukueva, E.V.; Vorobyeva, E.A.; Kudinova, E.S.; Chumakov, R.G.; Millet, P.; Grigoriev, S.A. Comparison of the Performance and Durability of PEM Fuel Cells with Different Pt-Activated Microporous Layers. Int. J. Hydrogen Energy 2021, 46, 18093–18106. [Google Scholar] [CrossRef]
- Jerkiewicz, G. Hydrogen Sorption ATIN Electrodes. Prog. Surf. Sci. 1998, 57, 137–186. [Google Scholar] [CrossRef]
- Armand, D.; Clavilier, J. Quantitative Analysis of the Distribution of the Hydrogen Adsorption States at Platinum Surfaces: Part I. Application to Pt (100) in Sulphuric Acid Medium. J. Electroanal. Chem. Interfacial Electrochem. 1987, 225, 205–214. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrocatalyst | ESA, m2 g–1 | d, nm |
|---|---|---|
| Pt black | 4 | 71.1 |
| Pt40/C | 45 | 6.2 |
| Pt20/C | 51 | 5.5 |
| Pt20/CNT | 43 | 6.5 |
| Pt20/CNF | 52 | 5.4 |
| Pt20/RGO | 35 | 8.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mensharapov, R.M.; Spasov, D.D.; Ivanova, N.A.; Zasypkina, A.A.; Smirnov, S.A.; Grigoriev, S.A. Screening of Carbon-Supported Platinum Electrocatalysts Using Frumkin Adsorption Isotherms. Inorganics 2023, 11, 103. https://doi.org/10.3390/inorganics11030103
Mensharapov RM, Spasov DD, Ivanova NA, Zasypkina AA, Smirnov SA, Grigoriev SA. Screening of Carbon-Supported Platinum Electrocatalysts Using Frumkin Adsorption Isotherms. Inorganics. 2023; 11(3):103. https://doi.org/10.3390/inorganics11030103
Chicago/Turabian StyleMensharapov, Ruslan M., Dmitry D. Spasov, Nataliya A. Ivanova, Adelina A. Zasypkina, Sergey A. Smirnov, and Sergey A. Grigoriev. 2023. "Screening of Carbon-Supported Platinum Electrocatalysts Using Frumkin Adsorption Isotherms" Inorganics 11, no. 3: 103. https://doi.org/10.3390/inorganics11030103
APA StyleMensharapov, R. M., Spasov, D. D., Ivanova, N. A., Zasypkina, A. A., Smirnov, S. A., & Grigoriev, S. A. (2023). Screening of Carbon-Supported Platinum Electrocatalysts Using Frumkin Adsorption Isotherms. Inorganics, 11(3), 103. https://doi.org/10.3390/inorganics11030103