
Phenolic 3° Phosphine Oxides as a Class of Metal-Free Catalysts for the Activation of C–O Bonds in Aliphatic Alcohols: Direct Synthesis of Catalyst Candidates, and Kinetic Studies

Abstract
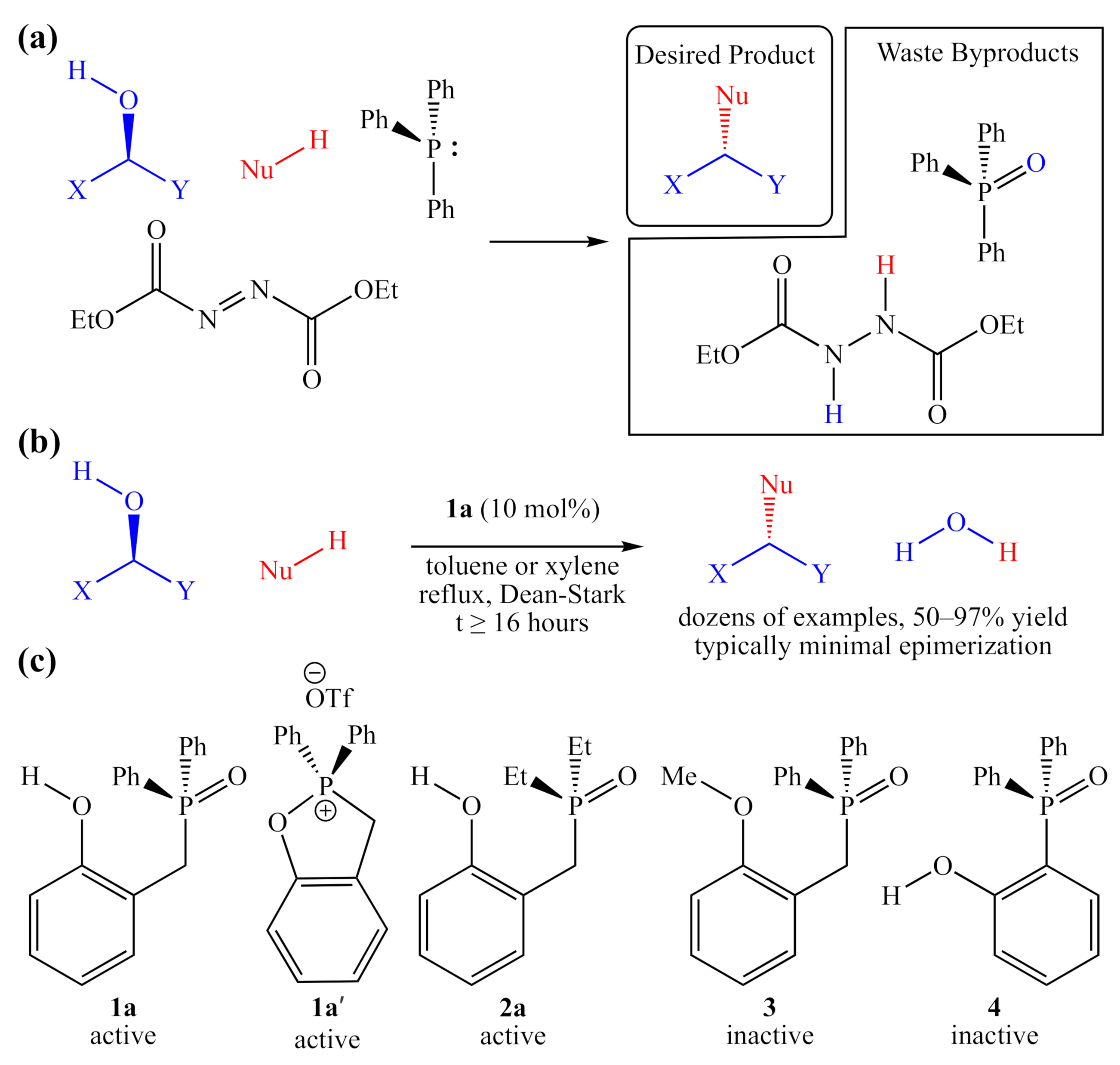
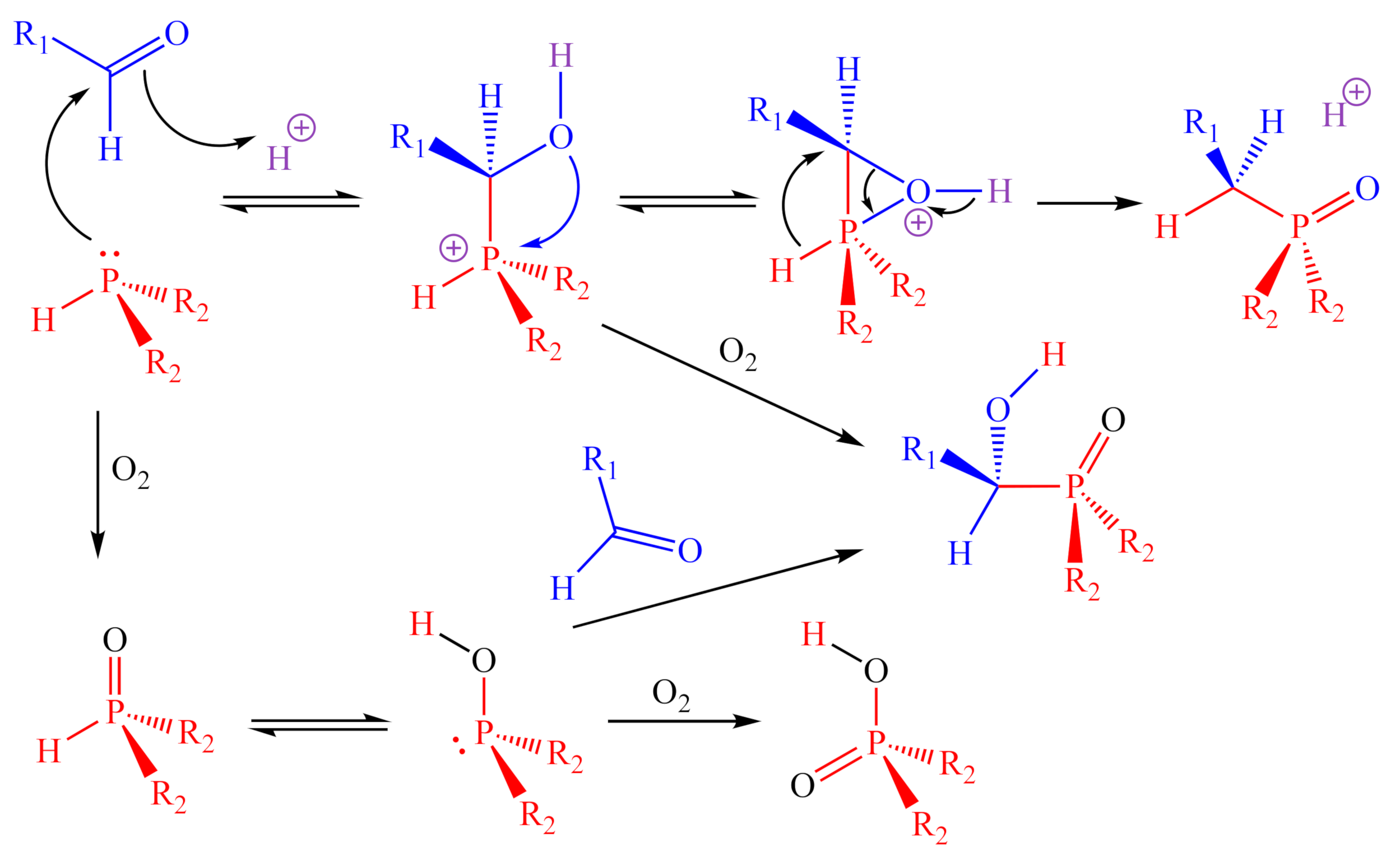
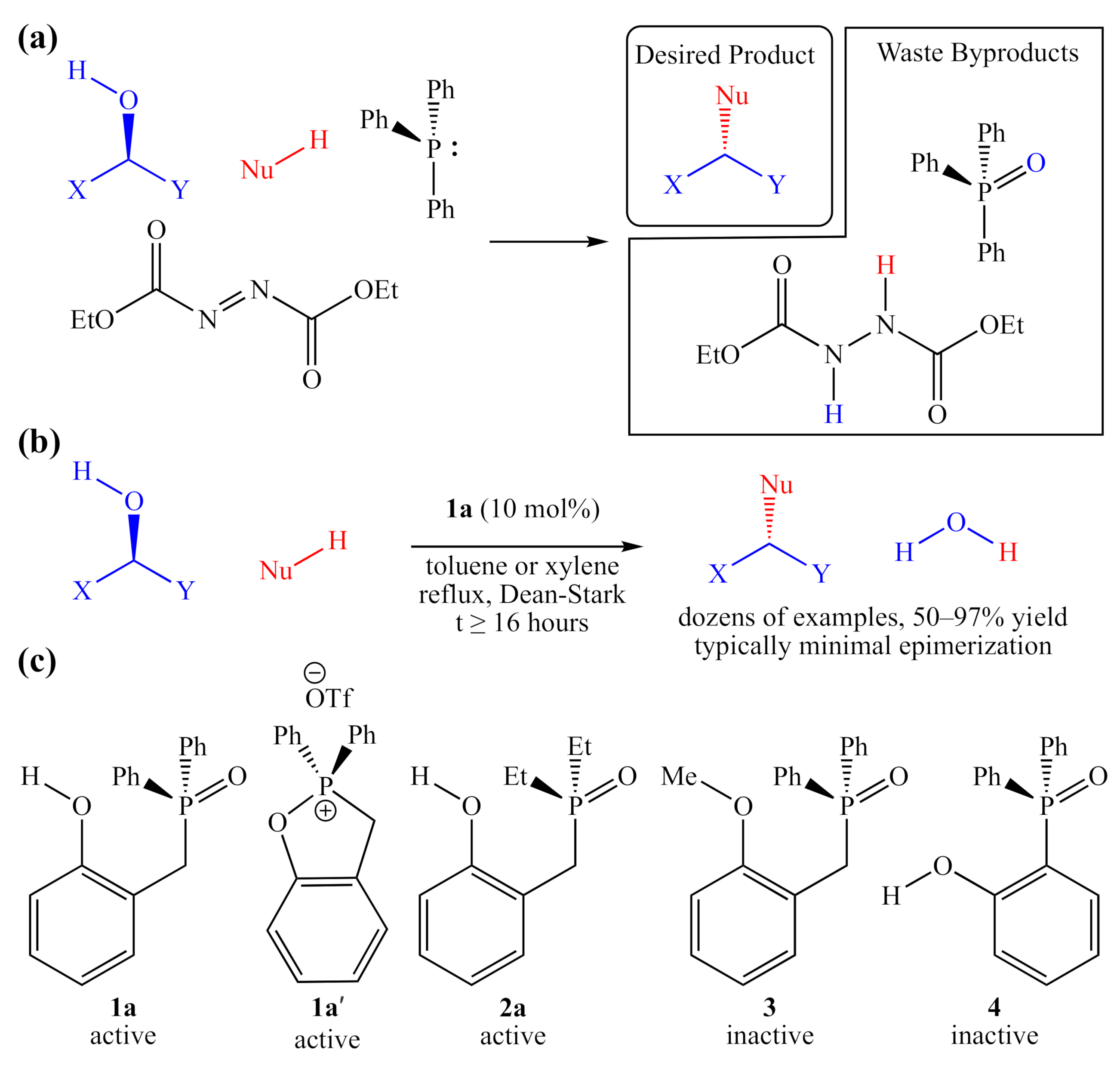
:1. Introduction
2. Results
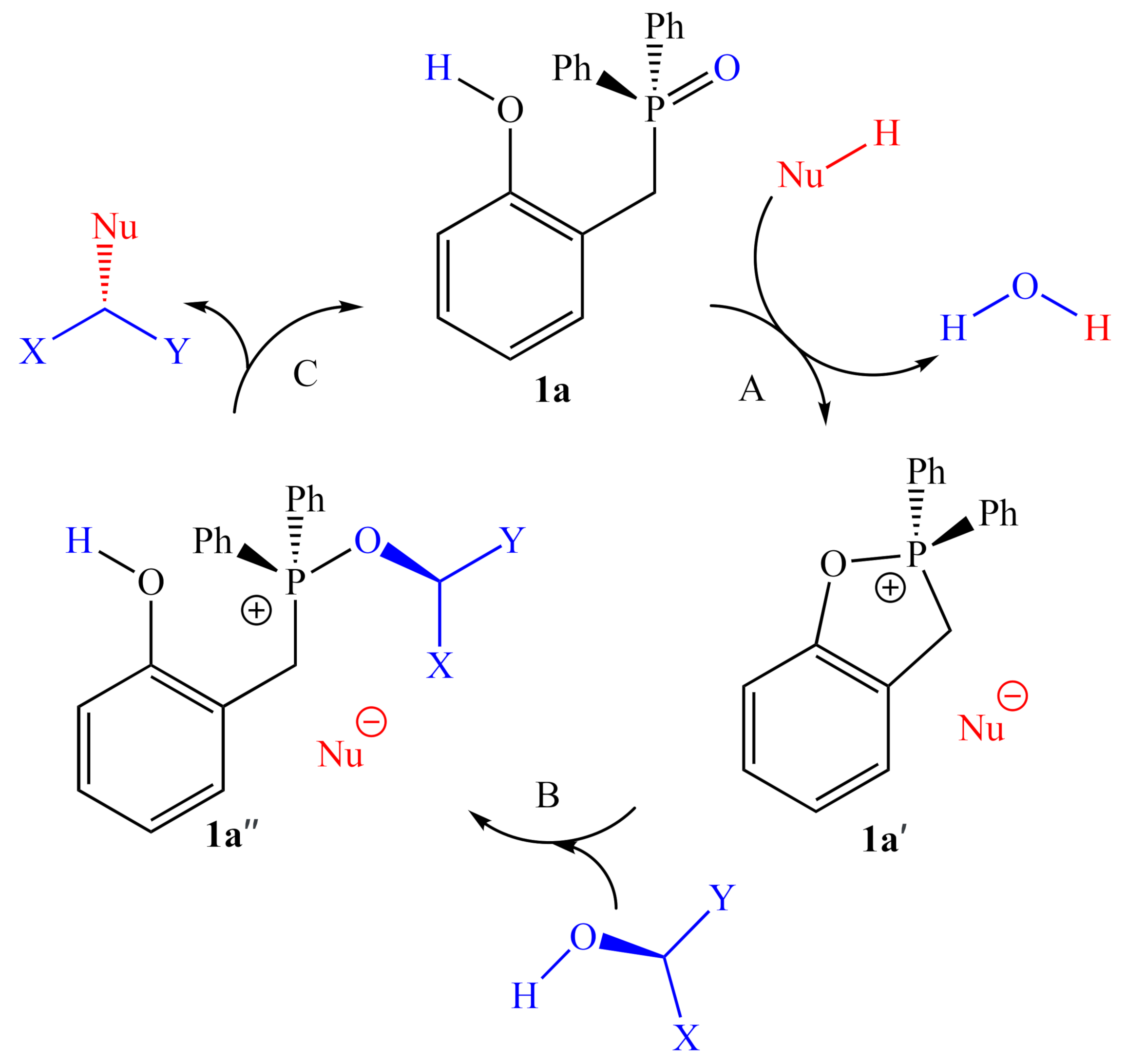
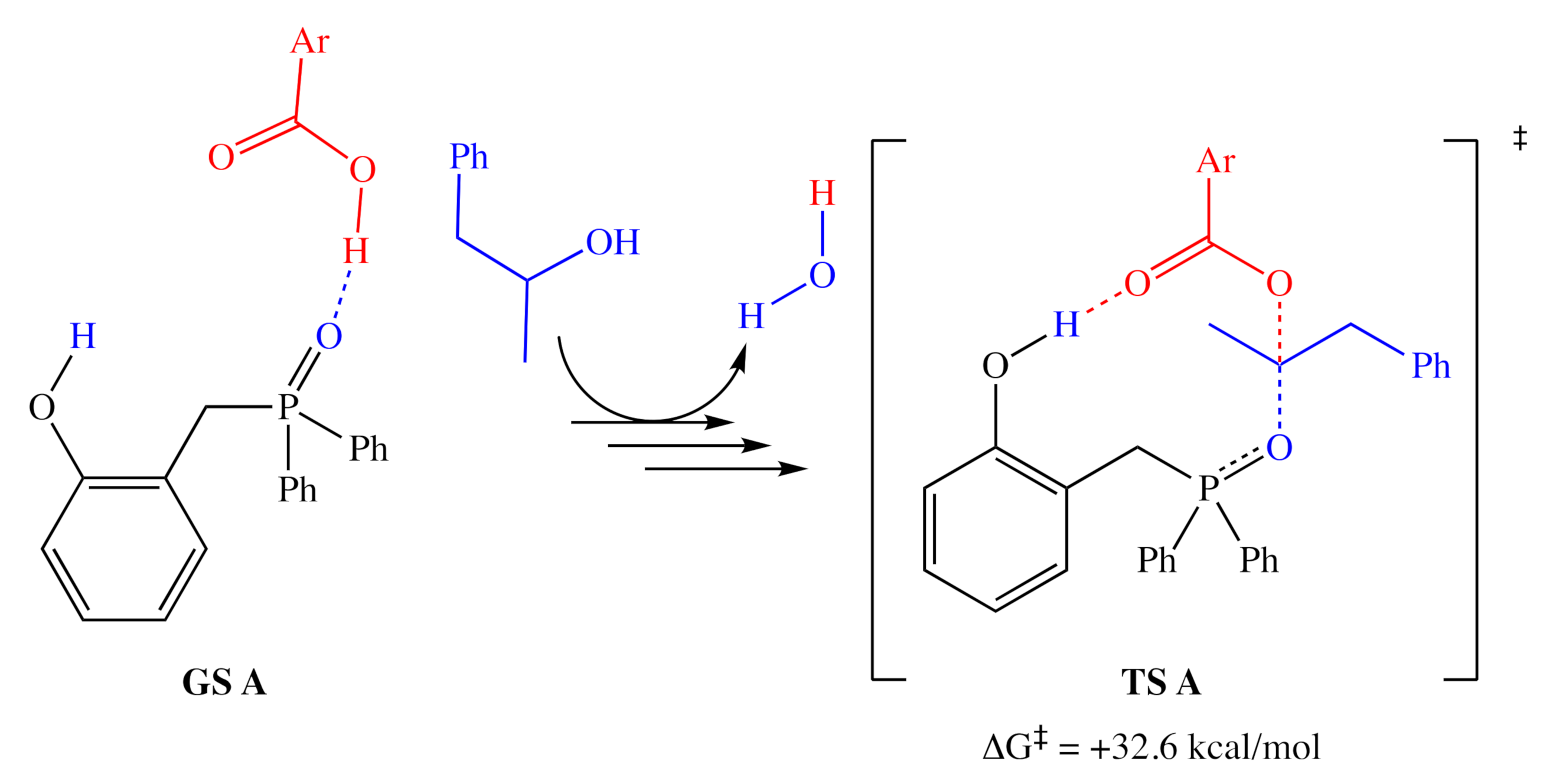
2.1. DFT Calculations
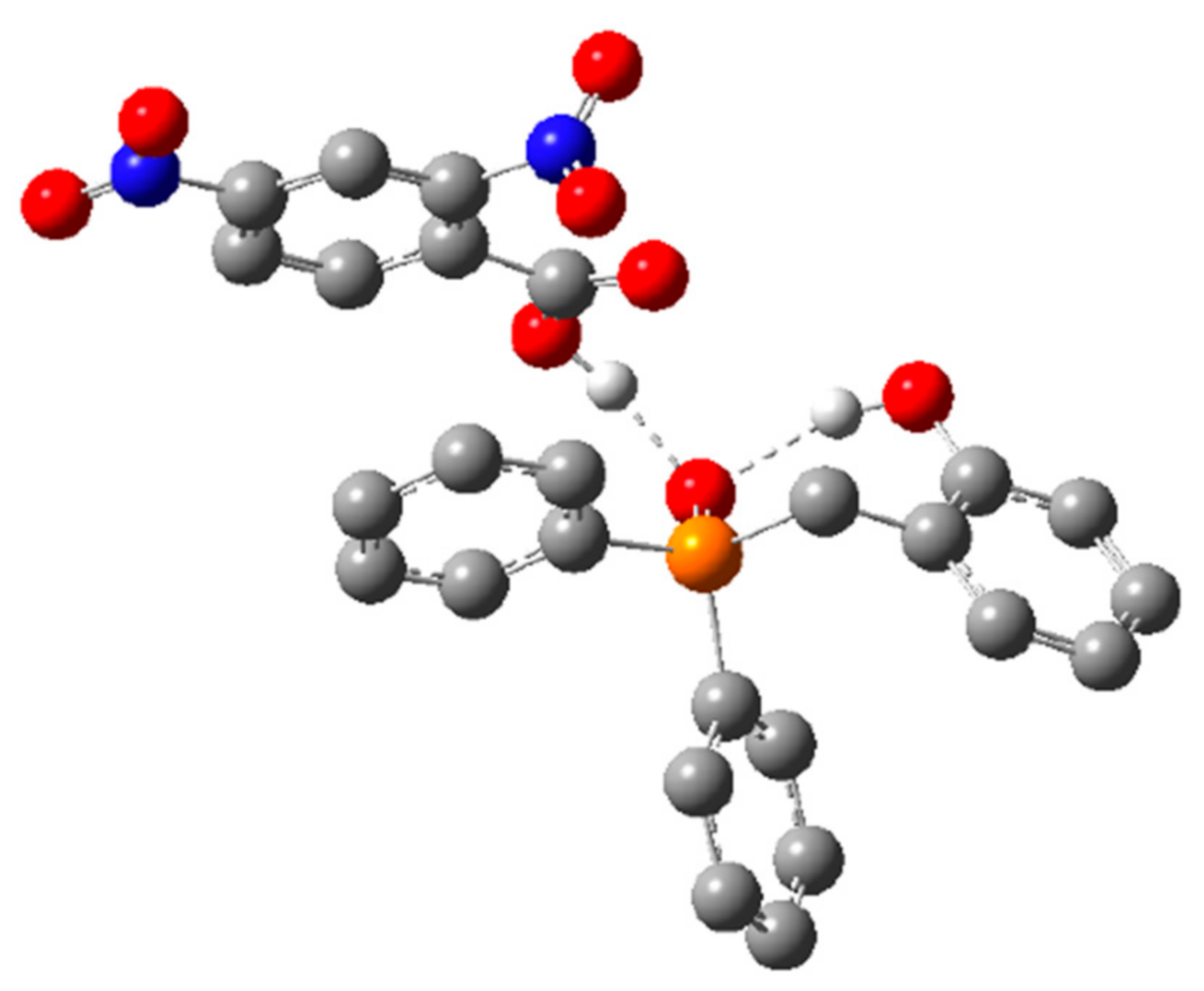
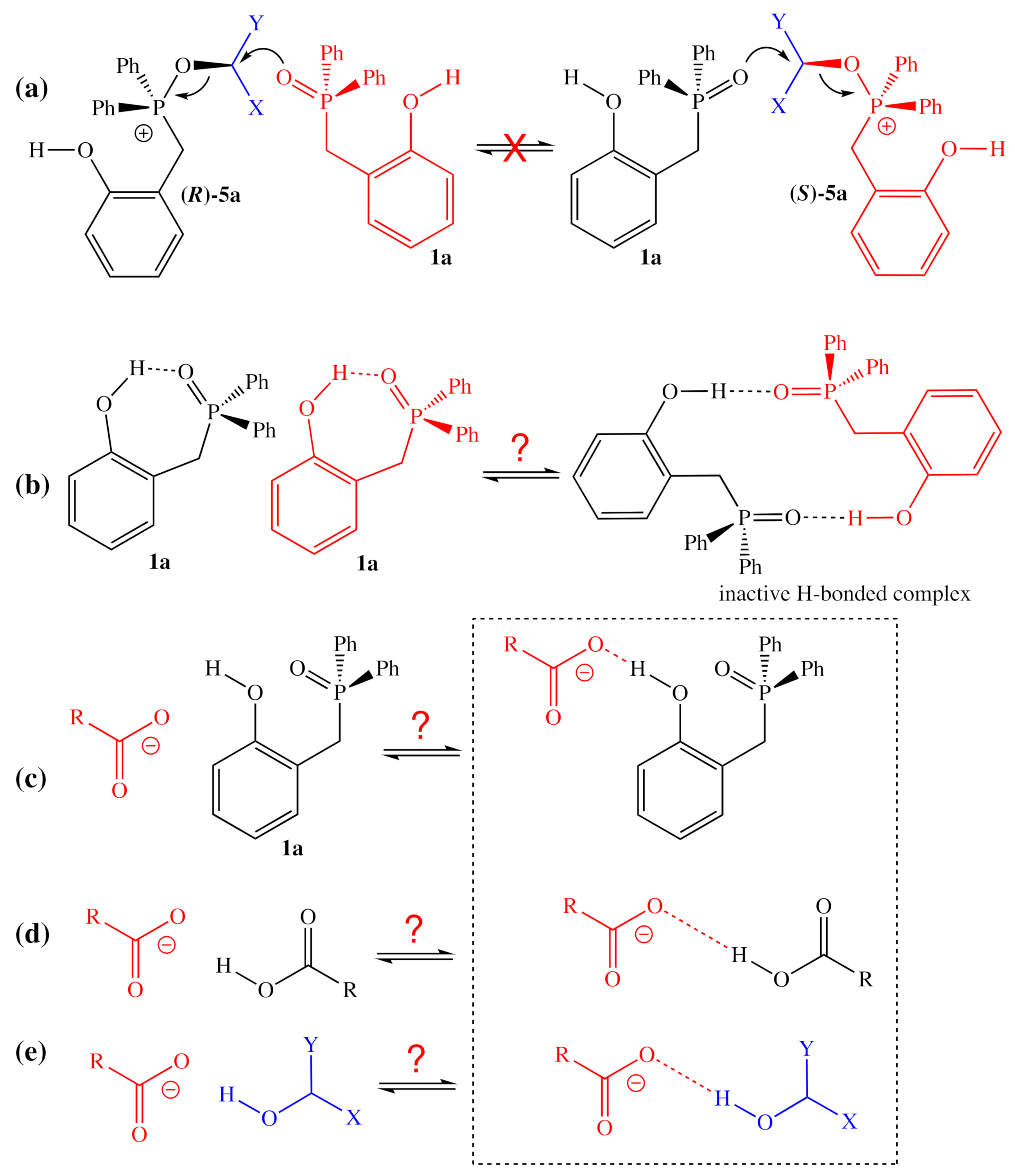
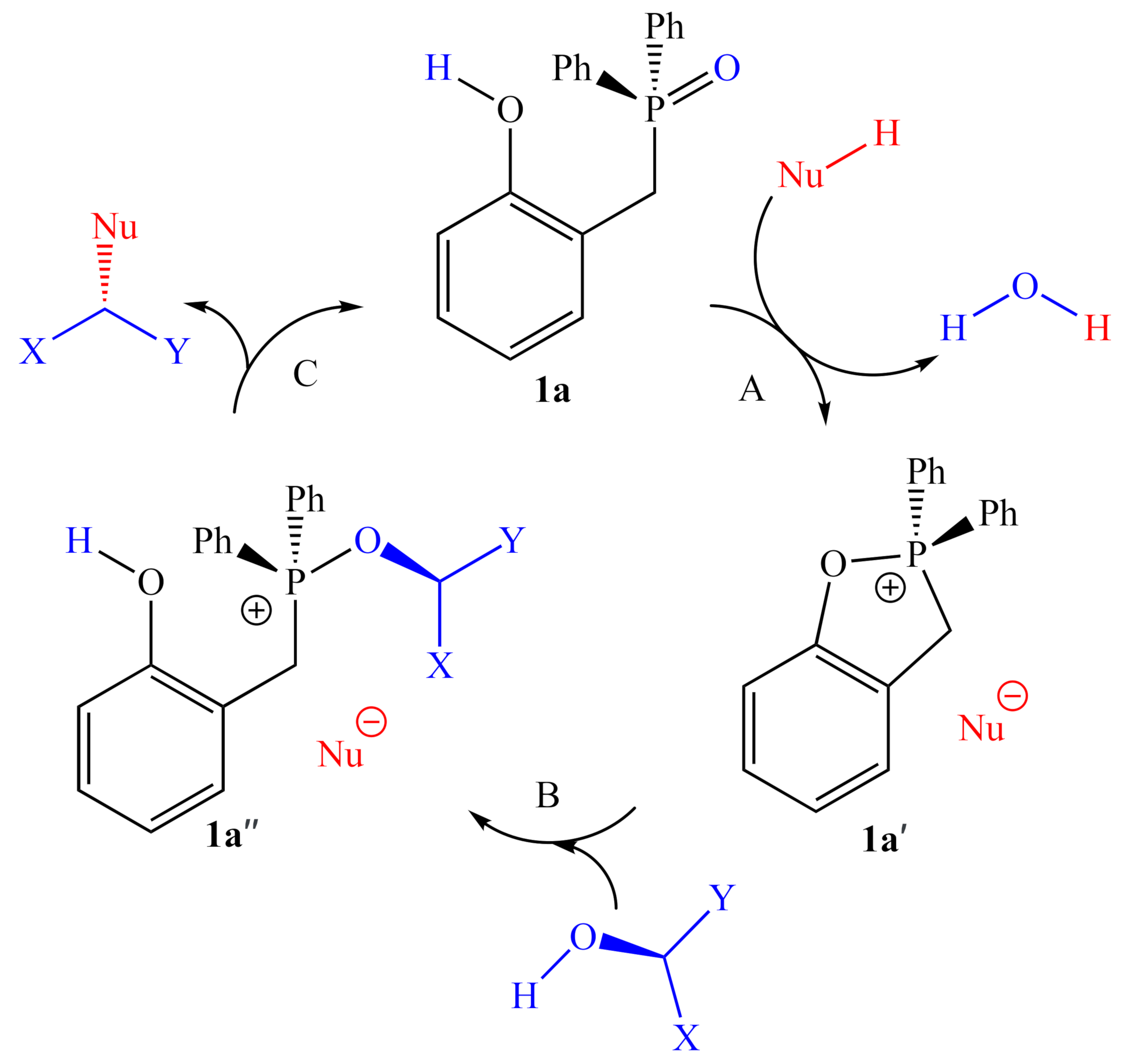
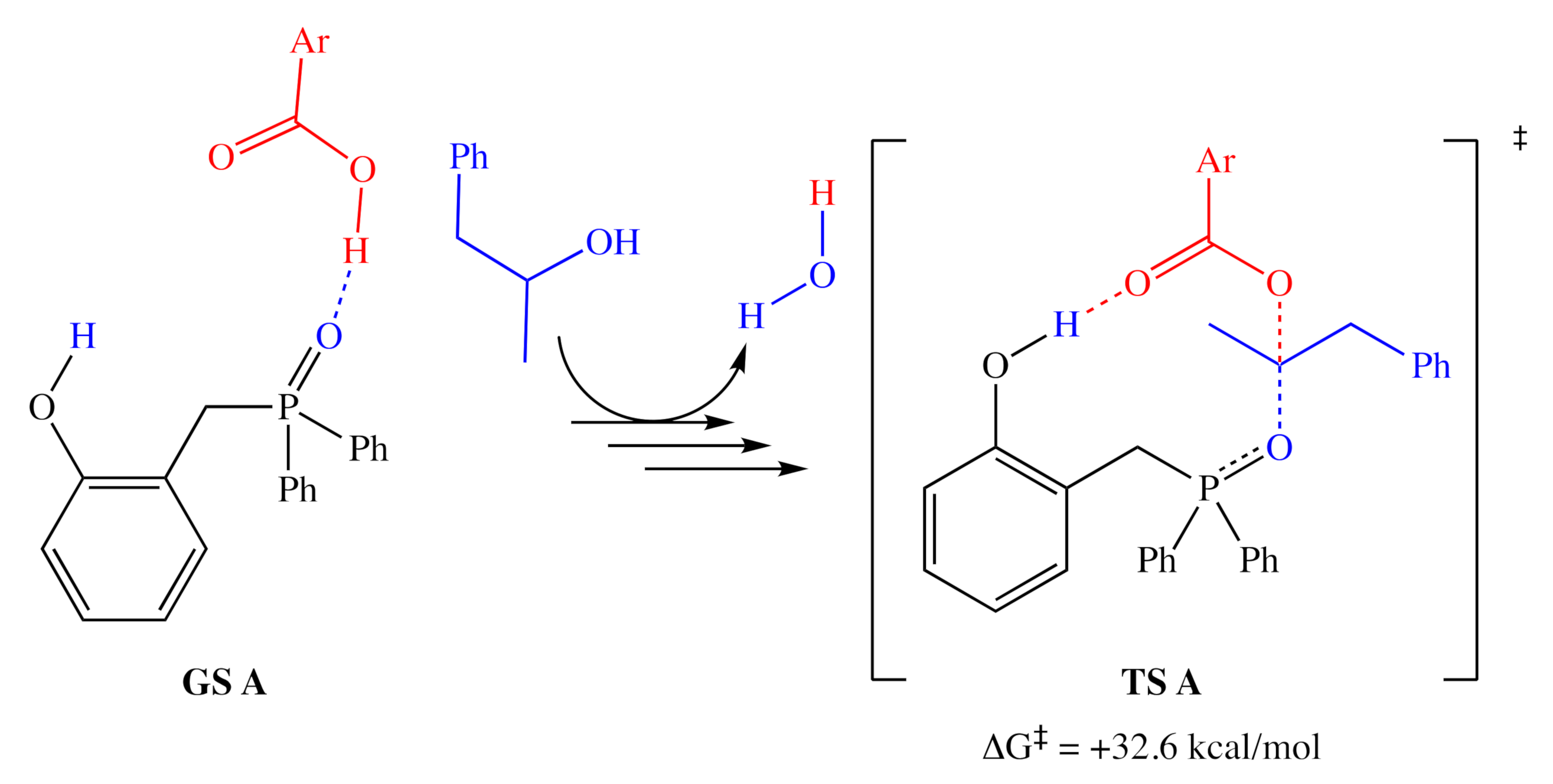
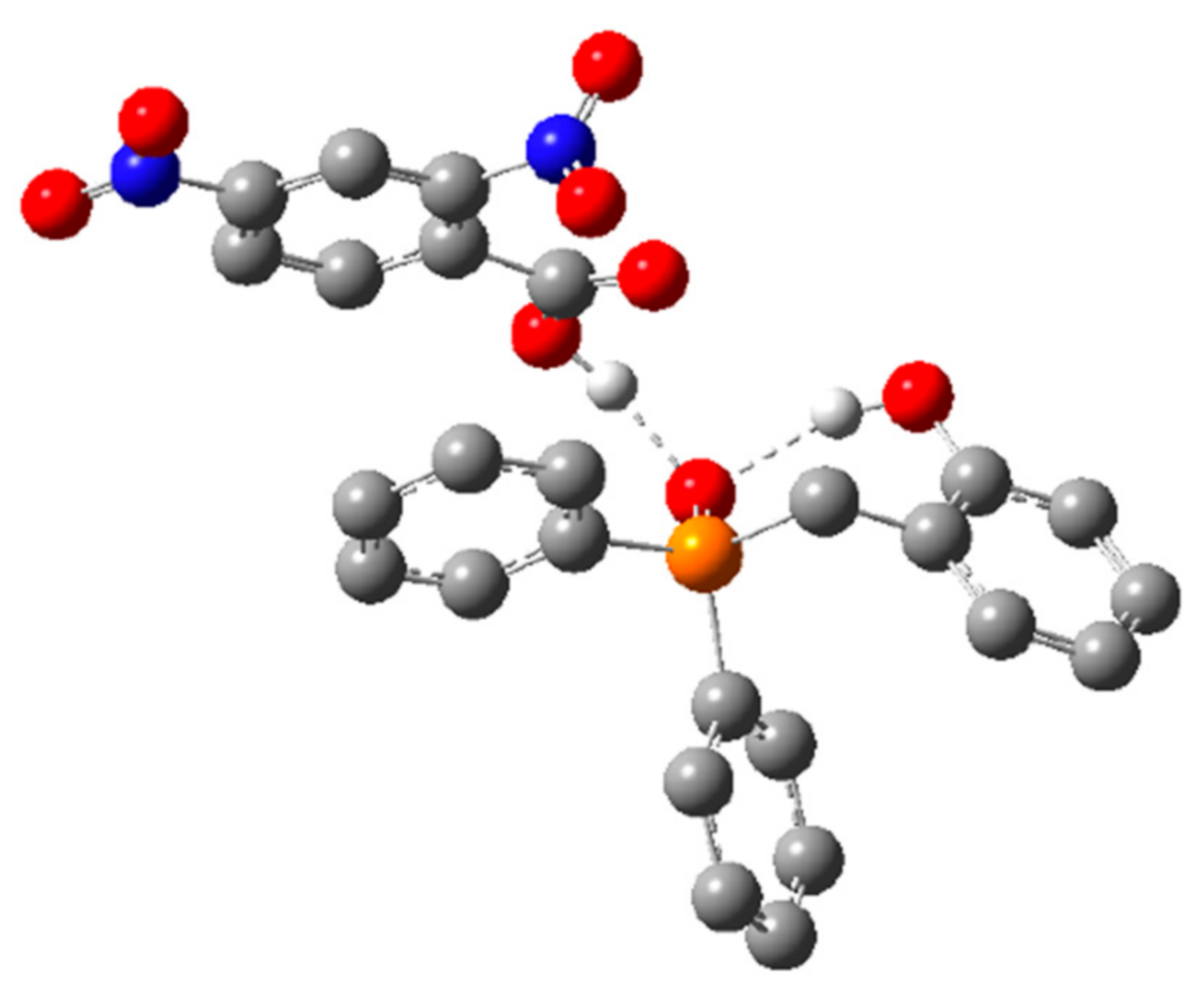
2.1.1. Alternative Resting State and Transition States
2.1.2. Structural Analogs
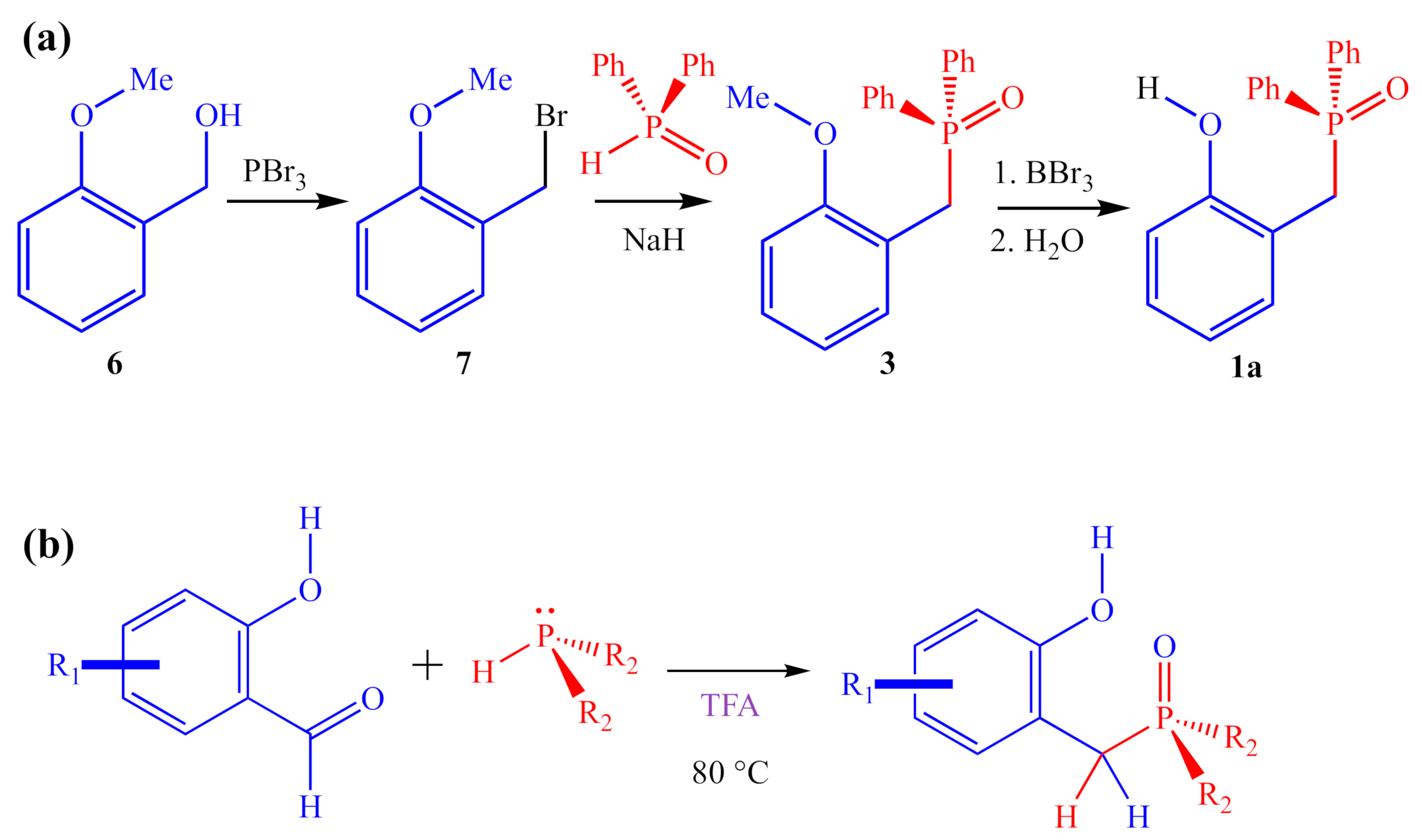
2.2. Synthesis of 2-HOBPO Catalyst Candidates
2.2.1. Small-Scale Syntheses (<20 mmol)
2.2.2. Large-Scale (30–50 mmol) Synthesis
2.3. Kinetics Studies
2.3.1. Rate Law of 2-Octanol and 2,4-DNBA Catalyzed by 1a
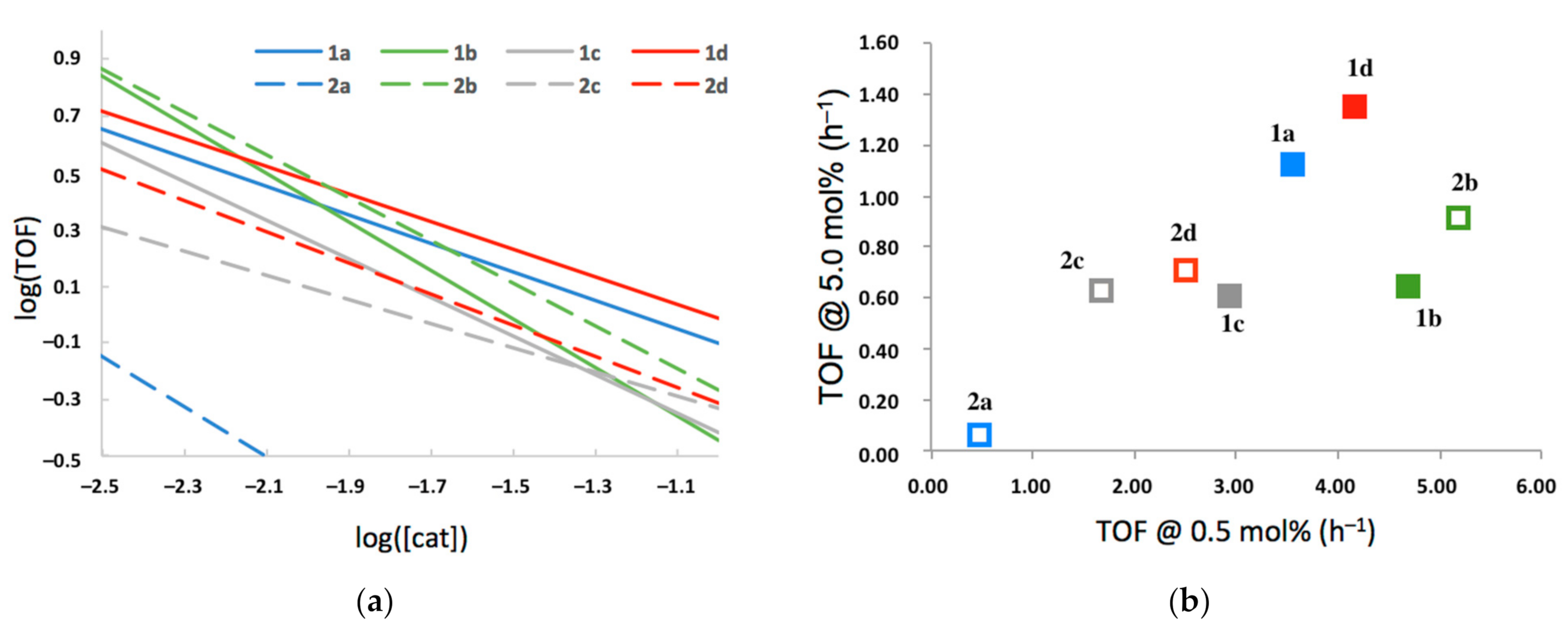
2.3.2. TOF as a Function of 2-HOBPO Loading
3. Discussion
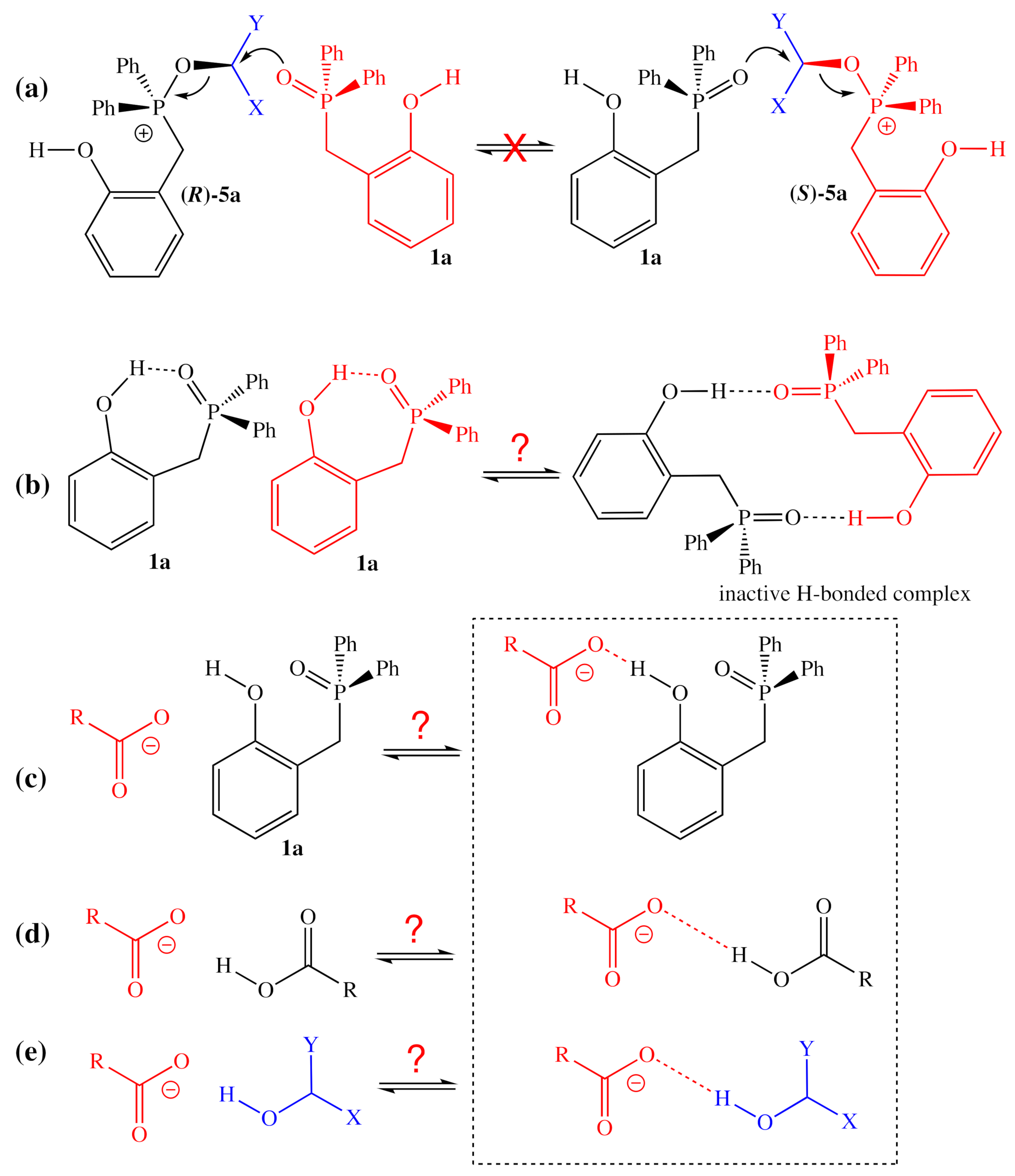
3.1. Interpretation of Rate Law Data
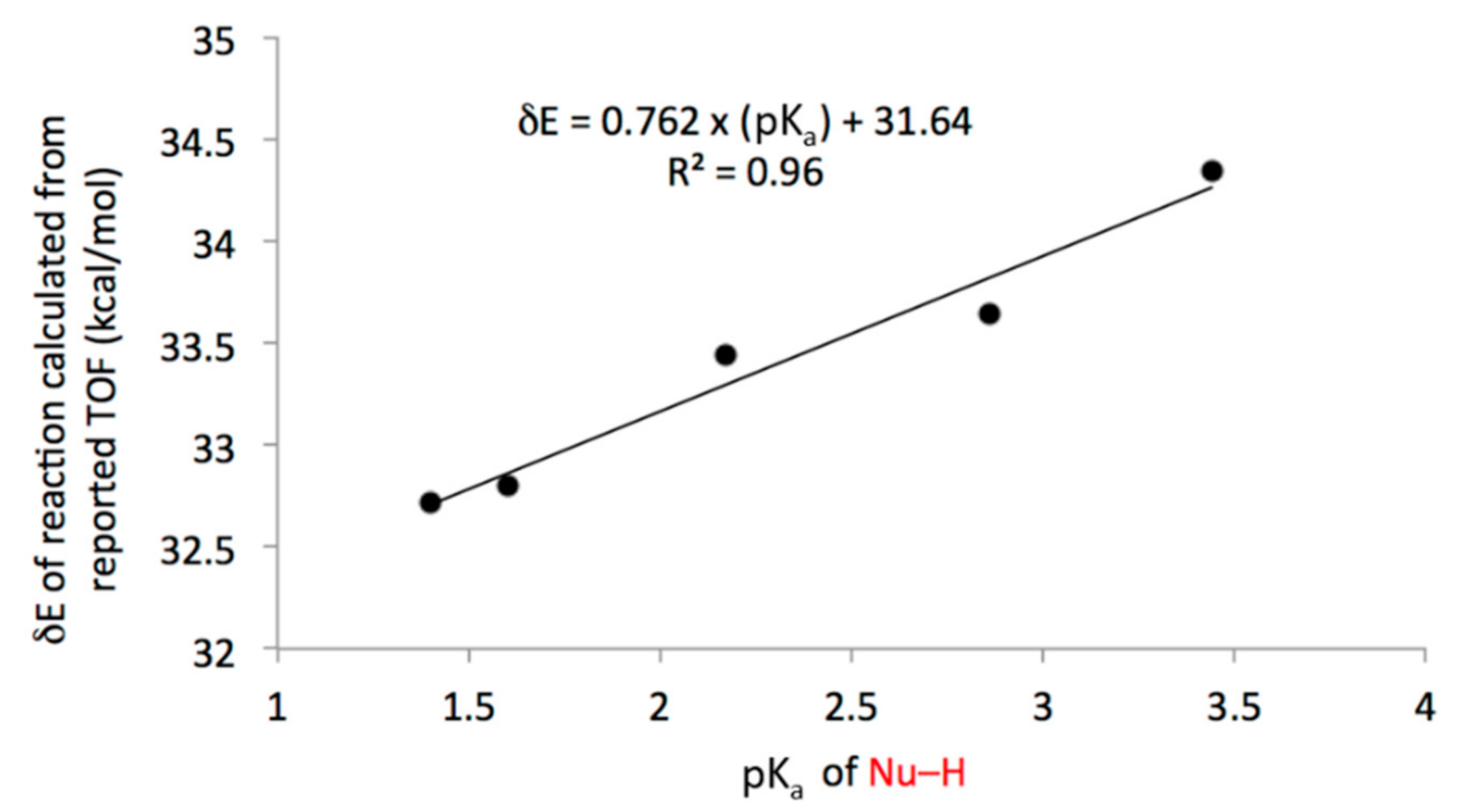
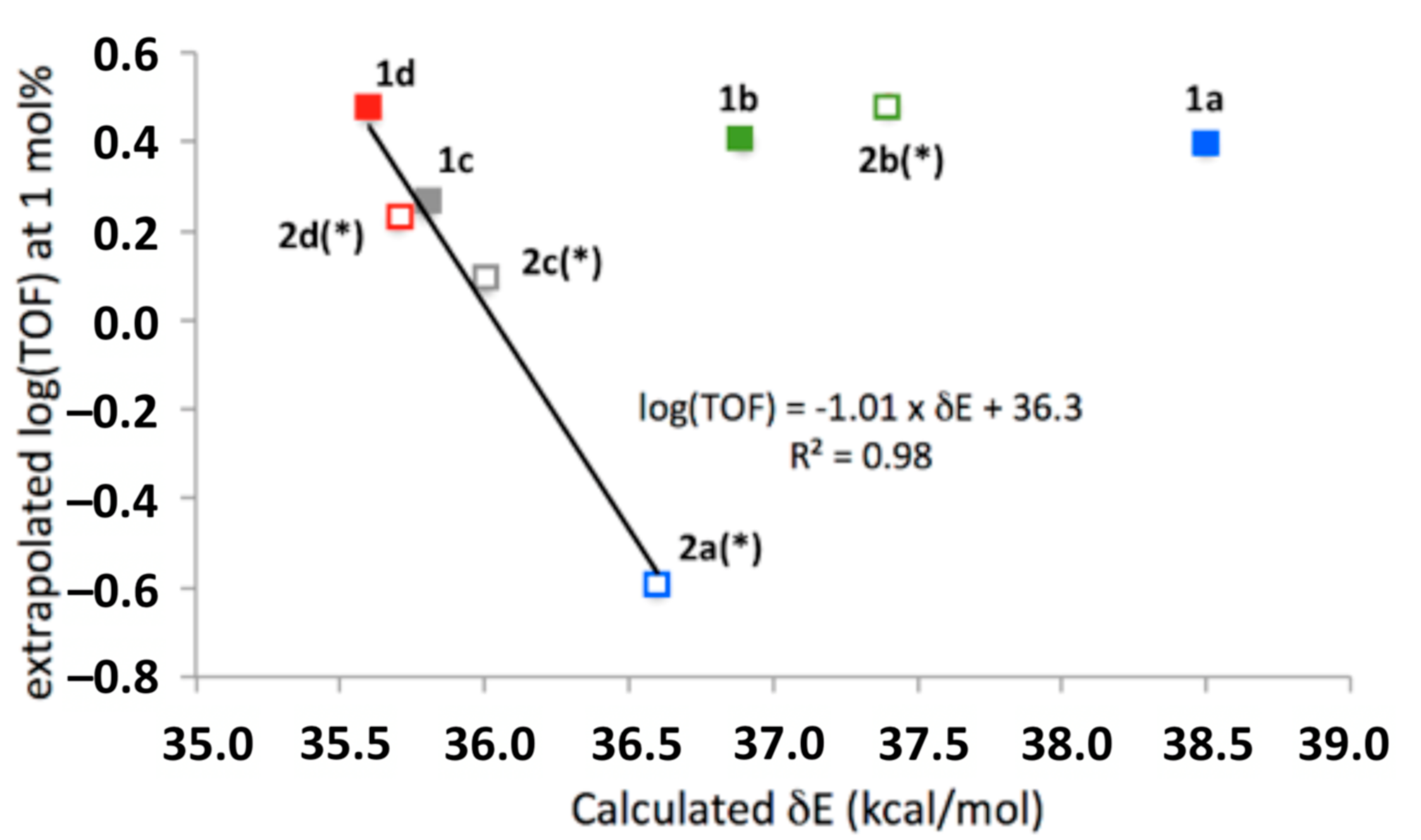
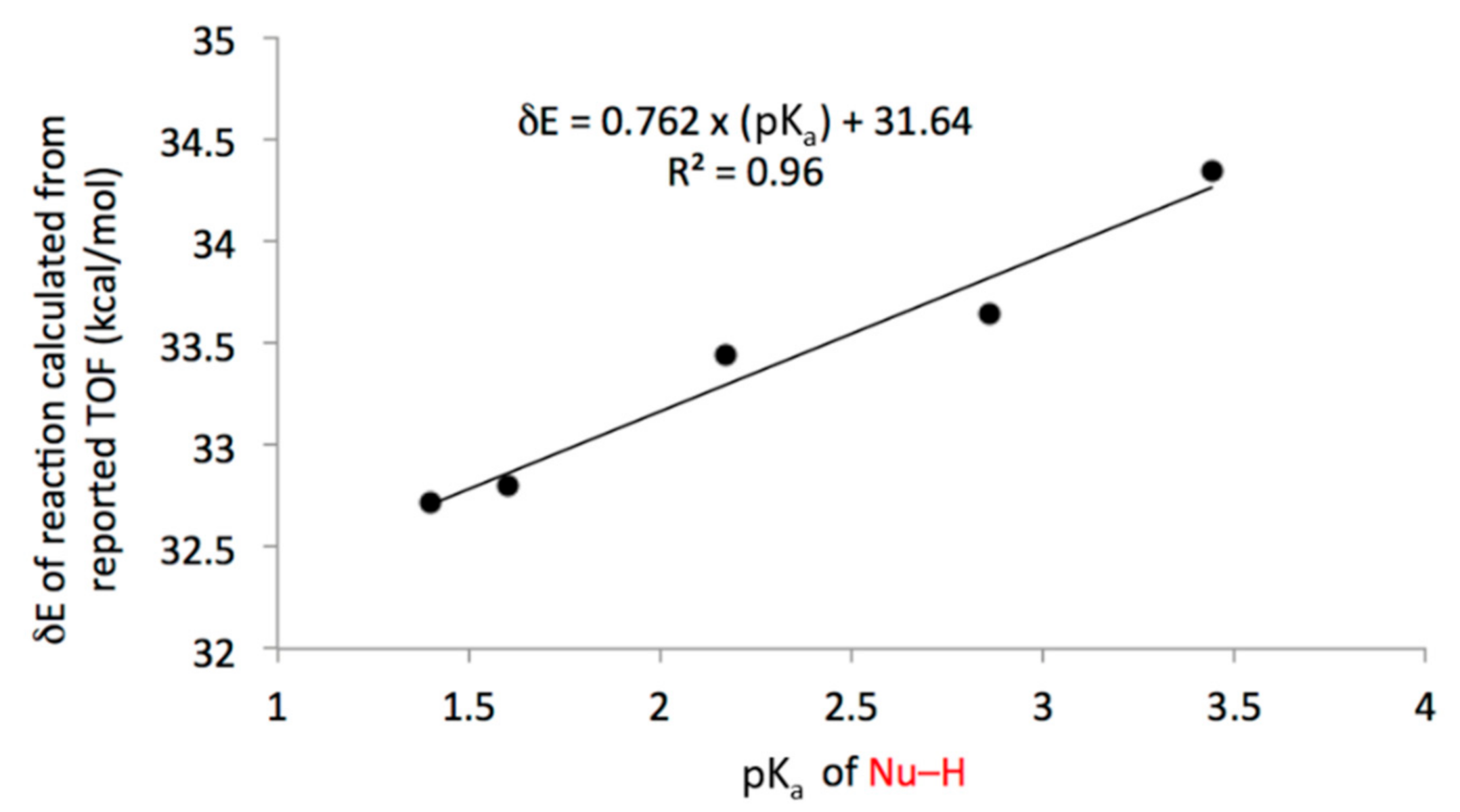
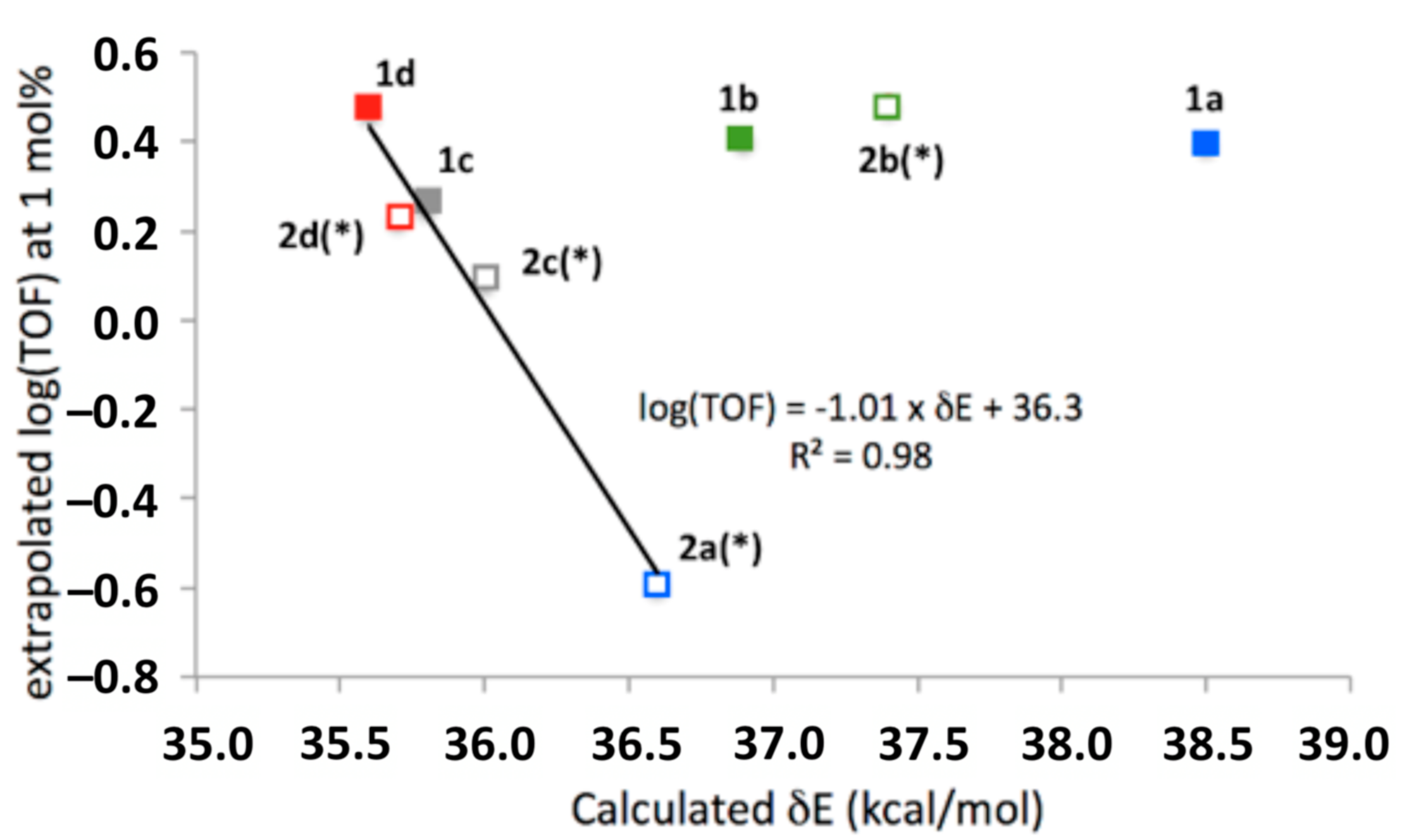
3.2. Comparison of Computational and Experimental Findings
3.3. Mechanistic Insight and SARs
- 2-HOBPO catalysts appear to be auto-inhibitory, with log(TOF) vs. log([2-HOBPO]) having slopes between −0.88 and −0.43.
- Both 2-HOBPOs containing aromatic substituents on the phosphine oxide (1x) and those containing aliphatic ones (2x) are competent catalysts.
- Only 1e and 2e, bearing trifluoromethyl substituents ortho to the phenol OH, appear to be completely inactive.
- The rate law appears to be nearly first order in alcohol and pronucleophile, consistent with a rate-determining transition state late in the catalytic cycle.
- There is no obvious relationship between the electron-donating or electron-withdrawing nature of the variable substituents, and the catalytic efficiency of the corresponding catalyst, but both 1b and 2b, with methyl substituents para to the phenol, appear to be the most active catalysts at low loadings, indicating significant levels of inherent activity, while at higher concentrations, 1a and 1d, with aromatic p substituents and electron-neutral or electron-poor phenol, appeared least inhibited at higher concentrations.
4. Materials and Methods
4.1. Materials
4.2. Generic Procedure for ARC Synthesis of 2-HOBPOs
4.3. Kinetic Studies
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Beddoe, R.H.; Andrews, K.G.; Magné, V.; Cathbertson, J.D.; Saska, J.; Shannon-Little, A.L.; Shanahan, S.E.; Sneddon, H.F.; Denton, R.M. Redox-neutral organocatalytic Mitsunobu reactions. Science 2019, 365, 910–914. [Google Scholar] [CrossRef] [PubMed]
- Swamy, K.C.K.; Kumar, N.N.B.; Balaraman, E.; Kumar, K.V.P.P. Mitsunobu and related reactions: Advances and applications. Chem. Rev. 2009, 109, 2551–2651. [Google Scholar] [CrossRef] [PubMed]
- Fletcher, S. The Mitsunobu reaction in the 21st century. Org. Chem. Front. 2015, 2, 739–752. [Google Scholar] [CrossRef]
- Tsunoda, T.; Yamamiya, Y.; Itô, S. 1,1′-(azodicarbonyl)dipiperidine-tributylphosphine, a new reagent system for mitsunobu reaction. Tetrahedron Lett. 1993, 34, 1639–1642. [Google Scholar] [CrossRef]
- Jackson, T.; Routledge, A. Synthesis and application of crown ether tagged triarylphosphines. Tetrahedron 2003, 44, 1305–1307. [Google Scholar] [CrossRef]
- Buonomo, J.A.; Aldrich, C.C. Mitsunobu reactions catalytic in phosphine and a fully catalytic system. Angew. Chem. Int. Ed. 2015, 54, 13041–13044. [Google Scholar] [CrossRef] [Green Version]
- But, T.Y.S.; Toy, P.H. Organocatalytic mitsunobu reactions. J. Am. Chem. Soc. 2006, 128, 9636–9637. [Google Scholar] [CrossRef]
- Zhou, L.; Perulli, S.; Mastandrea, M.M.; Llanes, P.; Lai, J.; Pericàs, M.A. Development of a robust immobilized organocatalyst for the redox-neutral mitsunobu reaction. Green Chem. 2021, 23, 8859–8864. [Google Scholar] [CrossRef]
- Zou, Y.; Wong, J.J.; Houk, K.N. Computational exploration of a redox-neutral organocatalytic mitsunobu reaction. J. Am. Chem. Soc. 2020, 142, 16403–16408. [Google Scholar] [CrossRef]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom-atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef] [Green Version]
- Kozuch, S.; Shaik, S. How to conceptualize catalytic cycles? The energetic span model. Acc. Chem. Res. 2011, 44, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Wittig, G.; Schöllkopf, U. Über Triphenyl-phosphine-methylene als olefinbildende Reagenzian. Chem. Ber. 1954, 87, 1318–1330. [Google Scholar] [CrossRef]
- Golobov, Y.G.; Zhmurova, I.N.; Kasukhin, L.F. Sixty years of Staudinger reaction. Tetrahedron 1981, 37, 437–472. [Google Scholar] [CrossRef]
- Matrosov, E.I.; Tsvetkov, E.N.; Mironova, Z.N.; Malevannaya, R.A.; Kabachnik, M.I. Acid-base properties of phosphine oxides in nitromethane. Izv. Akad. Nauk. SSSR Seriya Khimicheskaya 1975, 6, 1333–1337. [Google Scholar] [CrossRef]
- Gu, X.; Yuan, H.; Jiang, J.; Wu, Y.; Bai, W.-J. Catalytic asymmetric hydrophosphination of ortho-quinone methides. Org. Lett. 2018, 20, 7229–7233. [Google Scholar] [CrossRef]
- Tatarinov, D.A.; Kuznetsov, D.M.; Kostin, A.A.; Mironov, V.F. 2-Ethoxy-2,3-dihydro[d][1,2]oxaphosphole 2-Oxide in the Synthesis of Dialkyl(diaryl)2-hydroxybenzyl)phosphine Oxides. Zhurnal Obs. Khimii 2016, 86, 386–390. [Google Scholar] [CrossRef]
- Epstein, M.; Buckler, S.A. Reactions of primary and secondary phosphines with aldehydes and ketones. Tetrahedron 1962, 18, 1231–1242. [Google Scholar] [CrossRef]
- Chikkali, S.; Gudat, D. Hydrophosphination of phenolic aldehydes as facile synthetic approach to catechol-functionalized phosphane oxides and phosphanes. Eur. J. Inorg. Chem. 2006, 2006, 3005–3009. [Google Scholar] [CrossRef]
- Bloomfield, A.J.; Qian, J.M.; Herzon, S.B. Single-step synthesis of secondary phosphine oxides. Organometallics 2010, 29, 4193–4195. [Google Scholar] [CrossRef]
- Zhao, Y.; Tishchenko, O.; Truhlar, D.G. How well can density functional methods describe hydrogen bonds to π acceptors? J. Phys. Chem. B. 2005, 109, 19046–19051. [Google Scholar] [CrossRef]
- Trenkle, A.; Vahrenkamp, H.; Svoboda, J.; Brewer, L. 40. Dimethylphosphine. Inorg. Synth. 1982, 21, 180–181. [Google Scholar] [CrossRef]
- Zhou, Y.; Wu, J.; Lemmon, E.W. Thermodynamic properties of o-Xylene, m-Xylene, p-Xylene, and ethylbenzene. J. Phys. Chem. Ref. Data 2012, 41, 023103. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structural Representation 1 | Method | ∆H 2 | ∆G413.15 |
|---|---|---|---|
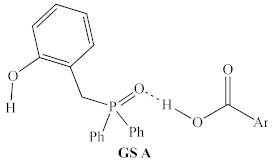 | B3LYP/ 6-31G | +8.4 kcal/mol | +6.9 kcal/mol |
| ωB97X-D/ 6-311G(d,p) | +5.2 kcal/mol | +6.0 kcal/mol | |
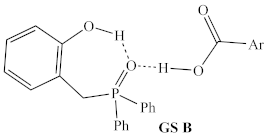 | B3LYP/ 6-31G | 0 kcal/mol | 0 kcal/mol |
| ωB97X-D/ 6-311G(d,p) | 0 kcal/mol | 0 kcal/mol | |
 | B3LYP/ 6-31G | +22.2 kcal/mol | +5.1 kcal/mol |
| ωB97X-D/ 6-311G(d,p) | +20.4 kcal/mol | +3.9 kcal/mol | |
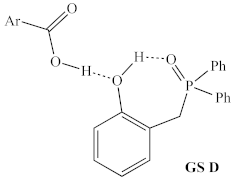 | B3LYP/ 6-31G | +4.4 kcal/mol | +4.0 kcal/mol |
| ωB97X-D/ 6-311G(d,p) | +5.0 kcal/mol | +5.2 kcal/mol | |
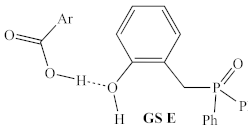 | B3LYP/ 6-31G | +16.1 kcal/mol | +13.1 kcal/mol |
| ωB97X-D/ 6-311G(d,p) | +9.0 kcal/mol | +9.6 kcal/mol | |
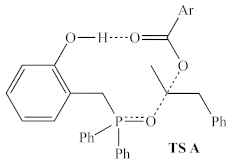 | ωB97X-D/ 6-311G(d,p) | +31.4 kcal/mol 3 | +38.5 kcal/mol 3 |
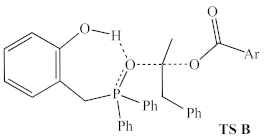 | ωB97X-D/ 6-311G(d,p) | +41.1 kcal/mol 3 | +44.2 kcal/mol 3 |
| Structure | R2 | Compound | ∆G‡ (TS A) (kcal/mol) | Imaginary Mode (cm−1) | ∆G‡ (TS B) (kcal/mol) | Imaginary Mode (cm−1) |
|---|---|---|---|---|---|---|
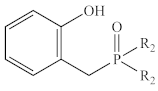 | Ph | 1a | 38.5 | −393 | 44.2 | −359 |
| Me | 2a* 1 | 36.6 | −386 | 44.1 | −329 | |
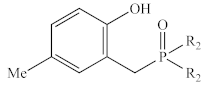 | Ph | 1b | 36.9 | −388 | 44.5 | −349 |
| Me | 2b* 1 | 37.4 | −386 | 44.4 | −331 | |
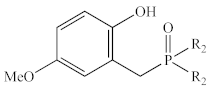 | Ph | 1c | 35.8 | −381 | 43.5 | −355 |
| Me | 2c* 1 | 36.0 | −388 | 43.3 | −323 | |
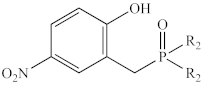 | Ph | 1d | 35.6 | −369 | 43.6 | −315 |
| Me | 2d* 1 | 35.7 | −382 | 47.0 | −301 |
| Structure | R2 | Compound | Yield 1 | 31P (δ) 2 |
|---|---|---|---|---|
 | Ph | 1a | 89% | 37.9 |
| Et | 2a | 67% | 58.5 | |
 | Ph | 1b | 88% | 38.1 |
| Et | 2b | 18% 3 | 58.0 | |
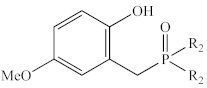 | Ph | 1c | 74% | 38.0 |
| Et | 2c | 76% | 58.6 | |
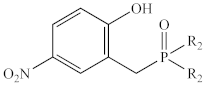 | Ph | 1d | 77% | 36.4 |
| Et | 2d | 92% | 61.6 | |
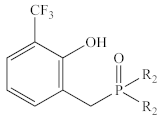 | Ph | 1e | 39% | 38.6 |
| Et | 2e | 37% 3 | 58.7 |
| Catalyst | TOF @ 0.5 mol% | TOF @ 5 mol% | log(TOF)/log([2-HOBPO]) |
|---|---|---|---|
| 1a | 3.56 h−1 | 1.12 h−1 | −0.50 |
| 2a | 0.47 h−1 | 0.06 h−1 | −0.88 |
| 1b | 4.66 h−1 | 0.65 h−1 | −0.86 |
| 2b | 5.17 h−1 | 0.91 h−1 | −0.75 |
| 1c | 2.94 h−1 | 0.61 h−1 | −0.68 |
| 2c | 1.67 h−1 | 0.62 h−1 | −0.43 |
| 1d | 4.16 h−1 | 1.36 h−1 | −0.49 |
| 2d | 2.52 h−1 | 0.71 h−1 | −0.55 |
| 1e 1 | N/A | N/A | N/A |
| 2e 1 | N/A | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, M.A.; Brown, S.L.; Beres, D.R.; Frederic, W.M.; Banks, A.M.; Bloomfield, A.J. Phenolic 3° Phosphine Oxides as a Class of Metal-Free Catalysts for the Activation of C–O Bonds in Aliphatic Alcohols: Direct Synthesis of Catalyst Candidates, and Kinetic Studies. Inorganics 2022, 10, 35. https://doi.org/10.3390/inorganics10030035
Martin MA, Brown SL, Beres DR, Frederic WM, Banks AM, Bloomfield AJ. Phenolic 3° Phosphine Oxides as a Class of Metal-Free Catalysts for the Activation of C–O Bonds in Aliphatic Alcohols: Direct Synthesis of Catalyst Candidates, and Kinetic Studies. Inorganics. 2022; 10(3):35. https://doi.org/10.3390/inorganics10030035
Chicago/Turabian StyleMartin, Matthew A., Sadie L. Brown, Danielle R. Beres, Wrebekah M. Frederic, Ashley M. Banks, and Aaron J. Bloomfield. 2022. "Phenolic 3° Phosphine Oxides as a Class of Metal-Free Catalysts for the Activation of C–O Bonds in Aliphatic Alcohols: Direct Synthesis of Catalyst Candidates, and Kinetic Studies" Inorganics 10, no. 3: 35. https://doi.org/10.3390/inorganics10030035
APA StyleMartin, M. A., Brown, S. L., Beres, D. R., Frederic, W. M., Banks, A. M., & Bloomfield, A. J. (2022). Phenolic 3° Phosphine Oxides as a Class of Metal-Free Catalysts for the Activation of C–O Bonds in Aliphatic Alcohols: Direct Synthesis of Catalyst Candidates, and Kinetic Studies. Inorganics, 10(3), 35. https://doi.org/10.3390/inorganics10030035