
Magnetostructural D-Correlations and Their Impact on Single-Molecule Magnetism




Abstract
:
1. Introduction
- (a)
- The axial zero-field splitting (ZFS) parameter D
- (b)
- The (ortho)rhombic ZFS parameter E
2. Spin-Hamiltonian Formalism
- The D-tensor (tensor of the spin–spin interaction)
- The g-tensor (tensor of the magnetogyric ratio)
- The κ-tensor (tensor of the temperature-independent paramagnetism)
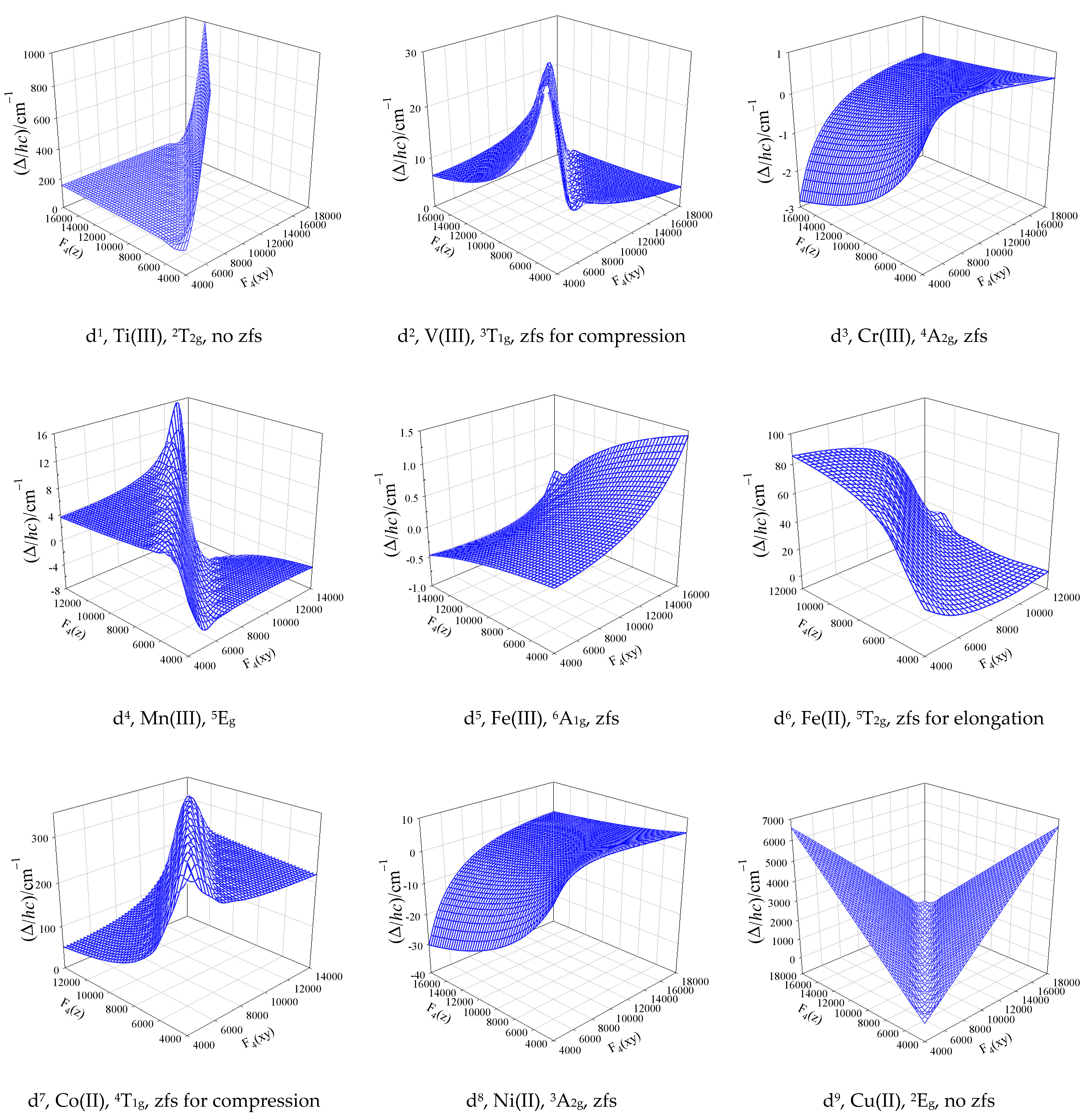
3. Electronic Structure of 3d-Complexes
- (a)
- The interelectron repulsion
- (b)
- The crystal-field potential
- (c)
- The spin–orbit interaction
- (d)
- The orbital Zeeman term
- (e)
- The spin Zeeman term
- (i)
- By diagonalizing the matrix
- (ii)
- By diagonalizing the matrix
- (iii)
- By diagonalizing the matrix
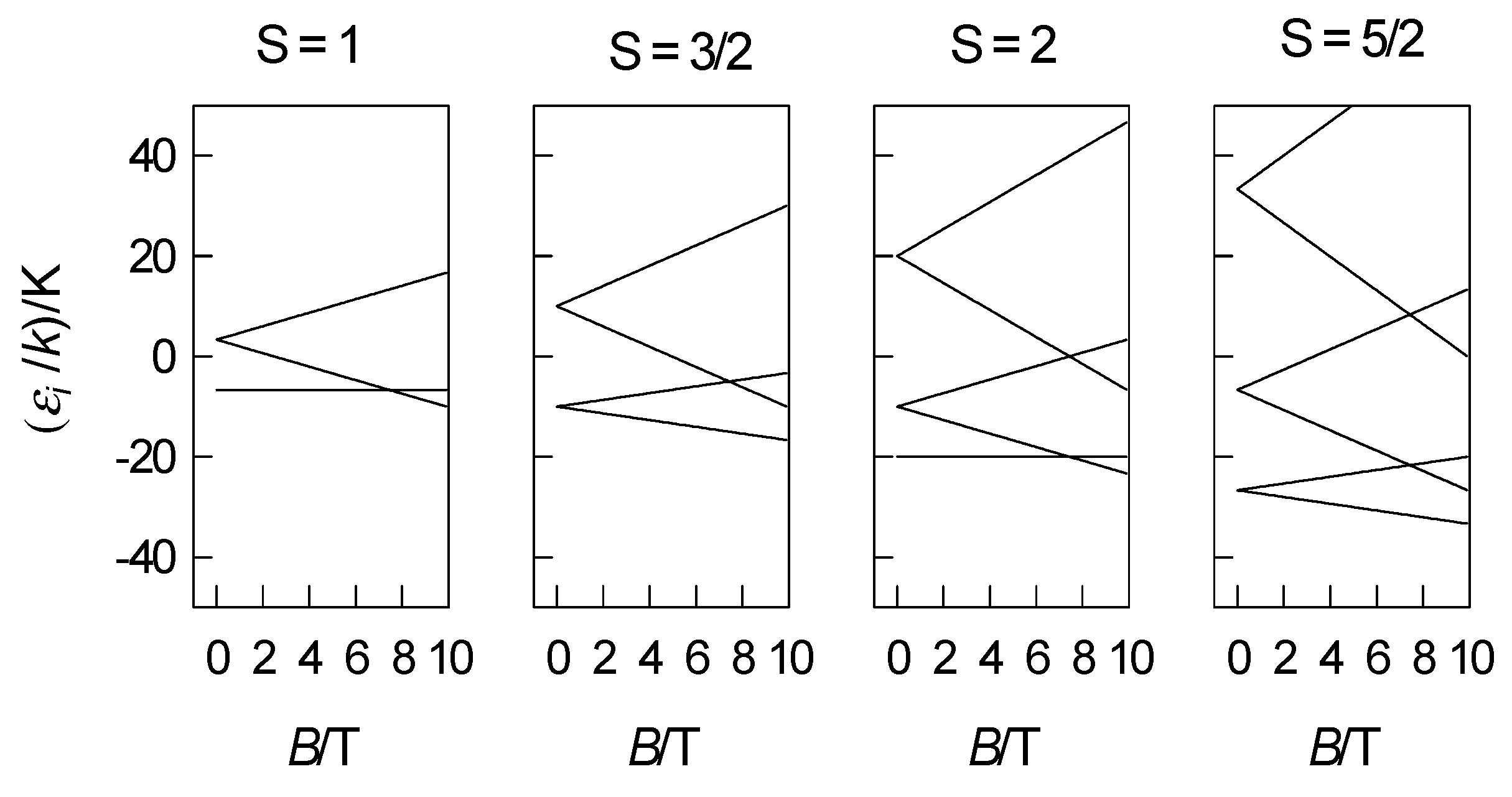
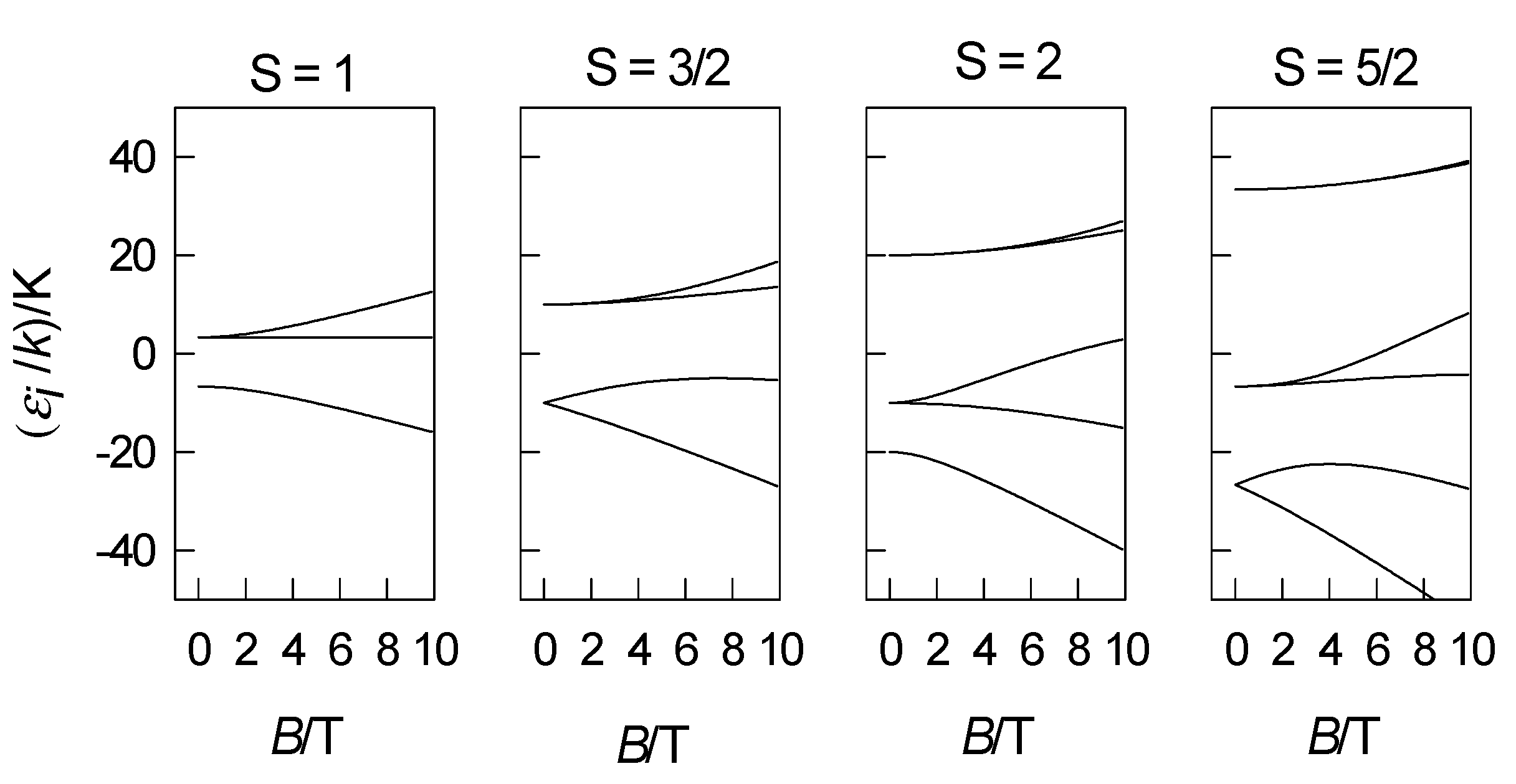
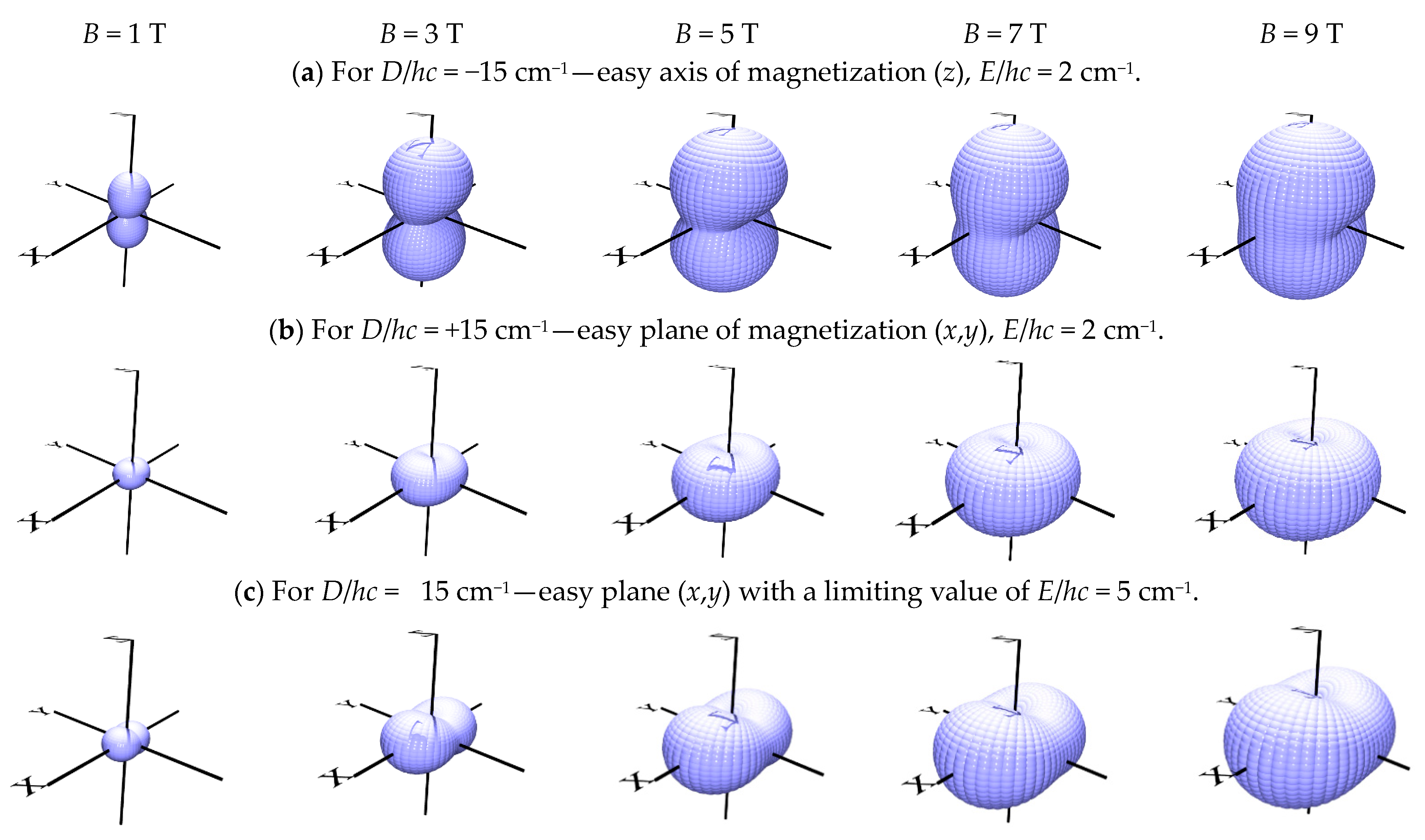
4. Zero-Field Splitting Parameters
5. Magnetostructural J-Correlations
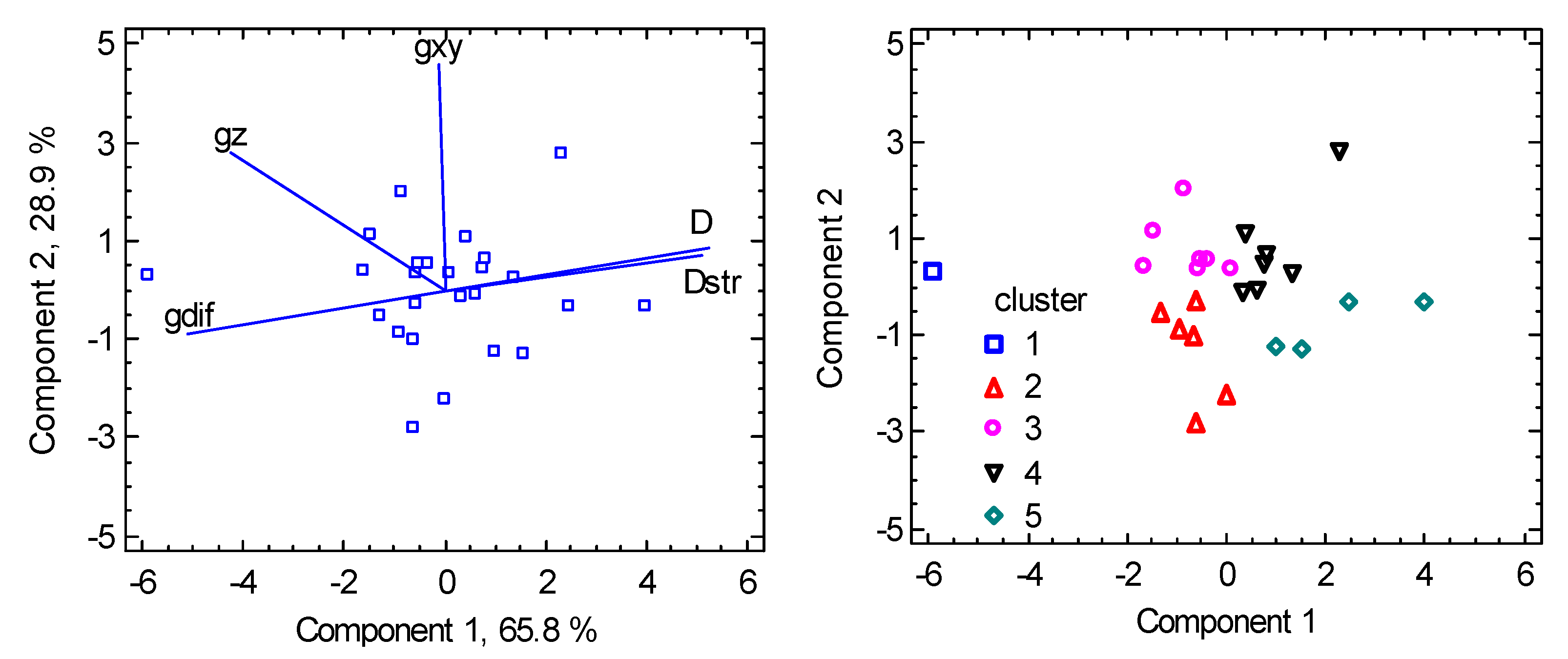
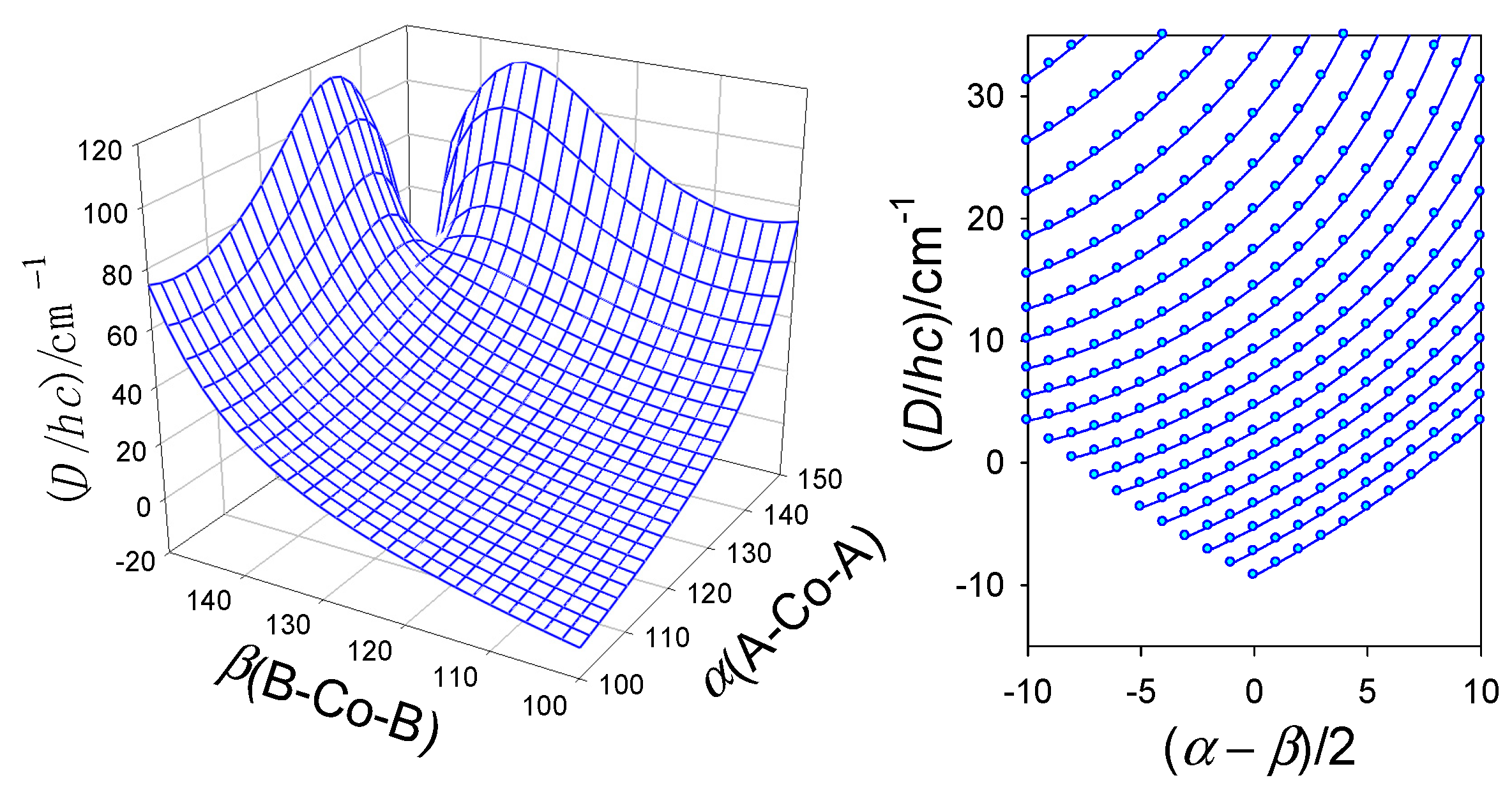
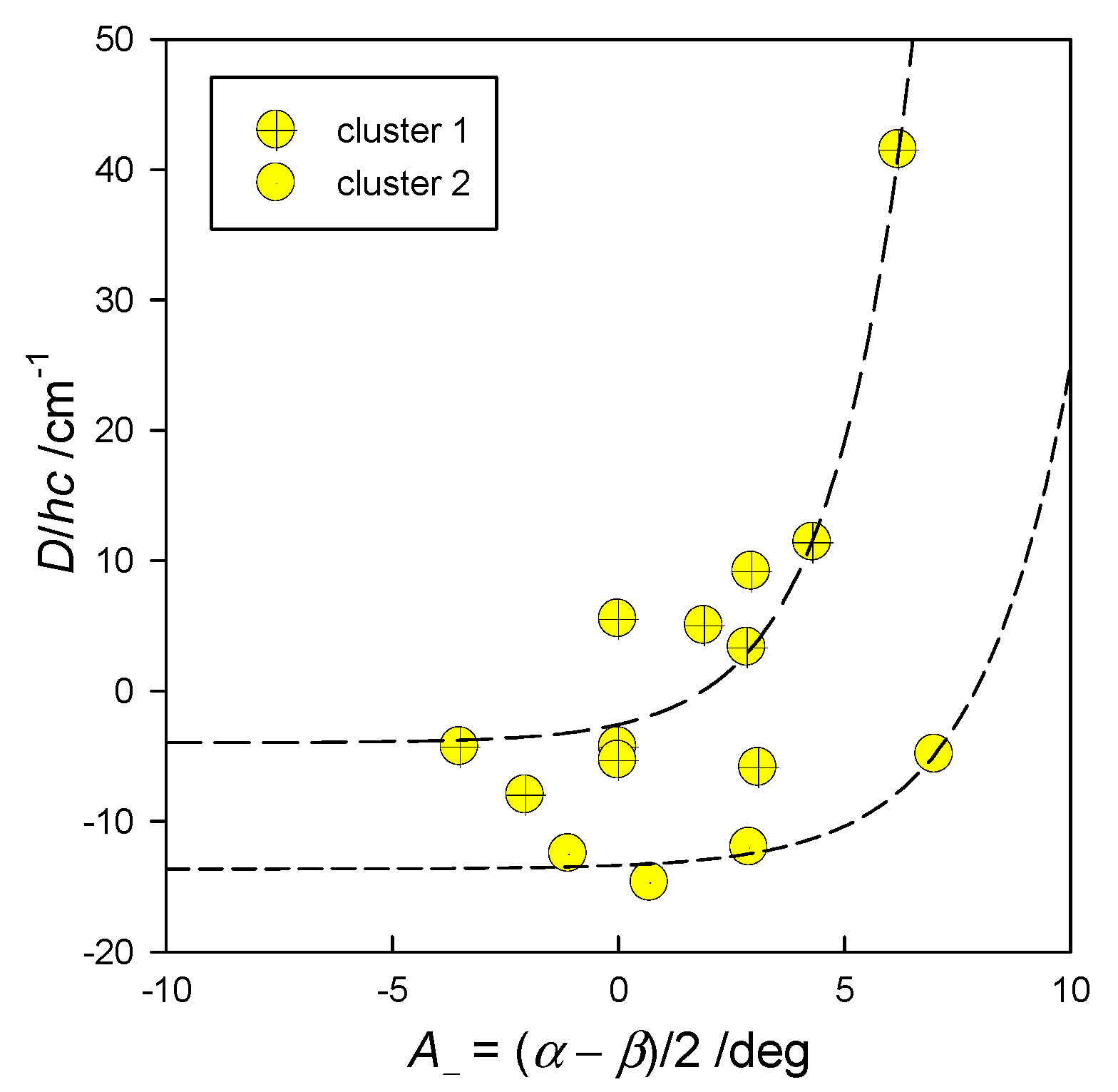
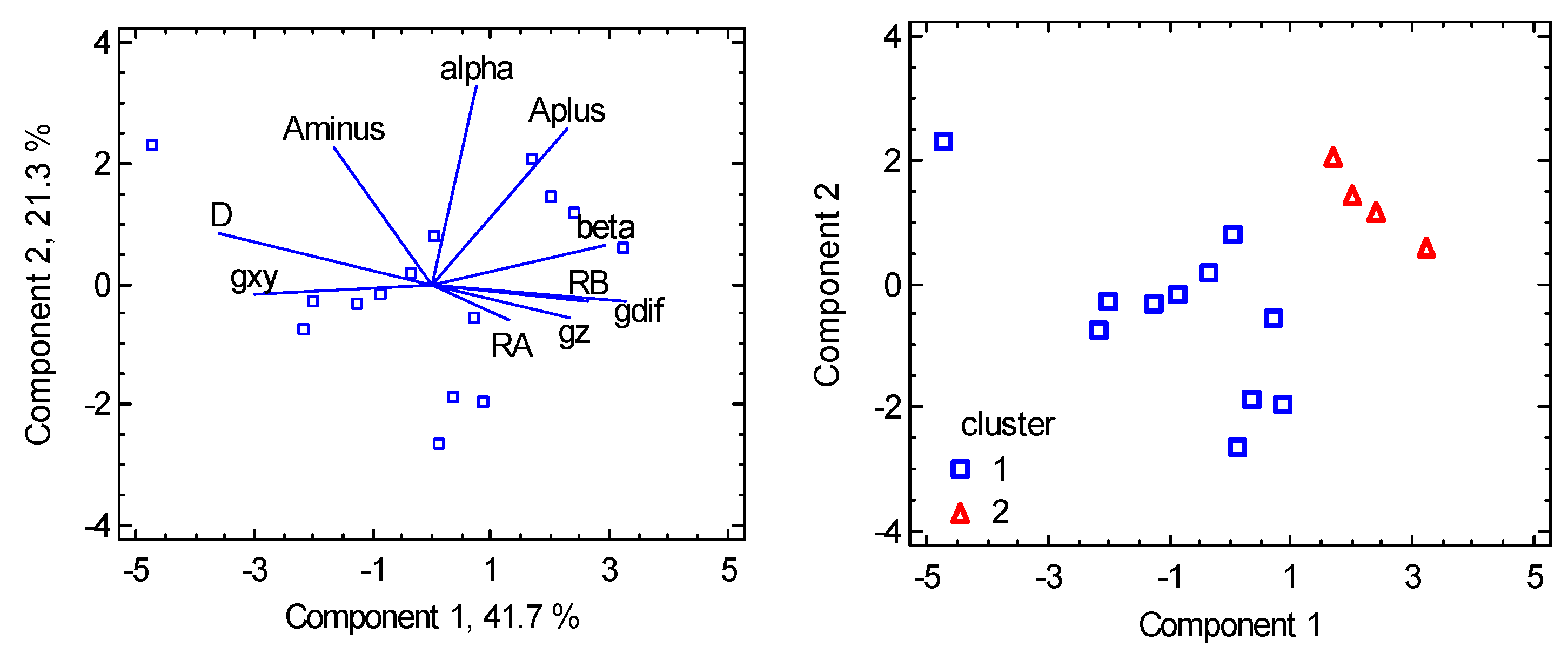
6. Magnetostructural D-Correlations
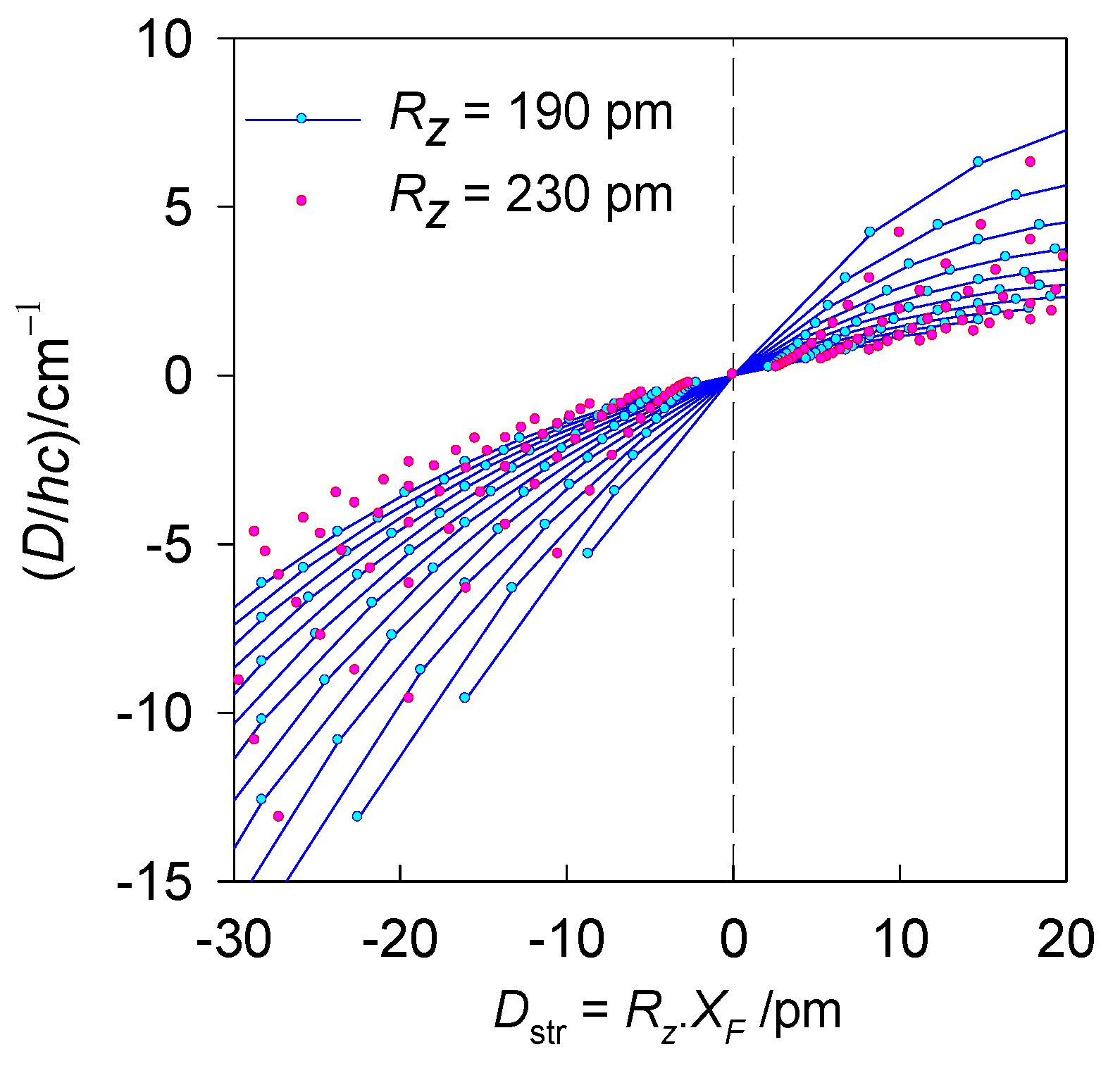
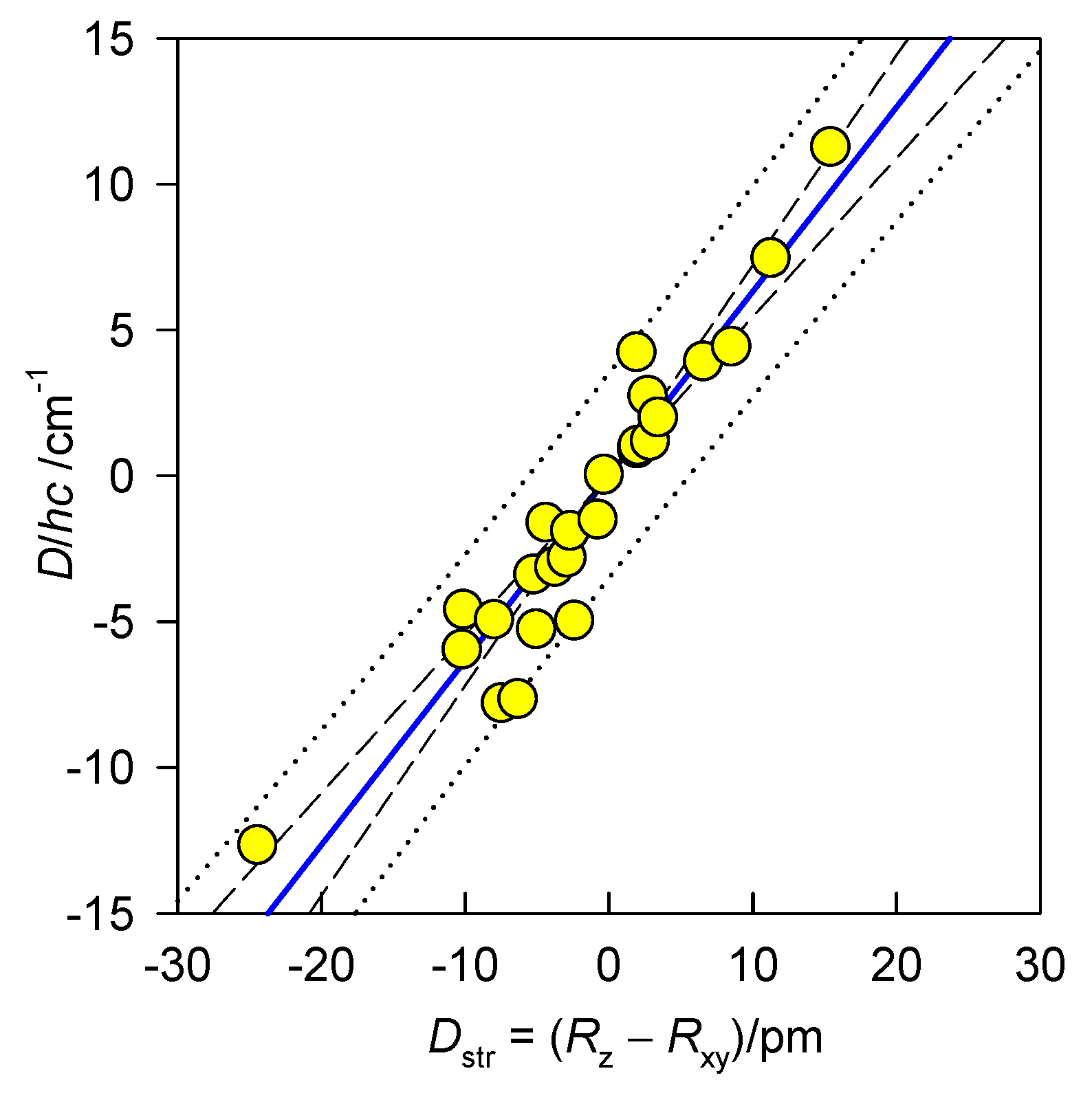
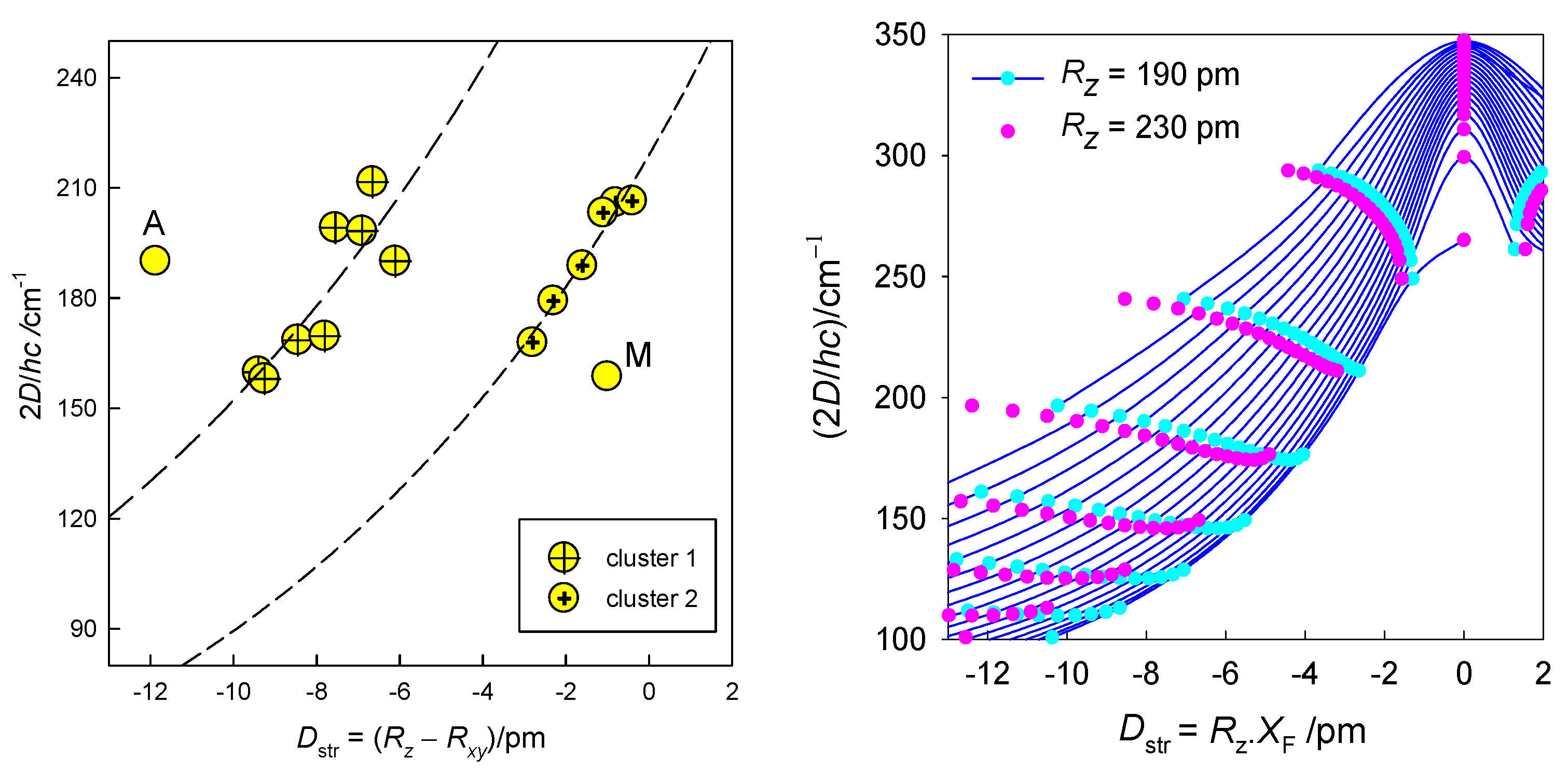
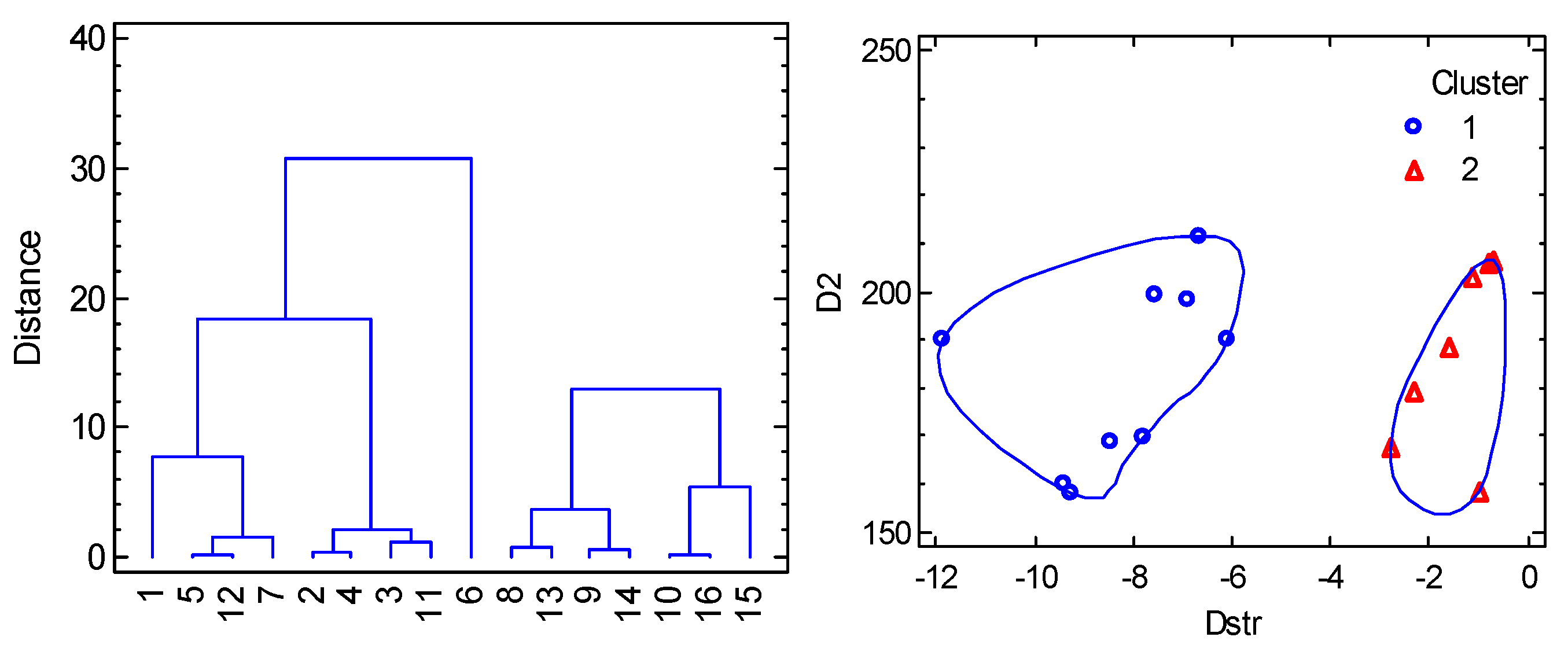
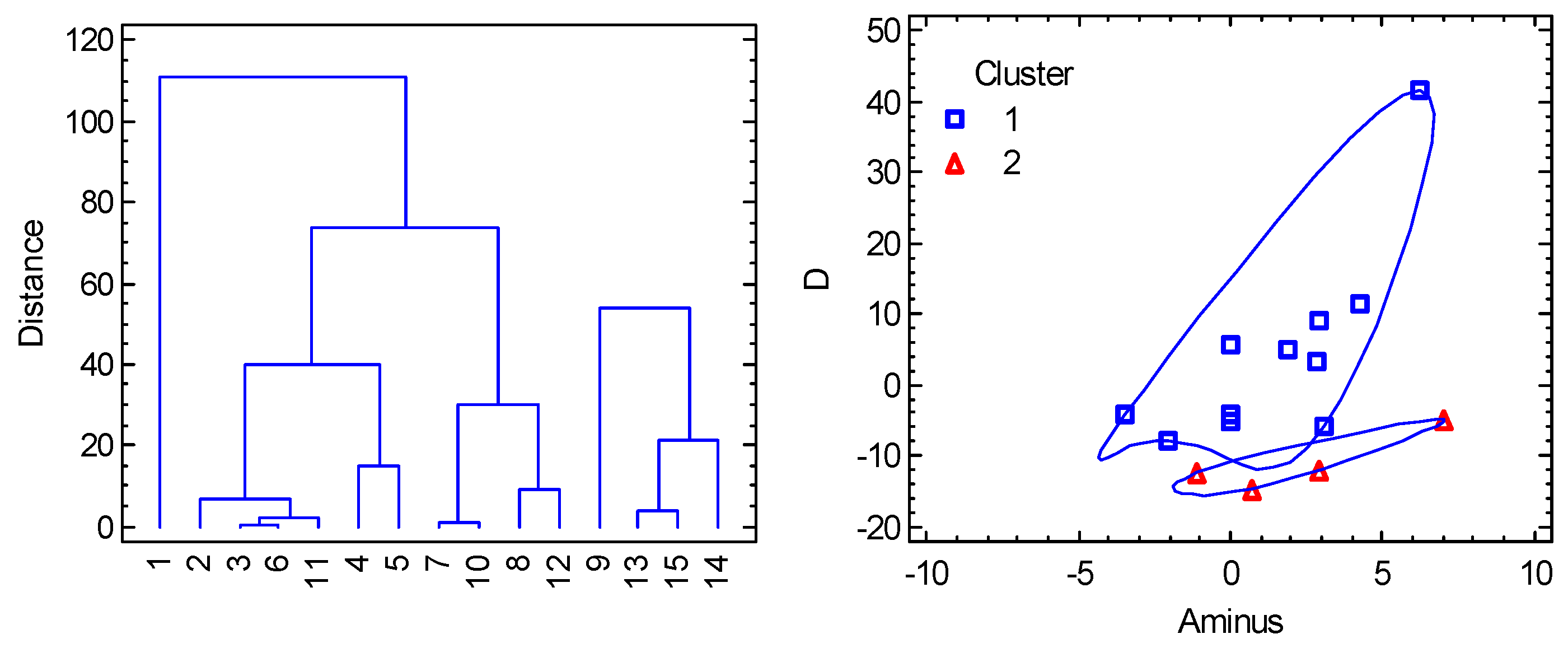
6.1. Hexacoordinate Ni(II) Complexes
6.2. Hexacoordinate Co(II) Complexes
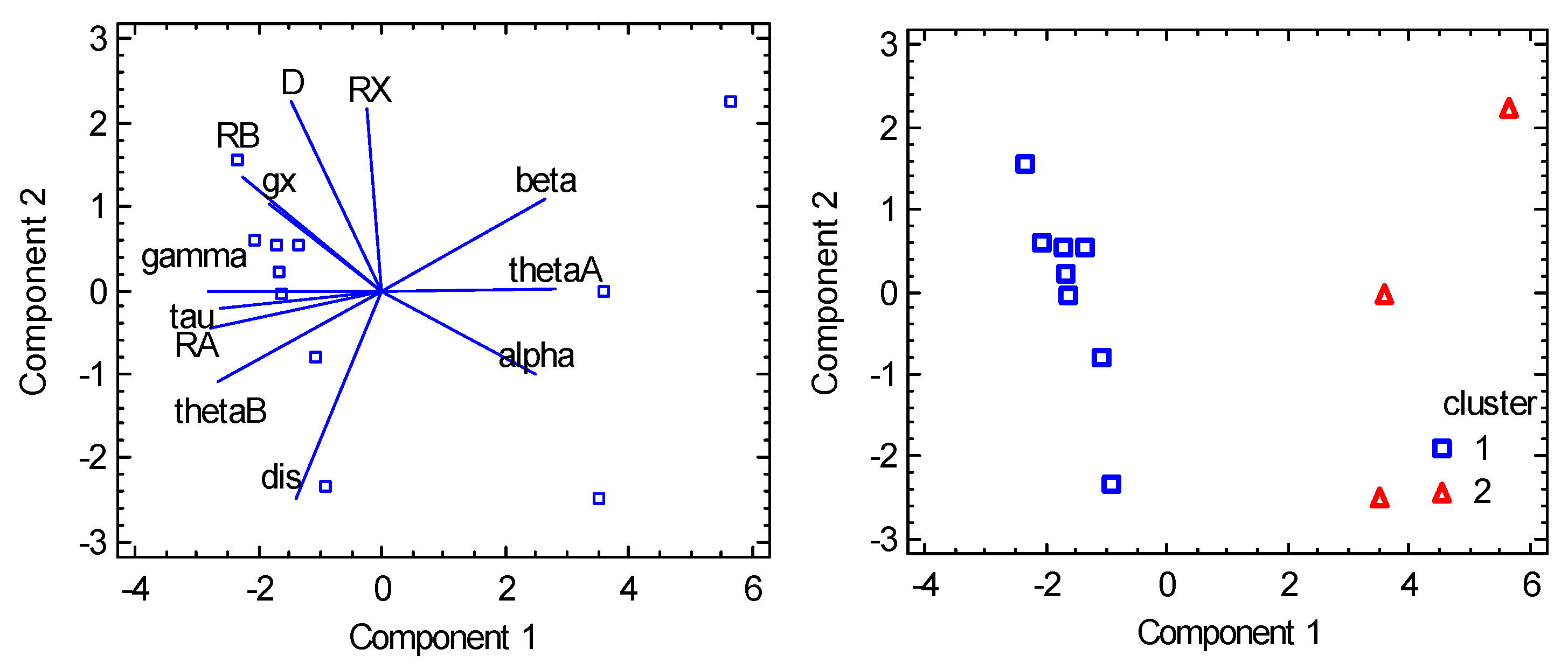
6.3. Tetracoordinate Co(II) Complexes
6.4. Pentacoordinate Co(II) Complexes
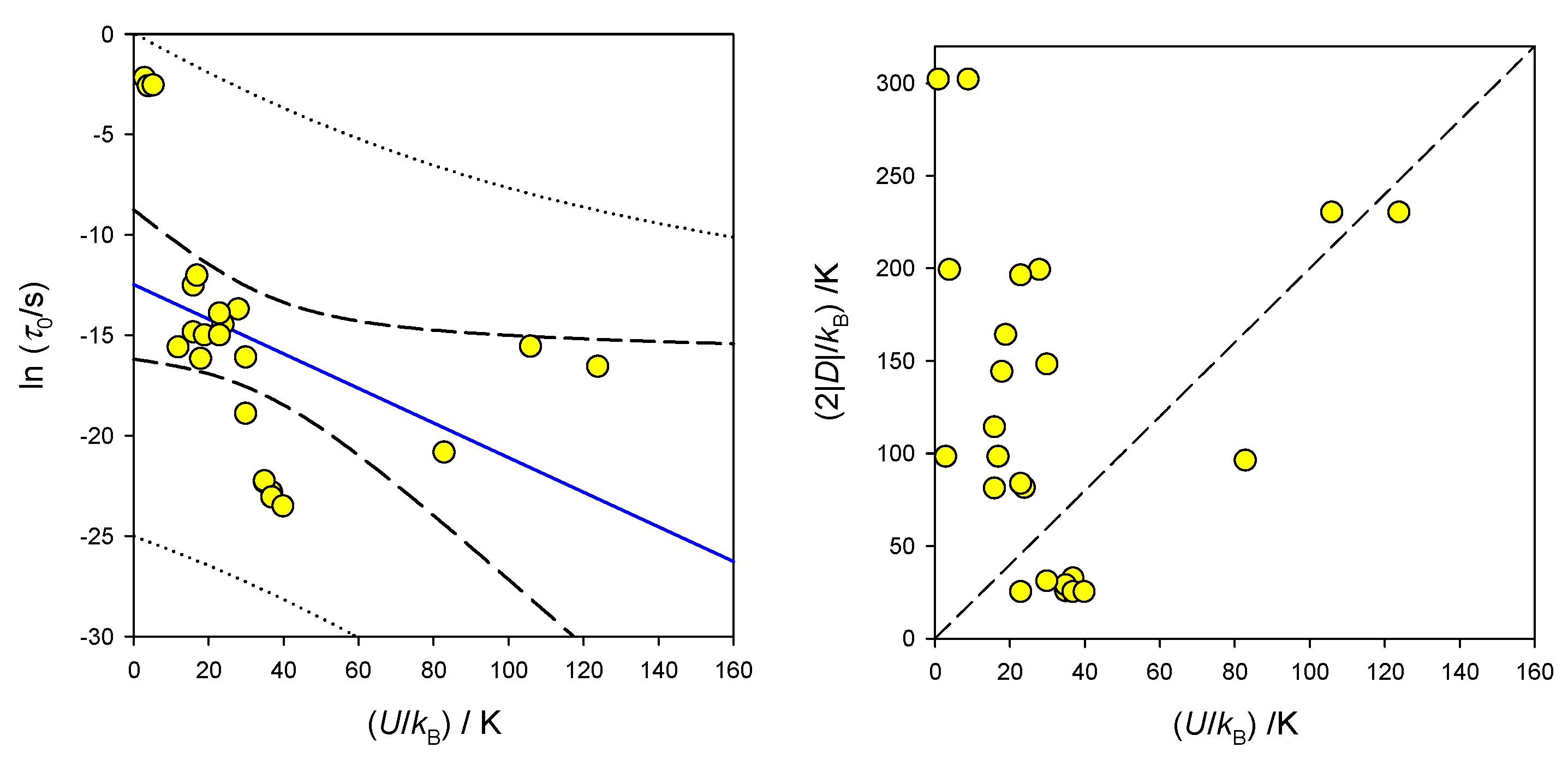
7. Selected Single-Molecule Magnets
8. Summary
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lis, T. Preparation, structure, and magnetic properties of a dodecanuclear mixed-valence manganese carboxylate. Acta Crystallogr. 1980, B36, 2042–2046. [Google Scholar] [CrossRef]
- Caneschi, A.; Gatteschi, D.; Sessoli, R.; Barra, A.L.; Brunel, L.C.; Guillot, L.C. Alternating current susceptibility, high field magnetization, and millimeter band EPR evidence for a ground S = 10 state in [Mn12O12(Ch3COO)16(H2O)4].2CH3COOH.4H2O. J. Am. Chem. Soc. 1991, 133, 5873–5874. [Google Scholar] [CrossRef]
- Aubin, S.M.J.; Wemple, M.W.; Adams, D.M.; Tsai, H.L.; Christou, G.; Hendrickson, D.N. Distorted MnIVMnIII3 Cubane Complexes as Single-Molecule Magnets. J. Am. Chem. Soc. 1996, 118, 7746–7754. [Google Scholar] [CrossRef]
- Gatteschi, D.; Sessoli, R.; Villain, J. Molecular Nanomagnets; Oxford University Press: Oxford, UK, 2006. [Google Scholar]
- Affronte, M. Molecular Nanomagnets for Information Technologies. J. Mater. Chem. 2009, 19, 1731–1737. [Google Scholar] [CrossRef]
- Lumetti, S.; Candini, A.; Godfrin, C.; Balestro, F.; Wernsdorfer, W.; Klyatskaya, S.; Ruben, M.; Affronte, M. Single-Molecule Devices with Graphene Electrodes. Dalton Trans. 2016, 45, 16570–16574. [Google Scholar] [CrossRef]
- Milios, C.J.; Winpenny, R.E.P. Cluster-Based Single-Molecule Magnets. Struct. Bond. 2015, 164, 1–109. [Google Scholar]
- Coronado, E. Molecular Magnetism: From Chemical Design to Spin Control in Molecules, Materials and Devices. Nat. Rev. Mater. 2020, 5, 87–104. [Google Scholar] [CrossRef]
- Shao, D.; Wang, X.Y. Development of Single-Molecule Magnets†. Chin. J. Chem. 2020, 38, 1005–1018. [Google Scholar] [CrossRef]
- Layfield, R.A. Organometallic Single-Molecule Magnets. Organometallics 2014, 33, 1084–1099. [Google Scholar] [CrossRef]
- Wernsdorfer, W.; Sessoli, R. Quantum Phase Interference and Parity Effects in Magnetic Molecular Clusters. Science 1999, 284, 133–135. [Google Scholar] [CrossRef]
- Coulon, C.; Miyasaka, H.; Clérac, R. Single-Chain Magnets: Theoretical Approach and Experimental Systems. Struct. Bonding 2006, 122, 163–206. [Google Scholar] [CrossRef]
- Mydosh, J.A. Spin Glasses: An Experimental Introduction; Taylor and Francis: London, UK, 1995. [Google Scholar]
- Winpenny, R. (Ed.) Single-Molecule Magnets and Related Phenomena, Structure and Bonding; Springer: Berlin/Heidelberg, Germany, 2006; p. 122. [Google Scholar]
- Fu, Z.; Qin, L.; Sun, K.; Hao, L.; Zheng, Y.-Z.; Lohstroh, W.; Günther, G.; Russina, M.; Liu, Y.; Xiao, Y.; et al. Low-temperature spin dynamics of ferromagnetic molecular ring {Cr8Y8}. NPJ Quantum Mater. 2020, 5, 32. [Google Scholar] [CrossRef]
- Garlatti, E.; Albring, M.A.; Baker, M.L.; Docherty, R.J.; Mutka, H.; Guidi, T.; Garcia, S.V.; Whitehead, G.F.; Pritchard, R.G.; Timco, G.A.; et al. A detailed study of the magnetism of chiral {Cr7M} rings: An investigation into parametrization and transferability of parameters. J. Am. Chem. Soc. 2014, 136, 9763–9772. [Google Scholar] [CrossRef]
- Low, D.M.; Rajaraman, G.; Helliwell, M.; Timco, G.; van Slageren, J.; Sessoli, R.; Ochsenbein, S.T.; Bircher, R.; Dobe, C.; Waldmann, O.; et al. A family of ferro- and antiferromagnetically coupled decametallic chromium(III) wheels. Chemistry 2006, 12, 1385–1396. [Google Scholar] [CrossRef]
- Miyasaka, H.; Madanbashi, T.; Saitoh, A.; Motokawa, N.; Ishikawa, R.; Yamashita, M.; Bahr, S.; Wernsdorfer, W.; Clérac, R. Cyano-Bridged MnIII-MIII Single-Chain Magnets with MIII=CoIII, FeIII, MnIII, and CrIII. Chem. Eur. J. 2012, 18, 3942–3954. [Google Scholar] [CrossRef]
- Boskovic, C.; Brechin, E.K.; Streib, W.E.; Folting, K.; Bollinger, J.C.; Hendrickson, D.N.; Christou, G. Single-molecule magnets: A new family of Mn12 clusters of formula [Mn12O8X4 (O2CPh)8L6]. J. Am. Chem. Soc. 2002, 124, 3725–3736. [Google Scholar] [CrossRef]
- Moushi, E.E.; Stamatatos, T.C.; Wernsdorfer, W.; Nastopoulos, V.; Christou, G.; Tasiopoulos, A.J. A Mn17 Octahedron with a Giant Ground-State Spin: Occurrence in Discrete Form and as Multidimensional Coordination Polymers. Inorg. Chem. 2009, 48, 5049–5051. [Google Scholar] [CrossRef]
- Ghulam Abbas, G.; Lan, Y.; Mereacre, V.; Wernsdorfer, W.; Clerac, R.; Buth, G.; Sougrati, M.T.; Grandjean, F.; Long, G.J.; Anson, C.E.; et al. Magnetic and 57Fe Mössbauer Study of the Single Molecule Magnet Behavior of a Dy3Fe7 Coordination Cluster. Inorg. Chem. 2009, 48, 9345–9355. [Google Scholar] [CrossRef]
- Morello, A.; Mettes, F.L.; Luis, F.; Fernández, F.J.; Krzystek, J.; Aromí, G.; Christou, G.; de Jongh, L.J. Long-range dipolar ferromagnetic ordering of high-spin molecular clusters. Phys. Rev. Lett. 2003, 90, 017206. [Google Scholar] [CrossRef]
- Burzuri, E.; Luis, F.; Barbara, B.; Ballou, R.; Ressouche, E.; Montero, O.; Campo, J.; Maegawa, S. Magnetic dipolar ordering and quantum phase transition in an Fe8 molecular magnet. Phys. Rev. Lett. 2011, 107, 097203. [Google Scholar] [CrossRef]
- Goodwin, J.C.; Sessoli, R.; Gatteschi, D.; Wernsdorfer, W.; Powell, A.K.; Heath, S.L. Towards nanostructured arrays of single molecule magnets: New Fe19 oxyhydroxide clusters displaying high ground state spins and hysteresis. J. Chem. Soc. Dalton Trans. 2020, 12, 1835–1840. [Google Scholar] [CrossRef]
- Affronte, M.; Lasjaunias, J.C.; Wernsdorfer, W.; Sessoli, R.; Gatteschi, D.; Heath, S.L.; Fort, A.; Rettori, A. Magnetic ordering in a high-spin Fe19 molecular nanomagnet. Phys. Rev. B 2002, 66, 064408. [Google Scholar] [CrossRef]
- Affronte, M.; Sessoli, R.; Gatteschi, D.; Wernsdorfer, W.; Lasjaunias, J.C.; Heath, S.L.; Powell, A.K.; Fort, A.; Rettori, A. Effects of intercluster coupling in high spin molecular magnets. J. Phys. Chem. Sol. 2004, 65, 745–748. [Google Scholar] [CrossRef]
- Bogani, L.; Wernsdorfer, W. Molecular spintronics using single-molecule magnets. Nat. Mater. 2008, 7, 179–186. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, H.; Clerac, R. Synthetic Strategy for Rational Design of Single-Chain Magnets. Bull. Chem. Soc. Jpn. 2005, 78, 1725–1748. [Google Scholar] [CrossRef]
- Miyasaka, H.H.; Takahashi, M.T.; Sugiura, K.; Clérac, R.; Nojiri, H. Cyano-Bridged MnIII3MIII (MIII = Fe, Cr) Complexes: Synthesis, Structure, and Magnetic Properties. Inorg. Chem. 2005, 44, 5969–5971. [Google Scholar] [CrossRef]
- Boča, R.; Rajnák, C. Unexpected behavior of single ion magnets. Coord. Chem. Rev. 2021, 430, 213657. [Google Scholar] [CrossRef]
- Craig, G.A.; Murrie, M. 3d single-ion magnets. Chem. Soc. Rev. 2015, 44, 2135–2147. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.-S.; Jiang, S.-D.; Wang, B.-W.; Gao, S. Understanding the Magnetic Anisotropy Toward Single-ion Magnets. Acc. Chem. Res. 2016, 49, 2381–2389. [Google Scholar] [CrossRef]
- Atzori, M.; Tesi, L.; Morra, E.; Chiesa, M.; Sorace, L.; Sessoli, R. Room-Temperature Quantum Coherence and Rabi Oscillations in Vanadyl Phthalocyanine: Toward Multifunctional Molecular Spin Qubits. J. Am. Chem. Soc. 2016, 138, 2154–2157. [Google Scholar] [CrossRef]
- Ding, M.; Cutsail, G.E.I.; Aravena, D.; Amoza, M.; Rouzières, M.; Dechambenoit, P.; Losovyj, Y.; Pink, M.; Ruiz, E.; Clérac, R. A low spin manganese(iv) nitride single molecule magnet. Chem. Sci. 2016, 7, 6132–6140. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, I.; Shaffer, D.W.; Yang, J.Y.; Shores, M.P. Single molecule magnet behaviour in a square planar S = 1/2 Co(ii) complex and spin-state assignment of multiple relaxation modes. Chem. Commun. 2020, 56, 6711–6714. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Song, J.; Zhao, W.; Yi, G.; Zhou, Z.; Yuan, A.; Song, Y.; Wang, Z.; Ouyang, Z.W. A mononuclear five-coordinate Co(ii) single molecule magnet with a spin crossover between the S = 1/2 and 3/2 states. Dalton Trans. 2018, 47, 16596–16602. [Google Scholar] [CrossRef] [PubMed]
- Boča, R. A Handbook of Magnetochemical Formulae; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Griffith, J.S. The Theory of Transition Metal Ions; Cambridge University Press: Cambridge, CA, USA, 1964. [Google Scholar]
- Figgis, B.N. Introduction to Ligand Fields; Wiley: New York, NY, USA, 1966. [Google Scholar]
- König, E.; Kremer, S. Magnetism Diagrams for Transition Metal Ions; Plenum Press: New York, NY, USA, 1979. [Google Scholar]
- König, E.; Kremer, S. Irreducible tensor operator methods in intermediate-field coupling. Int. J. Quant. Chem. 1974, 8, 347–362. [Google Scholar] [CrossRef]
- König, E.; Kremer, S. Irreducible Tensor Operator Methods in Strong-field Coupling. Int. J. Quant. Chem. 1977, 12, 1017–1031. [Google Scholar] [CrossRef]
- Boča, R. Program MIF&FIT; University of SS Cyril and Methodius: Trnava, Slovakia, 2022. [Google Scholar]
- Crawford, V.H.; Richardson, H.W.; Wasson, J.R.; Hodgson, D.J.; Hatfield, W.E. Relation between the singlet-triplet splitting and the copper-oxygen-copper bridge angle in hydroxo-bridged copper dimers. Inorg. Chem. 1976, 15, 2107–2110. [Google Scholar] [CrossRef]
- Hatfield, W.E.; Willett, R.D.; Gatteschi, D.; Kahn, O. Magneto-Structural Correlations in Exchange Coupled Systems; NATO ASI Series Reidel: Dordrecht, The Netherlands, 1985. [Google Scholar]
- Thompson, L.K.; Mandal, S.K.; Tandon, S.S.; Bridson, J.N.; Park, M.K. Magnetostructural Correlations in Bis(μ2-phenoxide)-bridged Macrocyclic Dinuclear Copper(ii) Complexes. Influence of Electron-withdrawing Substituents on Exchange Coupling. Inorg. Chem 1996, 35, 3117–3125. [Google Scholar] [CrossRef]
- Glerup, J.; Hodgson, D.J.; Pedersen, E. A Novel Correlation Between Magnetism and Structural Parameters in Superexchange Coupled Chromium(iii) Dimers. Acta. Chem. Scand. 1983, 37a, 161–164. [Google Scholar] [CrossRef]
- Law, N.A.; Kampf, J.W.; Pecoraro, V.L. A magneto-structural correlation between the Heisenberg constant, J, and the Mn-O-Mn angle in [MnIV(μ-O)]2 dimers. Inorg. Chim. Acta. 2000, 297, 252–264. [Google Scholar] [CrossRef]
- Gorun, S.M.; Lippard, S.J. Magnetostructural Correlations in Magnetically Coupled (.mu.-oxo)diiron(iii) Complexes. Inorg. Chem. 1991, 30, 1625–1630. [Google Scholar] [CrossRef]
- Werner, R.; Ostrovsky, S.; Griesar, K.; Haase, W. Magnetostructural Correlations in Exchange Coupled Phenoxo-, Alkoxo-, and Hydroxo-bridged Dinuclear Iron(iii) Compounds. Inorg. Chim. Acta 2001, 326, 78–88. [Google Scholar] [CrossRef]
- Mitchell, K.J.; Abboud, K.A.; Christou, G. Magnetostructural Correlation for High-Nuclearity Iron(III)/Oxo Complexes and Application to Fe5, Fe6, and Fe8 Clusters. Inorg. Chem. 2016, 55, 6597–6608. [Google Scholar] [CrossRef] [PubMed]
- Makohusová, M.; Mrázová, V.; Haase, W.; Boča, R. Magnetostructural J-correlations in complexes with tetrahedro-{Cu4} core. Polyhedron 2014, 81, 572–582. [Google Scholar] [CrossRef]
- Neese, F.; Solomon, R.I. Calculation of Zero-Field Splittings, g-Values, and the Relativistic Nephelauxetic Effect in Transition Metal Complexes. Application to High-Spin Ferric Complexes. Inorg. Chem. 1998, 37, 6568–6582. [Google Scholar] [CrossRef] [PubMed]
- Maganas, D.; Krzystek, J.; Ferentinos, E.; Whyte, A.M.; Robertson, N.; Psycharis, V.; Terzis, A.; Neese, F.; Kyritsis, P. Investigating Magnetostructural Correlations in the Pseudooctahedral Trans-[niii{(opph2)(epph2)n}2(sol)2] Complexes (E = S, Se; Sol = DMF, THF) by Magnetometry, HFEPR, and Ab Initio Quantum Chemistry. Inorg. Chem. 2012, 51, 7218–7231. [Google Scholar] [CrossRef] [PubMed]
- Duboc, C.; Ganyushin, D.; Sivalingam, K.; Collomb, M.-N.; Neese, F. Systematic Theoretical Study of the Zero-field Splitting in Coordination Complexes of Mn(iii). Density Functional Theory Versus Multireference Wave Function Approaches. J. Phys. Chem. A 2010, 114, 10750–10758. [Google Scholar] [CrossRef] [PubMed]
- Carlin, R.L. Magnetochemistry; Springer: Berlin/Heidelberg, Germany, 1986. [Google Scholar]
- Ganyushin, D.; Neese, F. First-principles calculations of zero-field splitting parameters. J. Chem. Phys. 2006, 125, 024103. [Google Scholar] [CrossRef]
- König, E.; Schnakig, R. Zero-field splitting of 6S(d5) ions in tetragonal and rhombic symmetry: I. Complete d-electron calculation of the crystal-field dependence of D and E. Phys. Status Solidi (b) 1976, 77, 657–666. [Google Scholar] [CrossRef]
- Neese, F. Importance of Direct Spin−spin Coupling and Spin-flip Excitations for the Zero-field Splittings of Transition Metal Complexes: A Case Study. J. Am. Chem. Soc. 2006, 128, 10213–10222. [Google Scholar] [CrossRef]
- SigmaPlot; Version 10.0; Systat Software, Inc.: San Jose, CA, USA, 2013.
- Boča, R.; Titiš, J. Coordination Chemistry Research Progress; Nova Science Publishers: New York, NY, USA, 2008; p. 247. [Google Scholar]
- Titiš, J.; Boča, R. Magnetostructural D Correlation in Nickel(II) Complexes: Reinvestigation of the Zero-Field Splitting. Inorg. Chem. 2010, 49, 3971–3973. [Google Scholar] [CrossRef]
- Statgraphics; Centurion XV Version 15.2.06.; StatPoint, Inc.: Herndon, VA, USA, 2007.
- Herchel, R.; Váhovská, L.; Potočňák, I.; Trávníček, Z. Slow Magnetic Relaxation in Octahedral Cobalt(II) Field-Induced Single-Ion Magnet with Positive Axial and Large Rhombic Anisotropy. Inorg. Chem. 2014, 53, 5896–5898. [Google Scholar] [CrossRef]
- Titiš, J.; Boča, R. Magnetostructural D Correlations in Hexacoordinated Cobalt(ii) Complexes. Inorg. Chem. 2011, 50, 11838–11845. [Google Scholar] [CrossRef]
- Papánková, B.; Boča, R.; Dlháň, Ľ.; Nemec, I.; Titiš, J.; Svoboda, I.; Fuess, H. Magneto-structural relationships for a mononuclear Co(II) complex with large zero-field splitting. Inorg. Chim. Acta 2010, 363, 147–156. [Google Scholar] [CrossRef]
- Idešicová, M.; Titiš, J.; Krzystek, J.; Boča, R. Zero-Field Splitting in Pseudotetrahedral Co(II) Complexes: A Magnetic, High-Frequency and -Field EPR, and Computational Study. Inorg. Chem. 2013, 52, 9409–9417. [Google Scholar] [CrossRef]
- Titiš, J.; Miklovič, J.; Boča, R. Magnetostructural study of tetracoordinate cobalt(II) complexes. Inorg. Chem. Commun. 2013, 35, 72–75. [Google Scholar] [CrossRef]
- Rajnák, C.; Titiš, J.; Šalitroš, I.; Boča, R.; Fuhr, O.; Ruben, M. Zero-field splitting in pentacoordinate Co(II) complexes. Polyhedron 2013, 65, 122–128. [Google Scholar] [CrossRef]
- Rajnák, C.; Titiš, J.; Fuhr, O.; Ruben, M.; Boča, R. Single-Molecule Magnetism in a Pentacoordinate Cobalt(II) Complex Supported by an Antenna Ligand. Inorg. Chem. 2014, 53, 8200–8202. [Google Scholar] [CrossRef]
- Makinen, M.V.; Kuo, L.C.; Yim, M.B.; Wells, G.B.; Fukuyama, J.M.; Kim, J.E. Ground Term Splitting of High-spin Cobalt(2+) Ion as a Probe of Coordination Structure. 1. Dependence of the Splitting on Coordination Geometry. J. Am. Chem. Soc. 1985, 107, 5245–5255. [Google Scholar] [CrossRef]
- Jurca, T.; Farghal, A.; Lin, P.-H.; Korobkov, I.; Murugesu, M.; Richeson, D.S. Single-Molecule Magnet Behavior with a Single Metal Center Enhanced through Peripheral Ligand Modifications. J. Am. Chem. Soc. 2011, 133, 15814–15817. [Google Scholar] [CrossRef]
- Ishikawa, R.; Miyamoto, R.; Nojiri, H.; Breedlove, B.K.; Yamashita, M. Slow Relaxation of the Magnetization of an MnIII Single Ion. Inorg. Chem. 2013, 52, 8300–8302. [Google Scholar] [CrossRef]
- Grigoropoulous, A.; Pissas, M.; Rapatolis, P.; Psycharis, V.; Kyritsis, P.; Sanakis, Y. Spin-relaxation Properties of a High-spin Mononuclear Mniiio6-containing Complex. Spin-Relaxation Properties of a High-Spin Mononuclear MnIIIO6-Containing Complex. Inorg. Chem. 2013, 52, 12869–12871. [Google Scholar] [CrossRef]
- Vallejo, J.; Pascual-Alvarez, A.; Cano, J.; Castro, I.; Julve, M.; Lloret, F.; Krzystek, J.; De Munno, G.; Armentano, D.; Wernsdorfer, W.; et al. Field-induced Hysteresis and Quantum Tunneling of the Magnetization in a Mononuclear Manganese(iii) Complex. Field-Induced Hysteresis and Quantum Tunneling of the Magnetization in a Mononuclear Manganese(III) Complex. Angew. Chem. Int. Ed. 2013, 125, 14325–14329. [Google Scholar] [CrossRef]
- Mossin, S.; Tran, B.L.; Adhikari, D.; Pink, M.; Heinemann, F.W.; Sutter, J.; Szilagyi, R.K.; Meyer, K.; Mindiola, D.J. A Mononuclear Fe(III) Single Molecule Magnet with a 3/2↔5/2 Spin Crossover. J. Am. Chem. Soc. 2012, 134, 13651–13661. [Google Scholar] [CrossRef] [PubMed]
- Harman, W.H.; Harris, T.D.; Freedman, D.E.; Fong, H.; Chang, A.; Rinehart, J.D.; Ozarowski, A.; Sougrati, M.T.; Grandjean, F.; Long, G.J.; et al. Slow Magnetic Relaxation in a Family of Trigonal Pyramidal Iron(ii) Pyrrolide Complexes. Slow Magnetic Relaxation in a Family of Trigonal Pyramidal Iron(II) Pyrrolide Complexes. J. Am. Chem. Soc. 2010, 132, 18115–18126. [Google Scholar] [CrossRef]
- Freedman, D.E.; Harman, W.H.; Harris, T.D.; Long, G.J.; Chang, C.J.; Long, J.R. Slow Magnetic Relaxation in a High-spin Iron(ii) Complex. J. Am. Chem. Soc. 2010, 132, 1224–1225. [Google Scholar] [CrossRef]
- Weismann, D.; Sun, Y.; Lan, Y.; Wolmershauser, G.; Powell, A.K.; Sitzmann, H. High-Spin Cyclopentadienyl Complexes: A Single-Molecule Magnet Based on the Aryl-Iron(II) Cyclopentadienyl Type. Chem. Eur. J. 2011, 17, 4700–4704. [Google Scholar] [CrossRef]
- Lin, P.-H.; Smythe, N.C.; Gorelsky, S.J.; Maguire, S.; Henson, N.J.; Korobkov, I.; Scott, B.L.; Gordon, J.C.; Baker, R.T.; Murugesu, M. Importance of Out-of-state Spin–orbit Coupling for Slow Magnetic Relaxation in Mononuclear Feii Complexes. J. Am. Chem. Soc. 2011, 133, 15806–15809. [Google Scholar] [CrossRef] [PubMed]
- Zadrozny, J.M.; Xiao, D.J.; Atanasov, M.; Long, G.J.; Grandjean, F.; Neese, F.; Long, J.R. Magnetic Blocking in a Linear Iron(i) Complex. Nature Chem. 2013, 5, 577–581. [Google Scholar] [CrossRef]
- Eichhofer, A.; Lan, Y.; Mereacre, V.; Bodenstein, T.; Weigend, F. Slow Magnetic Relaxation in Trigonal-planar Mononuclear Fe(ii) and Co(ii) Bis(trimethylsilyl)amido Complexes—A Comparative Study. Inorg. Chem. 2014, 55, 1962–1974. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Long, J.R. Slow Magnetic Relaxation at Zero Field in the Tetrahedral Complex [Co(SPh)4]2−. J. Am. Chem. Soc. 2011, 133, 20732–20734. [Google Scholar] [CrossRef]
- Zadrozny, J.M.; Liu, J.; Piro, N.A.; Chang, C.J.; Hill, S.; Long, J.R. Slow Magnetic Relaxation in a Pseudotetrahedral Cobalt(ii) Complex with Easy-plane Anisotropy. Chem. Commun. 2012, 48, 3927–3929. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Zhou, Q.; Zhang, Y.; Zeng, G.; Li, G.; Shi, Z.; Wang, B.; Feng, S. Inspiration from Old Molecules: Field-induced Slow Magnetic Relaxation in Three Air-stable Tetrahedral Cobalt(ii) Compounds. Chem. Commun. 2013, 49, 5289–5291. [Google Scholar] [CrossRef] [PubMed]
- Boča, R.; Miklovič, J.; Titiš, J. Simple Mononuclear Cobalt(ii) Complex: A Single-molecule Magnet Showing Two Slow Relaxation Processes. Inorg. Chem. 2014, 53, 2367–2369. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, J.; Castro, I.; Ruiz-Garcia, J.; Cano, J.; Julve, M.; Lloret, F.; De Munno, G.; Wernsdorfer, W.; Pardo, E. Field-induced Slow Magnetic Relaxation in a Six-coordinate Mononuclear Cobalt(ii) Complex with a Positive Anisotropy. J. Am. Chem. Soc. 2012, 134, 15704–15707. [Google Scholar] [CrossRef]
- Colacio, E.; Ruiz, K.; Ruiz, E.; Cremades, E.; Krzystek, J.; Carretta, S.; Cano, J.; Guidi, T.; Wernsdorfer, W.; Brechin, E.K. Slow Magnetic Relaxation in a Coii–yiii Single-ion Magnet with Positive Axial Zero-field Splitting. Angew. Chem. Int. Ed. 2013, 125, 9300–9304. [Google Scholar] [CrossRef]
- Zhu, Y.-Y.; Cui, C.; Zhang, Y.-Q.; Jia, J.-H.; Guo, X.; Gao, C.; Qian, K.; Jiang, S.-D.; Wang, B.-W.; Wang, Z.-M. Zero-field slow magnetic relaxation from single Co(ii) ion: A transition metal single-molecule magnet with high anisotropy barrier. Chem. Sci. 2013, 4, 1802–1806. [Google Scholar] [CrossRef]
- Habib, F.; Luca, O.R.; Vieru, V.; Shiddiq, M.; Korobkov, I.; Gorelsky, S.I.; Takase, M.K.; Chibotaru, L.F.; Hill, S.; Crabtree, R.H.; et al. Influence of the Ligand Field on Slow Magnetization Relaxation versus Spin Crossover in Mononuclear Cobalt Complexes. Angew. Chem. Int. Ed. 2013, 52, 11290–11293. [Google Scholar] [CrossRef]
- van Stapele, R.P.; Beljers, H.G.; Bongers, P.F.; Zijlstra, H. Ground State of Divalent Co Ions in Cs3CoCl5 and Cs3CoBr5. J. Chem. Phys. 1966, 44, 3719–3725. [Google Scholar] [CrossRef]
- Krzystek, J.; Zvyagin, S.A.; Ozarowski, A.; Fiedler, A.T.; Brunold, T.C.; Telser, J. Definitive Spectroscopic Determination of Zero-Field Splitting in High-Spin Cobalt(II). J. Am. Chem. Soc. 2004, 126, 2148–2155. [Google Scholar] [CrossRef]
- Kulik, H.J. Making machine learning a useful tool in the accelerated discovery of transition metal complexes. WIREs Comput. Mol. Sci. 2019, 10, e1439. [Google Scholar]
- Alvarez, S.; Llunell, M. Continuous symmetry measures of penta-coordinate molecules: Berry and non-Berry distortions of the trigonal bipyramid. J. Chem. Soc. Dalton Trans. 2000, 3288–3303. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C.n. | Chromophore | Geometry a | (D/hc)/cm−1 | (E/hc)/cm−1 | BDC/T | (U/kB)/K | τ0/s | Ref. |
| 3 | {CoN3} | −57 | 12.7 | 0.08 | 16 | 3.5 × 10−7 | [82] | |
| {CoN2O} | −72 | 13.5 | 0.06 | 18 | 9.3 × 10−8 | [82] | ||
| {CoN2P} | −82 | 0.075 | 19 | 3.0 × 10−7 | [82] | |||
| 4 | {CoS4} | −74 | 0.10 | 30 | 1.0 × 10−7 | [83] | ||
| {CoN3Cl} | +12.7 | 1.2 | 0.15 | 35 | 2 × 10−10 | [84] | ||
| {CoP2Cl2} | −16.2 | 0.9 | 0.10 | 37 | 1.2 × 10−10 | [85] | ||
| {CoP2Cl2} | −14.4 | 1.7 | 0.10 | 35 | 2.1 × 10−10 | [85] | ||
| {CoP2Cl2} | −15.4 | 1.3 | 0.10 | 30 | 6.0 × 10−9 | [85] | ||
| {CoP2Br2} | −12.5 | 0.10 | 37 | 9.4 × 10−11 | [86] | |||
| 0.20 | 40 | 6.0 × 10−11 | [86] | |||||
| 5 | {CoN3N2} | 4py | −40.5 | J > 0 | 0.20 | 16 | 3.6 × 10−6 | [86] |
| {CoN3N2} | 4py | −40.6 | 0.20 | 24 | 5.1 × 10−7 | [72] | ||
| {CoN3N2} | 3bpy | (+49.0), calc | 0.06 | 17 | 5.85 × 10−6 | [90] | ||
| 0.56 | 3 | 0.110 | [90] | |||||
| {CoN3Cl2} | 4py | (+99.5), calc | 0.06 | 28 | 1.1 × 10−6 | [90] | ||
| 0.56 | 4 | 0.074 | [90] | |||||
| {CoN3Cl2}2 | 4py | 151 | 11.6, J > 0 | 0.20 | 9 | 3.1 × 10−7 | [69] | |
| 0.20 | 1 | 0.27 | [69] | |||||
| 6 | {CoN4N2} | +48 | 13.0 | 0.30 | 83 | 8.7 × 10−10 | [64] | |
| {CoN4N2} | +98 | 8.4 | 0.10 | 23 | 3.0 × 10−7 | [72] | ||
| {CoO3N3} | +41.7 | 1.6 | 0.10 | 23 | 8.9 × 10−7 | [88] | ||
| {CoO6} | −115 | 2.8 | 0 | 106 | 1.7 × 10−7 | [89] | ||
| 0.15 | 124 | 6.3 × 10−8 | [89] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Titiš, J.; Rajnák, C.; Boča, R. Magnetostructural D-Correlations and Their Impact on Single-Molecule Magnetism. Inorganics 2023, 11, 452. https://doi.org/10.3390/inorganics11120452
Titiš J, Rajnák C, Boča R. Magnetostructural D-Correlations and Their Impact on Single-Molecule Magnetism. Inorganics. 2023; 11(12):452. https://doi.org/10.3390/inorganics11120452
Chicago/Turabian StyleTitiš, Ján, Cyril Rajnák, and Roman Boča. 2023. "Magnetostructural D-Correlations and Their Impact on Single-Molecule Magnetism" Inorganics 11, no. 12: 452. https://doi.org/10.3390/inorganics11120452
APA StyleTitiš, J., Rajnák, C., & Boča, R. (2023). Magnetostructural D-Correlations and Their Impact on Single-Molecule Magnetism. Inorganics, 11(12), 452. https://doi.org/10.3390/inorganics11120452