
Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors

 ,
,  ,
,
Abstract
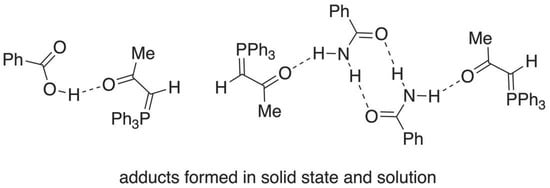
1. Introduction
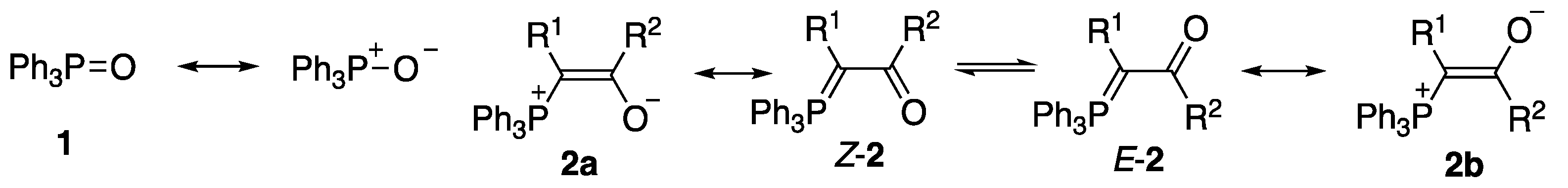
2. Results and Discussion
2.1. Initial Discovery
2.2. Scope of the Stabilised Ylide—Carboxylic Acid Interaction
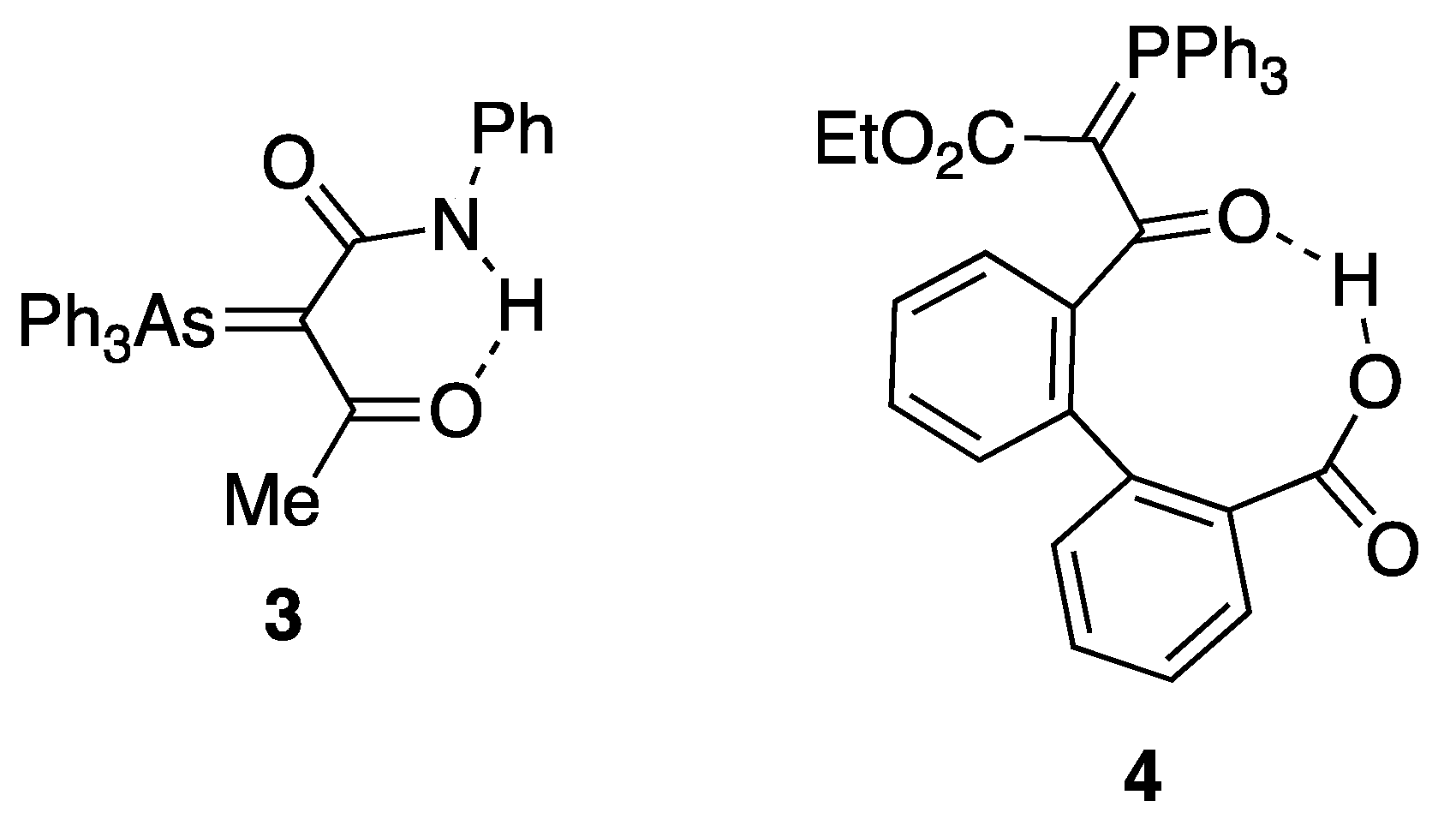
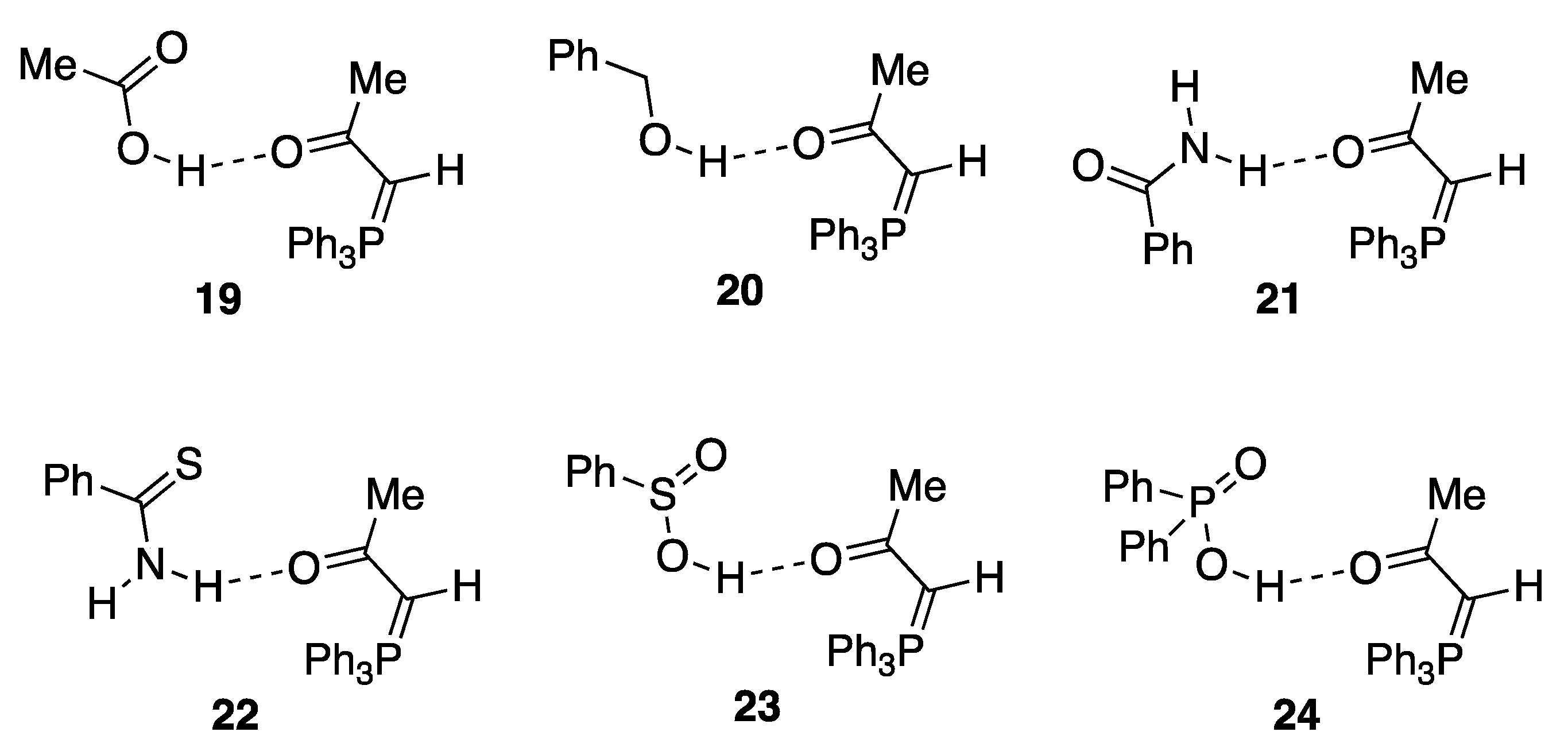
2.3. Expanding the Range of Hydrogen Bond Donors
2.4. Synthesis and Properties of a Bis(Stabilised Ylide)
3. Experimental Section
3.1. General Experimental Details
3.2. Preparation of Adducts with Methyl Triphenylphosphoranylidenepyruvate
3.2.1. Methyl Triphenylphosphoranylidenepyruvate—Benzoylpyruvic Acid Adduct 9
3.2.2. Methyl Triphenylphosphoranylidenepyruvate—Benzoic Acid Adduct 10
3.3. Preparation of Adducts with Triphenylphosphoranylideneacetone
3.3.1. Triphenylphosphoranylideneacetone—Benzoic Acid Adduct 11
3.3.2. Triphenylphosphoranylideneacetone—Phenylacetic Acid Adduct 13
3.3.3. Triphenylphosphoranylideneacetone—Diphenylacetic Acid Adduct 14
3.3.4. Triphenylphosphoranylideneacetone—2-Phenylbutyric Acid Adduct 15
3.4. Preparation of Adducts with Triphenylphosphoranylideneacetophenone
Triphenylphosphoranylideneacetophenone—Diphenylacetic Acid Adduct 17
3.5. Preparation of Adducts with 1-Phenyl-1-triphenylphosphoranylidenepropan-2-One
1-Phenyl-1-triphenylphosphoranylidenepropan-2-One—Benzoic Acid Adduct 12
3.6. Formation of Solution Adducts with Triphenylphosphoranylideneacetone
3.7. Preparation of Adducts with Triphenylphosphoranylideneacetone
3.7.1. Triphenylphosphoranylideneacetone—Benzamide Adduct 21
3.7.2. Triphenylphosphoranylideneacetone—Thiobenzamide Adduct 22
3.8. Preparation of Adducts with 1,3-Bis(triphenylphosphoranylideneacetyl)benzene 26
3.8.1. 1,3-Bis(triphenylphosphoranylideneacetyl)benzene 26
3.8.2. 1,3-Bis(triphenylphosphoranylideneacetyl)benzene—Benzoic Acid Adduct 27
3.9. X-ray Structure Determination of Adducts
3.9.1. Methyl Triphenylphosphoranylidenepyruvate—Benzoylpyruvic Acid Adduct 9
3.9.2. Triphenylphosphoranylideneacetone—Benzoic Acid Adduct 11
3.9.3. Triphenylphosphoranylideneacetone—Benzamide Adduct 21
3.9.4. 1,3-Bis(triphenylphosphoranylideneacetyl)benzene—Benzoic Acid Adduct 27
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Ramirez, F.; Dershowitz, S. Crystalline complexes of the phosphoryl group with polyphenols. J. Org. Chem. 1959, 24, 704–705. [Google Scholar] [CrossRef]
- Etter, M.C.; Baures, P.W. Triphenylphosphine oxide as a crystallization aid. J. Am. Chem. Soc. 1988, 110, 639–640. [Google Scholar] [CrossRef]
- Etter, M.C.; Reutzel, S.M. Hydrogen bond directed cocrystallization and molecular recognition properties of acyclic imides. J. Am. Chem. Soc. 1991, 113, 2586–2598. [Google Scholar] [CrossRef]
- Thierbach, D.; Huber, F. Darstellung sowie Krystall- und Molekülstruktur von 2 Ph3PO.(COOH)2. Z. Anorg. Allg. Chem. 1981, 477, 101–107. [Google Scholar] [CrossRef]
- Smith, G.; Lynch, D.E.; Byriel, K.A.; Kennard, C.H.L. Molecular cocrystals of carboxylic acids Part 24: Cocrystals involving triphenylphosphine oxide: Structures of unique adduct hydrates of triphenylphosphine oxide with adamantane carboxylic acid and terephthalic acid and the anhydrous adduct with o-phthalic acid. Z. Krystallogr. 1997, 212, 130–134. [Google Scholar] [CrossRef]
- Declerq, J.P.; Germain, G.; Putzeys, J.P.; Rona, S.; van Meerssche, M. Dimethylmalonic acid-triphenylphosphine oxide, C41H38O6P2. Cryst. Struct. Commun. 1974, 3, 579–582. [Google Scholar]
- Golic, L.; Kaucic, V. Triphenylphosphine oxide-trichloroacetic acid, (C6H5)3PO.HOOC-CCl3. Cryst. Struct. Commun. 1976, 5, 319–324. [Google Scholar]
- Al-Farhan, K.A. Triphenylphosphine oxide—3-chlorobenzoic acid (1/1). Acta Crystallogr. Sect. C 2003, 59, 179–180. [Google Scholar] [CrossRef]
- Lynch, D.E.; Smith, G.; Byriel, K.A.; Kennard, C.H.L.; Whittaker, A.K.; Hanna, J.V. Molecular cocrystals of carboxylic acids XVII. Spectral characterization of the adducts of triphenylphosphine oxide with substituted phenoxyacetic acids and the crystal structure of the 1:1 adduct with (4-chloro-2-methylphenoxy)acetic acid. Aust. J. Chem. 1994, 47, 1401–1411. [Google Scholar] [CrossRef]
- Lariucci, C.; de Almeida Santos, R.H.; Lechat, J.R. Structure of the 1:1 adduct formed by diphenylmethanol with triphenylphosphine oxide. Acta Crystallogr. Sect. C 1986, 42, 1825–1828. [Google Scholar] [CrossRef]
- Steiner, T. Triphenylmethanol-triphenylphosphine oxide (1/1). Acta Crystallogr. Sect. C 2000, 56, 1033–1034. [Google Scholar] [CrossRef]
- Aitken, R.A.; Karodia, N.; Lightfoot, P. The solid state conformation of oxo stabilised ylides: X-ray structures of four new polyoxo phosphorus ylides. J. Chem. Soc. Perkin Trans. 2 2000, 333–340. [Google Scholar] [CrossRef]
- Aitken, R.A.; Boubalouta, Y.; Chang, D.; Cleghorn, L.P.; Gray, I.P.; Karodia, N.; Reid, E.J.; Slawin, A.M.Z. The value of 2Jp–CO as a diagnostic parameter for the structure and reactivity of carbonyl-stabilised phosphonium ylides. Tetrahedron 2017, 73, 6275–6285. [Google Scholar] [CrossRef]
- Gosney, I.; Lloyd, D. Preparation and properties of some stable arsonium ylides. Tetrahedron 1973, 29, 1697–1710. [Google Scholar] [CrossRef]
- Ferguson, G.; Gosney, I.; Lloyd, D.; Ruhl, B.L. Crystal and molecular structures of some triphenylarsonium acetylylides. J. Chem. Res. (S) 1987, 8, 260–261. [Google Scholar]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. Sect. B. 1990, 46, 256–262. [Google Scholar] [CrossRef]
- Abell, A.D.; Morris, K.B.; McKee, V. A single-crystal X-ray analysis of a novel intramolecular hydrogen-bonded biphenyl phosphorane. A3ust. J. Chem. 1990, 43, 765–771. [Google Scholar] [CrossRef]
- Falvello, L.R.; Fernández, S.; Navarro, R.; Pascual, I.; Urriolabeitia, E.P. Oxygen vs. carbon coordination of α-keto-stabilized phosphorus ylides Ph3P=C(H)COR (R = Me, Ph or OMe) to palldium(II) cationic complexes. J. Chem. Soc. Dalton Trans. 1997, 763–771. [Google Scholar] [CrossRef]
- Navarro, R.; Urriolabeitia, E.P. α-Stabilized phosphoylides as versatile multifunctional ligands. J. Chem. Soc. Dalton Trans. 1999, 4111–4122. [Google Scholar] [CrossRef]
- Laavanya, P.; Venkatasubramanian, U.; Panchanatheswaran, K.; Krause Bauer, J.A. Subtlety in the reactivity of a diketo ylide towards mercuric halides: The unprecedented O-coordination of α-acetyl-α-benzoylmethylenetriphenylphosphorane to Hg(II). Chem. Commun. 2001, 1660–1661. [Google Scholar] [CrossRef]
- Buckle, J.; Harrison, P.G.; King, T.J.; Richards, J.A. Structural studies in main-group chemistry. Part IX. Crystal structure of chlorotrimethyl(triphenylphosphoranylideneacetone)tin(IV). J. Chem. Soc. Dalton Trans. 1975, 1552–1556. [Google Scholar] [CrossRef]
- Albanese, J.A.; Staley, D.L.; Rheingold, A.L.; Burmeister, J.L. Phosphorus ylides as hard donor ligands: Synthesis and characterization of MCl4(ylide-O)(THF) (M = Ti, Zr, Hf; ylide = (acetylmethylene)triphenylphosphorane, (benzoylmethylene)triphenylphosphorane). Molecular structure of trans-((Acetylmethylene)triphenylphosphorane-O)(tetrahydrofuran)tetrachlorotitanium(IV)-tetrahydrofuran. Inorg. Chem. 1990, 29, 2209–2213. [Google Scholar] [CrossRef]
- Sanehi, R.; Bansal, R.K.; Mehrotra, R.C. Organotin(IV) chloride complexes of bis-β-ketophosphonium ylides. J. Organomet. Chem. 1986, 303, 351–360. [Google Scholar] [CrossRef]
- Sanehi, R.; Bansal, R.K.; Mehrotra, R.C. Preparation & characterization of some new bisphosphonium ylides & their palladium(II) & mercury(II) complexes. Indian J. Chem A 1985, 24, 398–402. [Google Scholar]
- Aitken, R.A.; Kozminykh, E.N.; Kozminykh, V.O.; Lightfoot, P. Solution and solid state structure of highly delocalised 5-triphenylphosphoranylidenecyclopentene-3,4-diones. Phosphorus Sulfur Silicon Relat. Elem. 1998, 134, 487–492. [Google Scholar] [CrossRef]
- Aliev, Z.G.; Shurov, S.N.; Nekrasov, D.D.; Podvintsev, I.B.; Atovmyan, L.O. Character of enolization of the β-dicarbonyl fragment in α,γ-dioxocarboxylic acids. Crystal and molecular structure of benzoyl- and cinnamoylpyruvic acids. J. Struct. Chem. 2000, 41, 1041–1045. [Google Scholar] [CrossRef]
- Berclas, T.; Bernardinelli, G.; Geoffroy, M.; Rao, G.; Tancic, Z. EPR/ENDOR study of an X-irradiated single crystal of 1-triphenylphosphoranylidene-2-propanone: The role of hydrogen bonds in the trapping of radiogenic radicals. Radiation Phys. Chem. 1999, 56, 539–545. [Google Scholar] [CrossRef]
- Aitken, R.A.; Cleghorn, L.P.; Leitch, R.M.; Morrill, L.C.; Slawin, A.M.Z. Unexpected rearrangement leading to formation of a 1,3-bis(triphenylphosphonio)prop-1-en-3-idyl carboxylate. Eur. J. Org. Chem. 2010, 2010, 3211–3214. [Google Scholar] [CrossRef]
- Aitken, R.A.; Karodia, N. Flash vacuum pyrolysis of stabilized phosphorus ylides, 9. Preparation and pyrolysis of β,γ-dioxo ylides, β,β’,γ,γ’-tetraoxo ylides and hexaoxo bis(ylides). Liebigs Ann./Recueil 1997, 1997, 779–783. [Google Scholar] [CrossRef]
- Brömme, E.; Claisen, L. Ueber die Einwirkung des Oxaläthers auf Acetophenon. Ber. Dtsch. Chem. Ges. 1888, 21, 1131–1135. [Google Scholar] [CrossRef]
- Ramirez, F.; Dershowitz, S. Phosphinemethylenes II. Triphenylphosphineacylmethylenes. J. Org. Chem. 1957, 22, 41–45. [Google Scholar] [CrossRef]
- Aitken, R.A.; Cadogan, J.I.G.; Gosney, I. Convenient preparation of unsymmetrically substituted benzils by permanganate oxidation of β-oxo phosphorus ylides. Phosphorus Sulfur Silicon Relat. Elem. 1995, 101, 281–286. [Google Scholar] [CrossRef]
- Sheldrick, G.M. A short history of SHELXL. Acta Crystallogr. Sect. A 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | D–H...A | D–H | H...A | D...A | D–H...A |
|---|---|---|---|---|---|
| 9 | O(7)–H(7)...O(3) | 0.98(8) | 1.62(9) | 2.490(9) | 145(6) |
| 9 | O(9)–H(9)...O(11) | 0.98(6) | 1.59(6) | 2.501(8) | 153(6) |
| 9 | O(47)–H(47)...O(43) | 0.98(4) | 1.51(2) | 2.471(8) | 165(7) |
| 9 | O(49)–H(49)...O(51) | 0.98(9) | 1.58(8) | 2.507(9) | 156(8) |
| 11 | O(6)–H(6)...O(3) | 0.93(5) | 1.53(5) | 2.501(6) | 173(6) |
| 11 | O(36)–H(36)...O(33) | 0.98(5) | 1.53(5) | 2.506(6) | 171(6) |
| 21 | N(5)–H(5A)...O(3) | 0.86(3) | 2.04(3) | 2.889(3) | 169(2) |
| 21 | N(5)–H(5B)...O(6) | 0.94(3) | 1.97(3) | 2.901(4) | 173(3) |
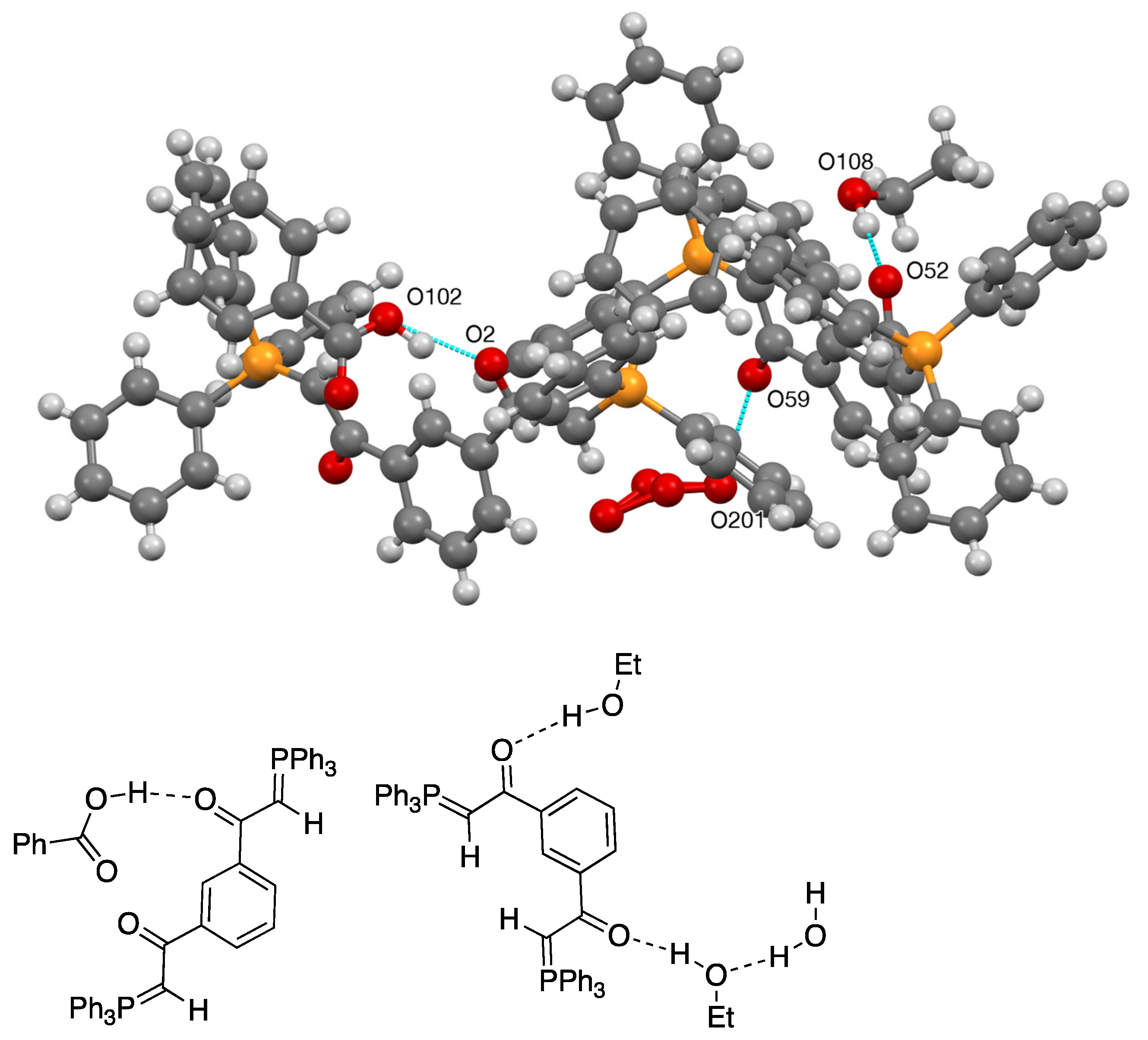
| 27 | O(102)–H(10B)...O(2) | 0.980(4) | 1.639(3) | 2.555(5) | 153.83(19) |
| 27 | O(108)–H(10M)...O(52) | 0.980(3) | 1.803(3) | 2.783(4) | 177.8(2) |
| Adduct | CO2H (δ) | P=C (δ) | ||||
|---|---|---|---|---|---|---|
| Adduct | Free Acid | Δδ | Adduct | Free Ylide | Δδ | |
| 10 | 170.2 | 172.6 | −2.4 | 58.8 | 57.0 | +1.8 |
| 11 | 169.6 | 172.6 | −3.0 | 56.3 | 51.5 | +4.8 |
| 12 | 169.7 | 172.6 | −2.9 | 75.1 | 71.0 | +4.1 |
| 13 | 174.6 | 178.2 | −3.6 | 56.1 | 51.5 | +4.6 |
| 14 | 175.2 | 179.0 | −3.8 | 56.0 | 51.5 | +4.5 |
| 15 | 177.0 | 180.5 | −3.5 | 55.4 | 51.5 | +3.9 |
| 16 | 174.5 | 178.2 | −3.7 | 52.7 | 50.6 | +2.1 |
| 17 | 174.9 | 179.0 | −4.1 | 52.5 | 50.6 | +1.9 |
| 18 | 177.7 | 180.5 | −2.8 | 53.5 | 50.6 | +2.9 |
| Solvent | CO2H (δ) | P=C (δ) | ||||
|---|---|---|---|---|---|---|
| Adduct | Free Acid | Δδ | Adduct | Free Ylide | Δδ | |
| C6D6 | 168.8 | 168.9 | −0.1 | 55.9 | 50.2 | +5.7 |
| CDCl3 | 169.6 | 172.6 | −3.0 | 56.3 | 51.5 | +4.8 |
| CD3COCD3 | 168.1 | 168.0 | +0.1 | 54.4 | 51.7 | +2.7 |
| CD3OD | 170.7 | 169.8 | +0.9 | 54.1 | 51.8 | +2.3 |
| CD3SOCD3 | 167.4 | 167.3 | +0.1 | 50.2 | 49.7 | +0.5 |
| Adduct | Donor | δC Adduct | δC Free Ylide | Δδ |
|---|---|---|---|---|
| 19 | MeCO2H | 56.5 | 51.5 | +5.0 |
| 20 | PhCH2OH | 58.0 | 51.5 | +6.5 |
| 21 | PhCONH2 | 56.2 | 51.5 | +4.7 |
| 22 | PhCSNH2 | 54.5 | 51.5 | +3.0 |
| 23 | PhSO2H | 57.2 | 51.5 | +5.7 |
| 24 | Ph2P(=O)OH | 59.1 | 51.5 | +7.6 |
| - | PhCH2NH2 | 51.8 | 51.5 | +0.3 |
| - | PhSO3H | - | 51.5 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aitken, R.A.; Cleghorn, L.P.; Dawson, G.; Gray, I.P.; Lashtabeg, A.; Slawin, A.M.Z. Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors. Inorganics 2023, 11, 50. https://doi.org/10.3390/inorganics11020050
Aitken RA, Cleghorn LP, Dawson G, Gray IP, Lashtabeg A, Slawin AMZ. Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors. Inorganics. 2023; 11(2):50. https://doi.org/10.3390/inorganics11020050
Chicago/Turabian StyleAitken, R. Alan, Lee P. Cleghorn, Graham Dawson, Ian P. Gray, Anna Lashtabeg, and Alexandra M. Z. Slawin. 2023. "Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors" Inorganics 11, no. 2: 50. https://doi.org/10.3390/inorganics11020050
APA StyleAitken, R. A., Cleghorn, L. P., Dawson, G., Gray, I. P., Lashtabeg, A., & Slawin, A. M. Z. (2023). Oxo-Stabilised Phosphonium Ylides as Hydrogen Bond Acceptors. Inorganics, 11(2), 50. https://doi.org/10.3390/inorganics11020050