
Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction


Abstract
1. Introduction
2. Experimental Details
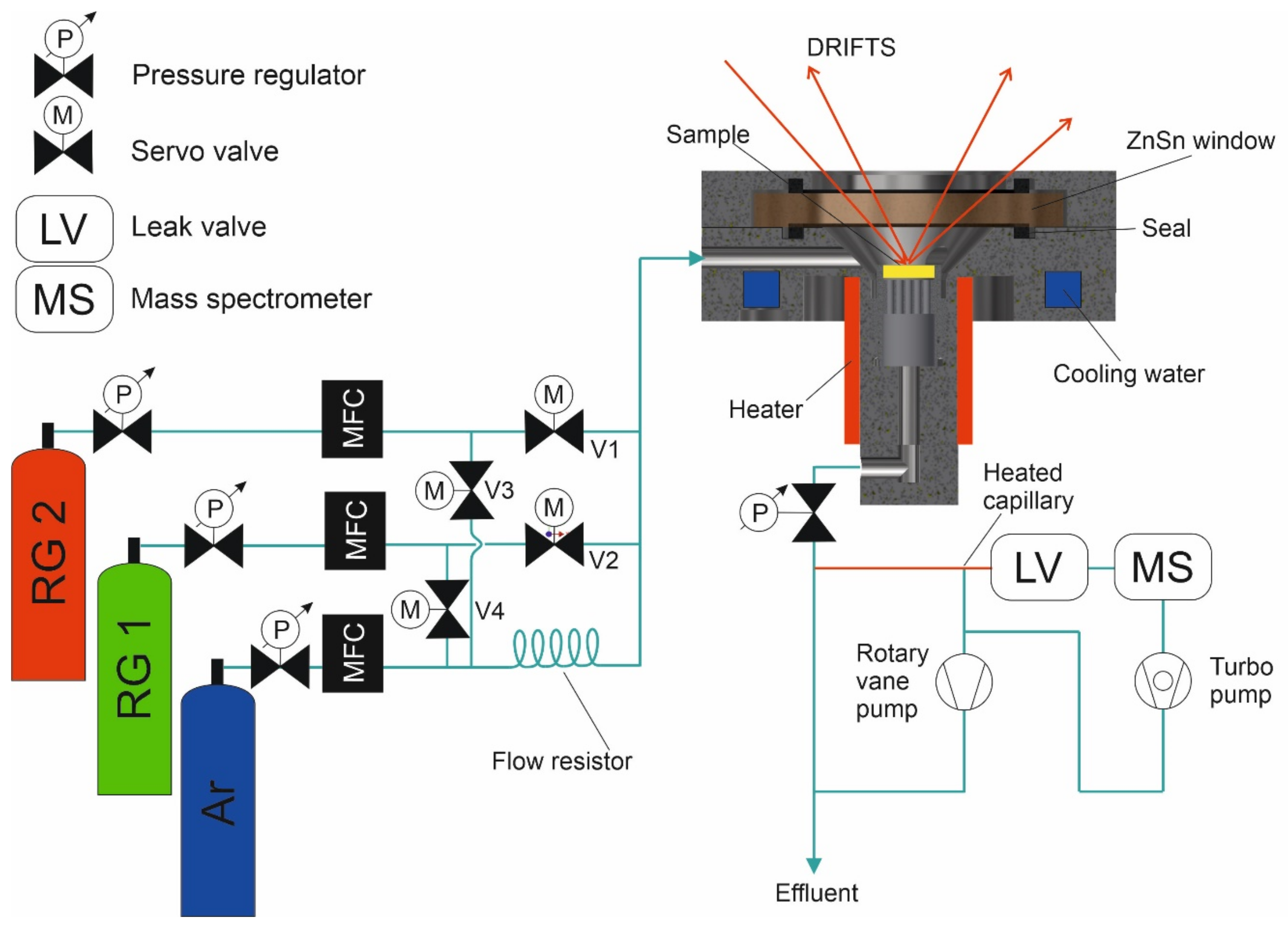
2.1. In Situ DRIFTS Cell
2.2. Sample Preparation and Characterization
2.3. Reaction Conditions
3. Experimental Results
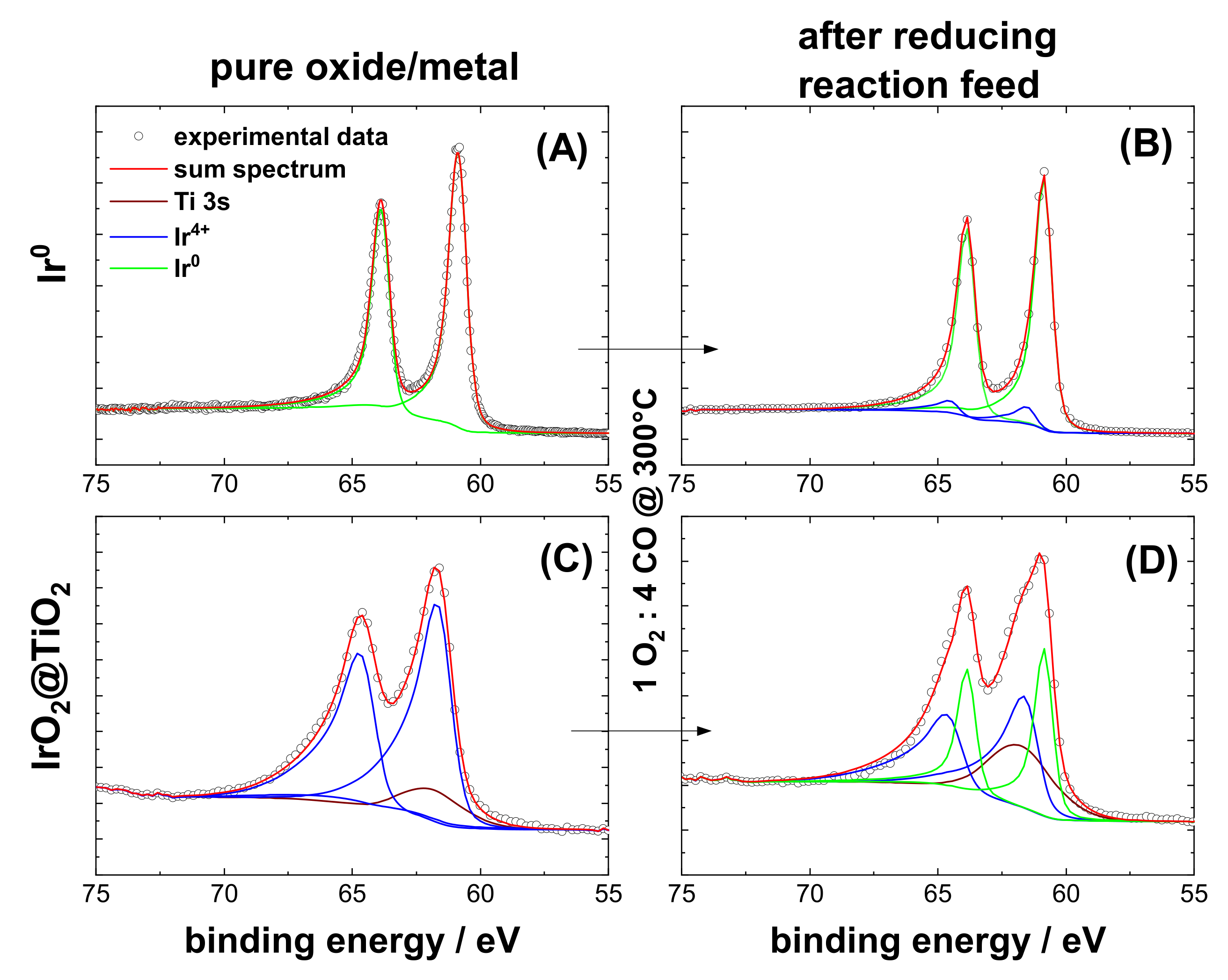
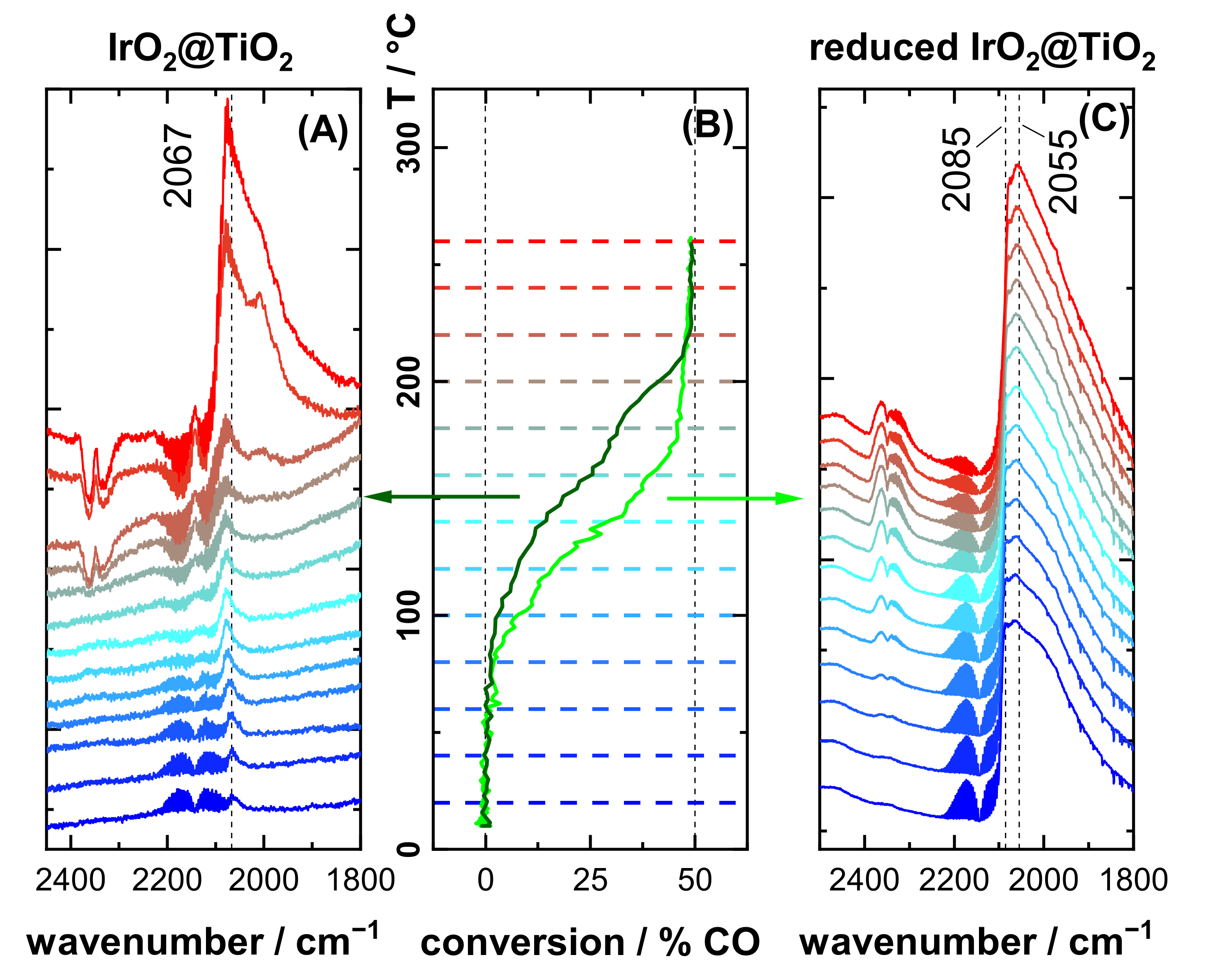
3.1. Characterization of Pre-Oxidized and Pre-Reduced IrO2@TiO2 and Ir0 + TiO2 Samples
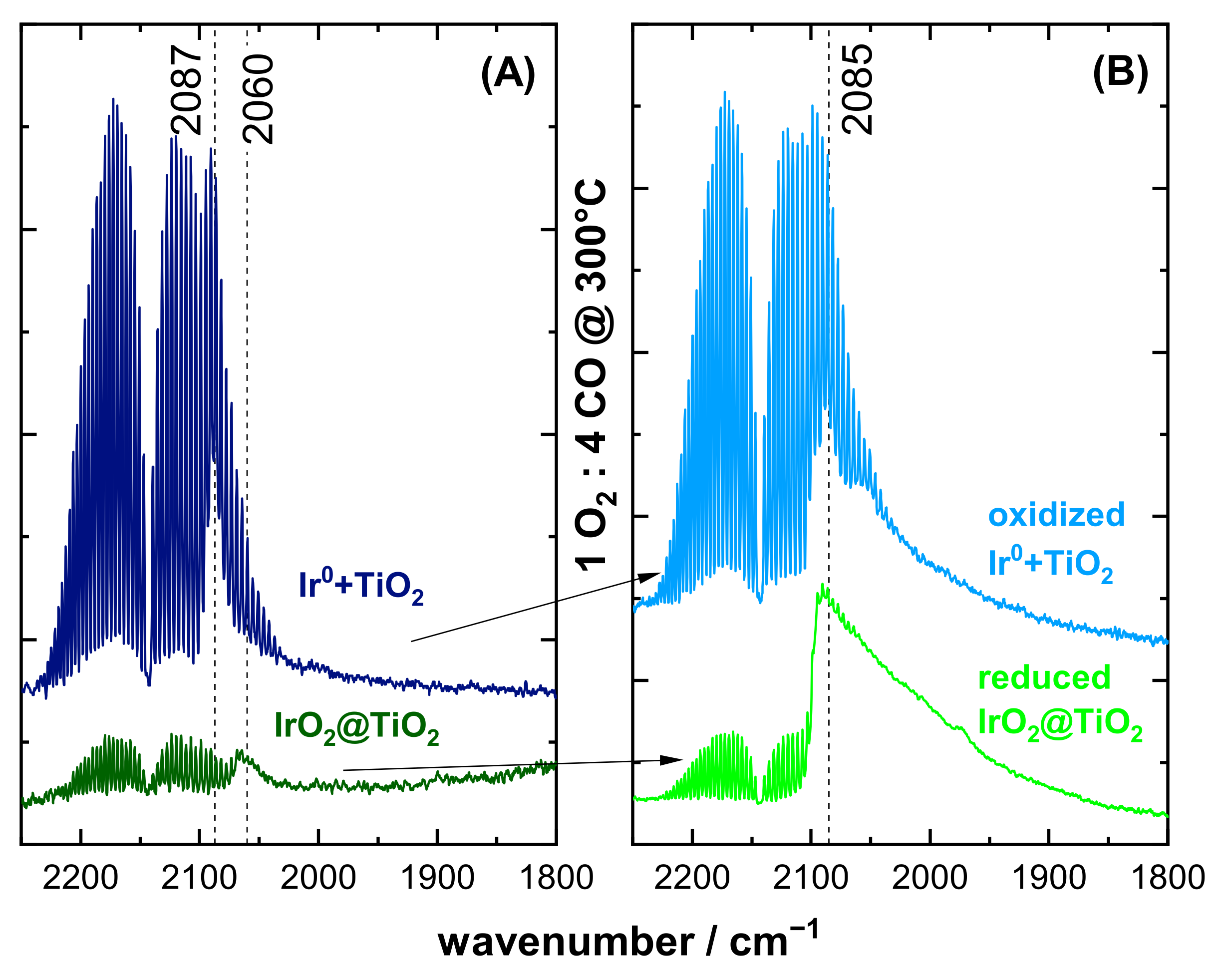
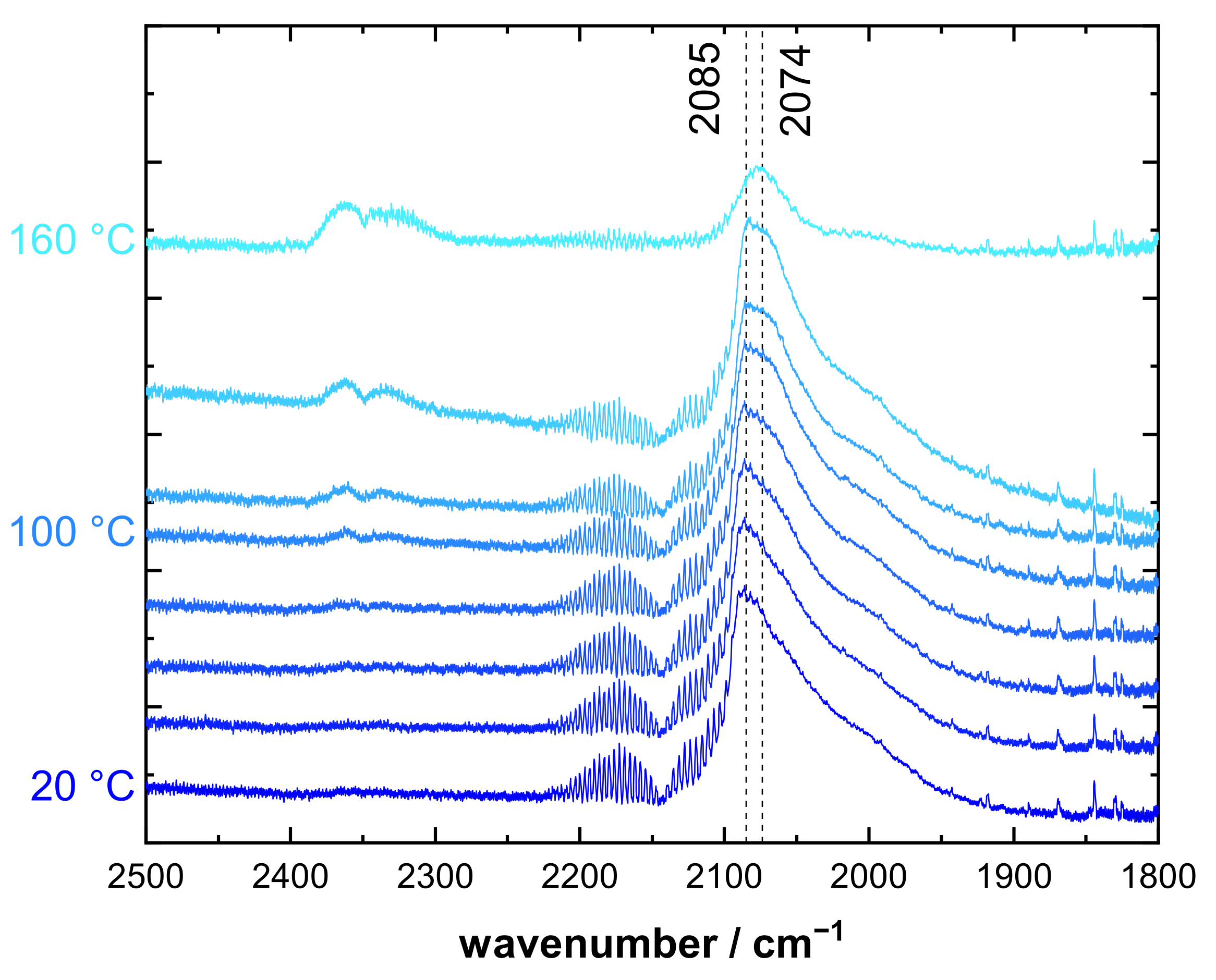
3.2. CO DRIFTS Experiments of Oxidized and Reduced IrO2@TiO2 and Ir0 + TiO2 Samples
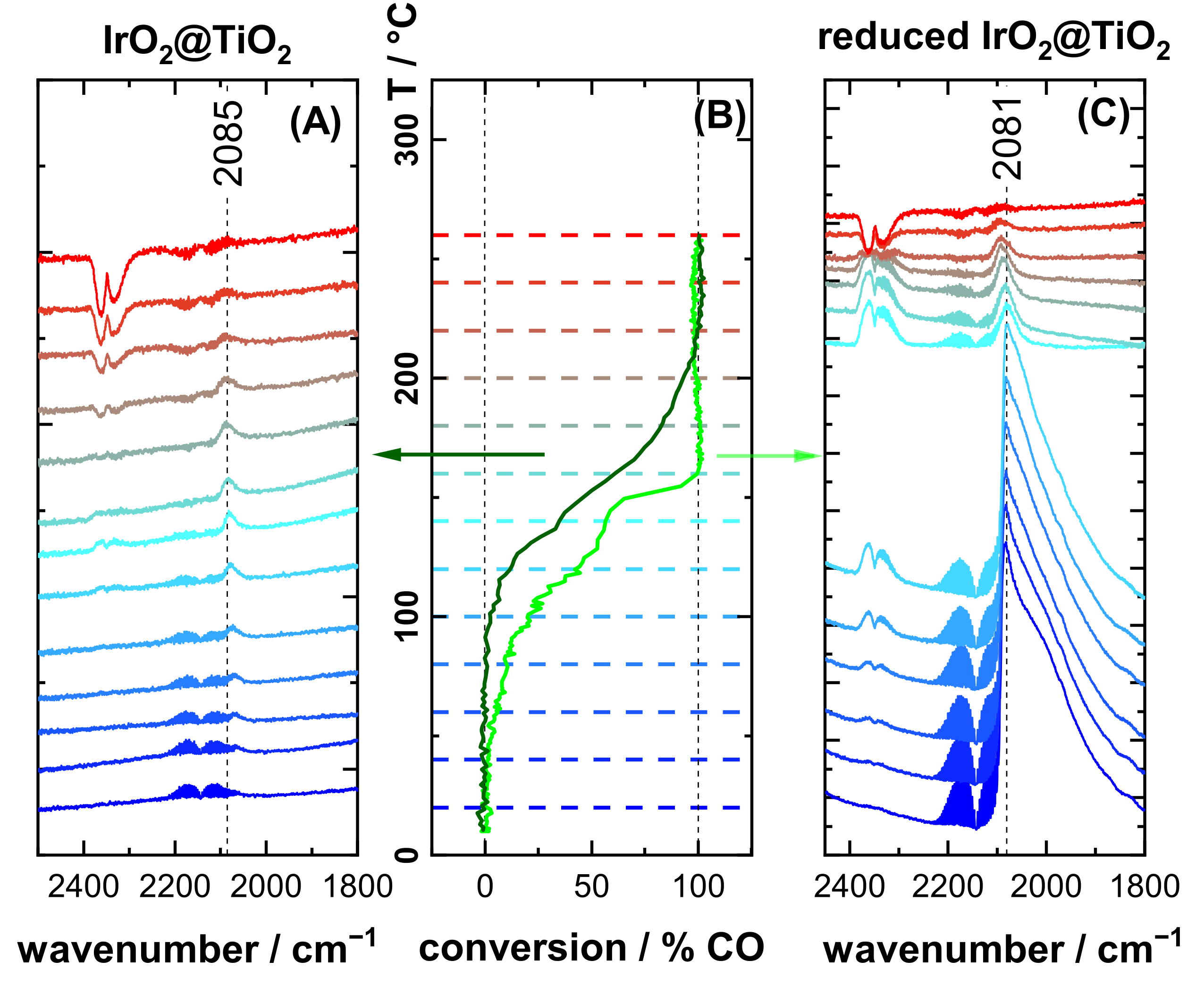
3.3. CO Oxidation Experiments of IrO2@TiO2 Samples
4. Discussion
4.1. CO as a Probe Molecule for the Surface Oxidation State of IrO2
4.2. Case Study: Catalytic CO Oxidation
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Over, H.; Balmes, O.; Lundgren, E. Direct Comparison of the Reactivity of the Non-Oxidic Phase of Ru(0001) and the RuO2 Phase in the CO Oxidation Reaction. Surf. Sci. 2009, 603, 298–303. [Google Scholar] [CrossRef]
- Gao, F.; Goodman, D.W. CO Oxidation over Ruthenium: Identification of the Catalytically Active Phases at near-Atmospheric Pressures. Phys. Chem. Chem. Phys. 2012, 14, 6688–6697. [Google Scholar] [CrossRef]
- Gustafson, J.; Balmes, O.; Zhang, C.; Shipilin, M.; Schaefer, A.; Hagman, B.; Merte, L.R.; Martin, N.M.; Carlsson, P.A.; Jankowski, M.; et al. The Role of Oxides in Catalytic CO Oxidation over Rhodium and Palladium. ACS Catal. 2018, 8, 4438–4445. [Google Scholar] [CrossRef]
- Martin, R.; Kim, M.; Lee, C.J.; Mehar, V.; Albertin, S.; Hejral, U.; Merte, L.R.; Asthagiri, A.; Weaver, J.F. Isothermal Reduction of IrO2(110) Films by Methane Investigated Using in Situ x-Ray Photoelectron Spectroscopy. ACS Catal. 2021, 11, 5004–5016. [Google Scholar] [CrossRef]
- Weaver, J.F. Surface Chemistry of Late Transition Metal Oxides. Chem. Rev. 2013, 113, 4164–4215. [Google Scholar] [CrossRef]
- Schlögl, R. Heterogeneous Catalysis. Angew. Chemie Int. Ed. 2015, 54, 3465–3520. [Google Scholar] [CrossRef]
- Weckhuysen, B.M. Determining the Active Site in a Catalytic Process: Operando Spectroscopy Is More than a Buzzword. Phys. Chem. Chem. Phys. 2003, 5, 4351–4360. [Google Scholar] [CrossRef]
- Grunwaldt, J.D.; Baiker, A. In Situ Spectroscopic Investigation of Heterogeneous Catalysts and Reaction Media at High Pressure. Phys. Chem. Chem. Phys. 2005, 7, 3526–3539. [Google Scholar] [CrossRef]
- Ryczkowski, J. IR Spectroscopy in Catalysis. Catal. Today 2001, 68, 263–381. [Google Scholar] [CrossRef]
- Zaera, F. Infrared Absorption Spectroscopy of Adsorbed CO: New Applications in Nanocatalysis for an Old Approach. ChemCatChem 2012, 4, 1525–1533. [Google Scholar] [CrossRef]
- Meunier, F.C. Relevance of IR Spectroscopy of Adsorbed CO for the Characterization of Heterogeneous Catalysts Containing Isolated Atoms. J. Phys. Chem. C 2021, 125, 21810–21823. [Google Scholar] [CrossRef]
- Hoffman, F.M. Infrared Reflection-Absorption Spectroscopy of Adsorbed Molecules. Surf. Sci. Rep. 1983, 3, 107–192. [Google Scholar] [CrossRef]
- Hollins, P.; Pritchard, J. Infrared Studies of Chemisorbed Layers on Single Crystals. Prog. Surf. Sci. 1985, 19, 275–349. [Google Scholar] [CrossRef]
- Villegas, I.; Weaver, M.J. Modeling Electrochemical Interfaces in Ultrahigh Vacuum: Molecular Roles of Solvation in Double-Layer Phenomena. J. Phys. Chem. B 1997, 101, 10166–10177. [Google Scholar] [CrossRef]
- Van Santen, R.A.; Tranca, I.; Hensen, E.J.M. Theory of Surface Chemistry and Reactivity of Reducible Oxides. Catal. Today 2015, 244, 63–84. [Google Scholar] [CrossRef]
- Khalid, O.; Spriewald Luciano, A.; Drazic, G.; Over, H. Mixed RuxIr1−xO2 Supported on Rutile TiO2: Catalytic Methane Combustion, a Model Study. ChemCatChem 2021, 13, 3983–3994. [Google Scholar] [CrossRef]
- Martin, R.; Lee, C.J.; Mehar, V.; Kim, M.; Asthagiri, A.; Weaver, J.F. Catalytic Oxidation of Methane on IrO2 (110) Films Investigated Using Ambient-Pressure X-Ray Photoelectron Spectroscopy. ACS Catal. 2022, 12, 2840–2853. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, W.; Khalid, O.; Weber, T.; Luciano, A.S.; Zhan, W.; Smarsly, B.M.; Over, H. Supported RuxIr1-xO2 Mixed Oxides Catalysts for Propane Combustion: Resistance Against Water Poisoning. ChemCatChem 2022, 14, e202200149. [Google Scholar] [CrossRef]
- Assmann, J.; Narkhede, V.; Breuer, N.A.; Muhler, M.; Seitsonen, A.P.; Knapp, M.; Crihan, D.; Farkas, A.; Mellau, G.; Over, H. Heterogeneous Oxidation Catalysis on Ruthenium: Bridging the Pressure and Materials Gaps and Beyond. J. Phys. Condens. Matter 2008, 20, 184017. [Google Scholar] [CrossRef]
- Khalid, O.; Weber, T.; Drazic, G.; Djerdj, I.; Over, H. Mixed RuxIr1-xO2 Oxide Catalyst with Well-Defined and Varying Composition Applied to CO Oxidation. J. Phys. Chem. C 2020, 124, 18670–18683. [Google Scholar] [CrossRef]
- Trasatti, S. Electrocatalysis: Understanding the Success of DSA®. Electrochim. Acta 2000, 45, 2377–2385. [Google Scholar] [CrossRef]
- Kötz, R.; Stucki, S. Stabilization of RuO2 by IrO2 for Anodic Oxygen Evolution in Acid Media. Electrochim. Acta 1986, 31, 1311–1316. [Google Scholar] [CrossRef]
- Over, H. Atomic Scale Insights into Electrochemical versus Gas Phase Oxidation of HCl over RuO2-Based Catalysts: A Comparative Review. Electrochim. Acta 2013, 93, 314–333. [Google Scholar] [CrossRef]
- Over, H. Fundamental Studies of Planar Single-Crystalline Oxide Model Electrodes (RuO2, IrO2) for Acidic Water Splitting. ACS Catal. 2021, 11, 8848–8871. [Google Scholar] [CrossRef]
- Fabbri, E.; Habereder, A.; Waltar, K.; Kötz, R.; Schmidt, T.J. Developments and Perspectives of Oxide-Based Catalysts for the Oxygen Evolution Reaction. Catal. Sci. Technol. 2014, 4, 3800–3821. [Google Scholar] [CrossRef]
- Drochner, A.; Fehlings, M.; Krauß, K.; Vogel, H. A New DRIFTS Cell for the In-Situ Investigation of Heterogeneously Catalyzed Reactions. Chem. Eng. Technol. 2000, 23, 319–322. [Google Scholar] [CrossRef]
- Meunier, F.C. Pitfalls and Benefits of: In Situ and Operando Diffuse Reflectance FT-IR Spectroscopy (DRIFTS) Applied to Catalytic Reactions. React. Chem. Eng. 2016, 1, 134–141. [Google Scholar] [CrossRef]
- Freund, H.J.; Meijer, G.; Scheffler, M.; Schlögl, R.; Wolf, M. CO Oxidation as a Prototypical Reaction for Heterogeneous Processes. Angew. Chemie Int. Ed. 2011, 50, 10064–10094. [Google Scholar] [CrossRef]
- Abb, M.J.S.; Weber, T.; Glatthaar, L.; Over, H. Growth of Ultrathin Single-Crystalline IrO2(110) Films on a TiO2(110) Single Crystal. Langmuir 2019, 35, 7720–7726. [Google Scholar] [CrossRef]
- He, Y.; Langsdorf, D.; Li, L.; Over, H. Versatile Model System for Studying Processes Ranging from Heterogeneous to Photocatalysis: Epitaxial RuO2(110) on TiO2(110). J. Phys. Chem. C 2015, 119, 2692–2702. [Google Scholar] [CrossRef]
- Mamaca, N.; Mayousse, E.; Arrii-Clacens, S.; Napporn, T.W.; Servat, K.; Guillet, N.; Kokoh, K.B. Electrochemical Activity of Ruthenium and Iridium Based Catalysts for Oxygen Evolution Reaction. Appl. Catal. B Environ. 2012, 111–112, 376–380. [Google Scholar] [CrossRef]
- Terezo, A.J.; Pereira, E.C. Preparation and Characterization of Ti/RuO2 Anodes Obtained by Sol-Gel and Conventional Routes. Mater. Lett. 2002, 53, 339–345. [Google Scholar] [CrossRef]
- Reksten, A.; Sunde, S.; Seland, F.; Moradi, F. Iridium-Ruthenium Mixed Oxide for Oxygen Evolution Reaction Prepared By Pechini Synthesis. ECS Meet. Abstr. 2013, MA2013-02, 80. [Google Scholar] [CrossRef]
- Rosario, A.V.; Bulhoões, L.O.S.; Pereira, E.C. Investigation of Pseudocapacitive Properties of RuO2 Film Electrodes Prepared by Polymeric Precursor Method. J. Power Sources 2006, 158, 795–800. [Google Scholar] [CrossRef]
- Freakley, S.J.; Ruiz-Esquius, J.; Morgan, D.J. The X-Ray Photoelectron Spectra of Ir, IrO2 and IrCl3 Revisited. Surf. Interface Anal. 2017, 49, 794–799. [Google Scholar] [CrossRef]
- Abb, M.J.S.; Weber, T.; Langsdorf, D.; Koller, V.; Gericke, S.M.; Pfaff, S.; Busch, M.; Zetterberg, J.; Preobrajenski, A.; Grönbeck, H.; et al. Thermal Stability of Single-Crystalline IrO2(110) Layers: Spectroscopic and Adsorption Studies. J. Phys. Chem. C 2020, 124, 15324–15336. [Google Scholar] [CrossRef]
- Over, H. Surface Chemistry of Ruthenium Dioxide in Heterogeneous Catalysis and Electrocatalysis: From Fundamental to Applied Research. Chem. Rev. 2012, 112, 3356–3426. [Google Scholar] [CrossRef]
- Morgan, D.J. Resolving Ruthenium: XPS Studies of Common Ruthenium Materials. Surf. Interface Anal. 2015, 47, 1072–1079. [Google Scholar] [CrossRef]
- Solymosi, F.; Novák, É.; Molnár, A. Infrared Spectroscopic Study on CO-Induced Structural Changes of Iridium on an Alumina Support. J. Phys. Chem. 1990, 94, 7250–7255. [Google Scholar] [CrossRef]
- Solymosi, F.; Raskó, J. An Infrared Study of CO and NO Adsorption on Alumina-Supported Iridium Catalyst. J. Catal. 1980, 62, 253–263. [Google Scholar] [CrossRef]
- Lauterbach, J.; Boyle, R.W.; Schick, M.; Mitchell, W.J.; Meng, B.; Weinberg, W.H. The Adsorption of CO on Ir(111) Investigated with FT-IRAS. Surf. Sci. 1996, 350, 32–44. [Google Scholar] [CrossRef]
- Fujitani, T.; Nakamura, I.; Kobayashi, Y.; Takahashi, A.; Haneda, M.; Hamada, H. Adsorption and Reactions of NO on Clean and CO-Precovered Ir(111). J. Phys. Chem. B 2005, 109, 17603–17607. [Google Scholar] [CrossRef] [PubMed]
- Farkas, A.; Mellau, G.C.; Over, H. Novel Insight in the CO Oxidation on RuO2(110) by in Situ Reflection-Absorption Infrared Spectroscopy. J. Phys. Chem. C 2009, 113, 14341–14355. [Google Scholar] [CrossRef]
- Farkas, A.; Hess, F.; Over, H. “First-Principles” Kinetic Monte Carlo Simulations Revisited: CO Oxidation over RuO2(110). J. Comput. Chem. 2011, 33, 757–766. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reaction Conditions | Ar/% | O2/% | CO/% |
|---|---|---|---|
| Oxidizing | 96 | 2 | 2 |
| Reducing | 95 | 1 | 4 |
| CO only | 96 | - | 4 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Timmer, P.; Weber, T.; Glatthaar, L.; Over, H. Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction. Inorganics 2023, 11, 102. https://doi.org/10.3390/inorganics11030102
Timmer P, Weber T, Glatthaar L, Over H. Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction. Inorganics. 2023; 11(3):102. https://doi.org/10.3390/inorganics11030102
Chicago/Turabian StyleTimmer, Phillip, Tim Weber, Lorena Glatthaar, and Herbert Over. 2023. "Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction" Inorganics 11, no. 3: 102. https://doi.org/10.3390/inorganics11030102
APA StyleTimmer, P., Weber, T., Glatthaar, L., & Over, H. (2023). Operando CO Infrared Spectroscopy and On-Line Mass Spectrometry for Studying the Active Phase of IrO2 in the Catalytic CO Oxidation Reaction. Inorganics, 11(3), 102. https://doi.org/10.3390/inorganics11030102