Flexible Multiplane Structured Illumination Microscope with a Four-Camera Detector


Abstract
1. Introduction
2. Materials and Methods
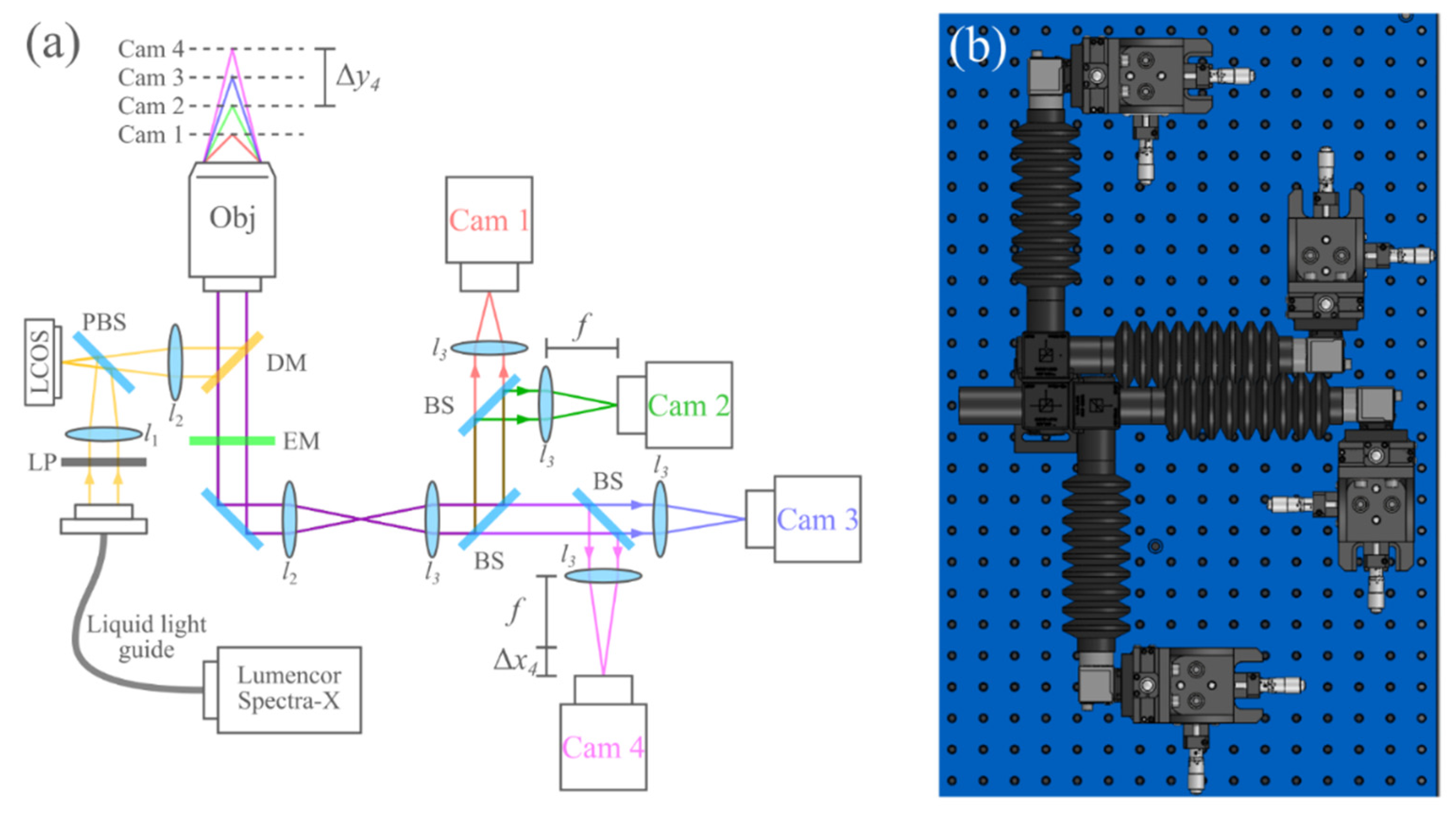
2.1. Microscope Setup and Data Acquisition
2.2. Cell Lines and Reagents
2.3. Preparation of Samples for Imaging
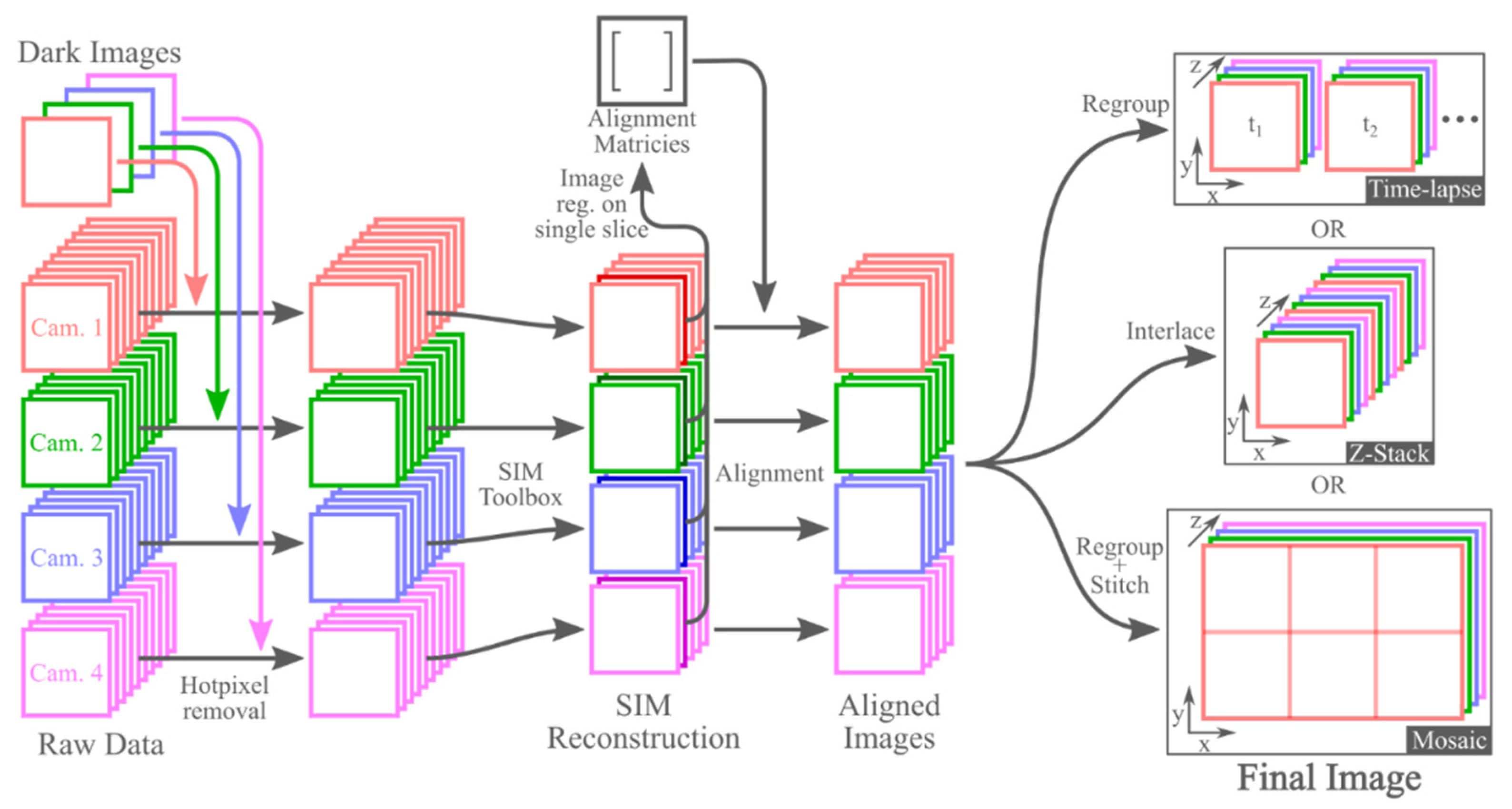
2.4. Image Preprocessing
2.5. SIM Data Processing
2.6. Image Registration and Assembly/Stitching
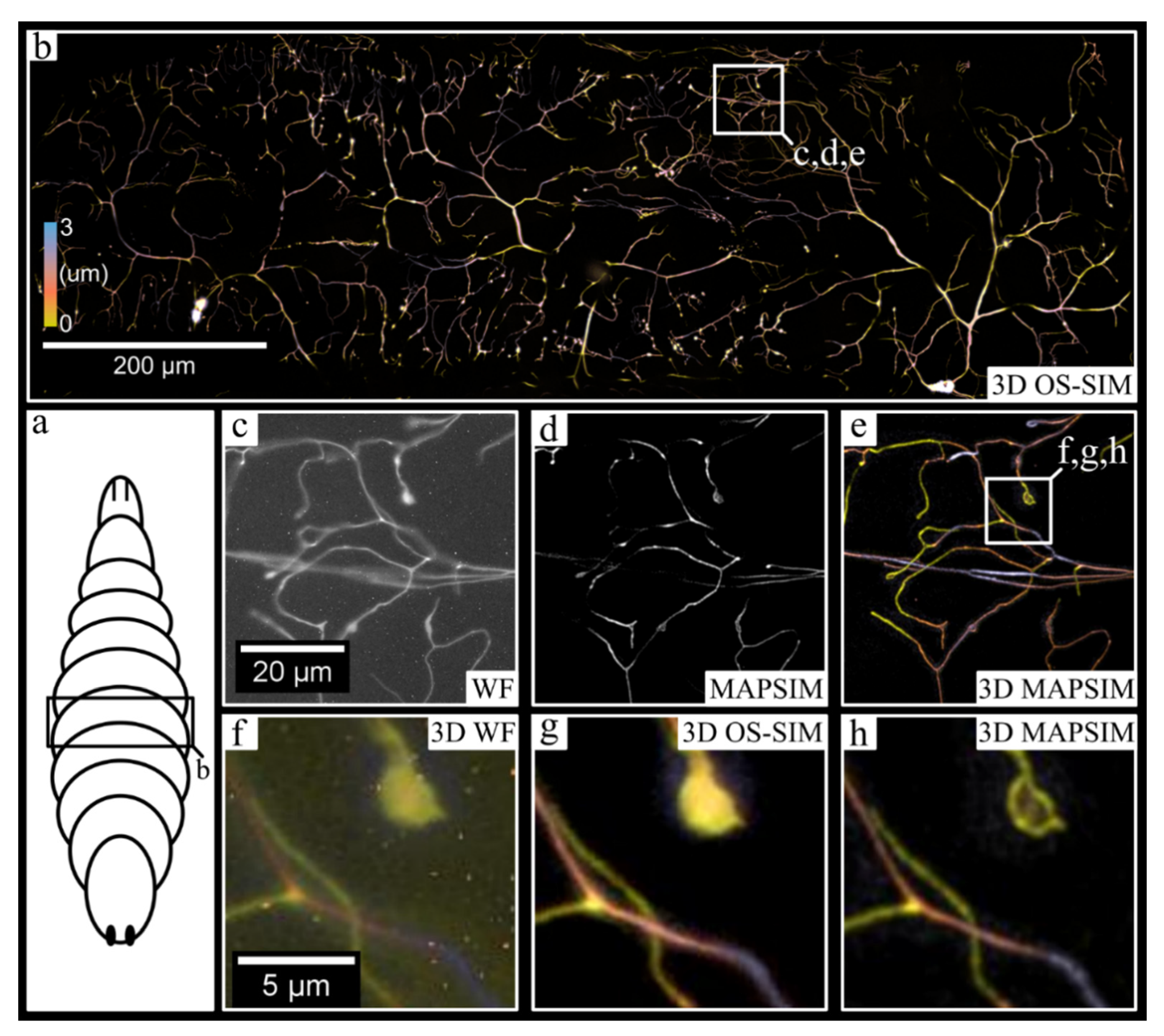
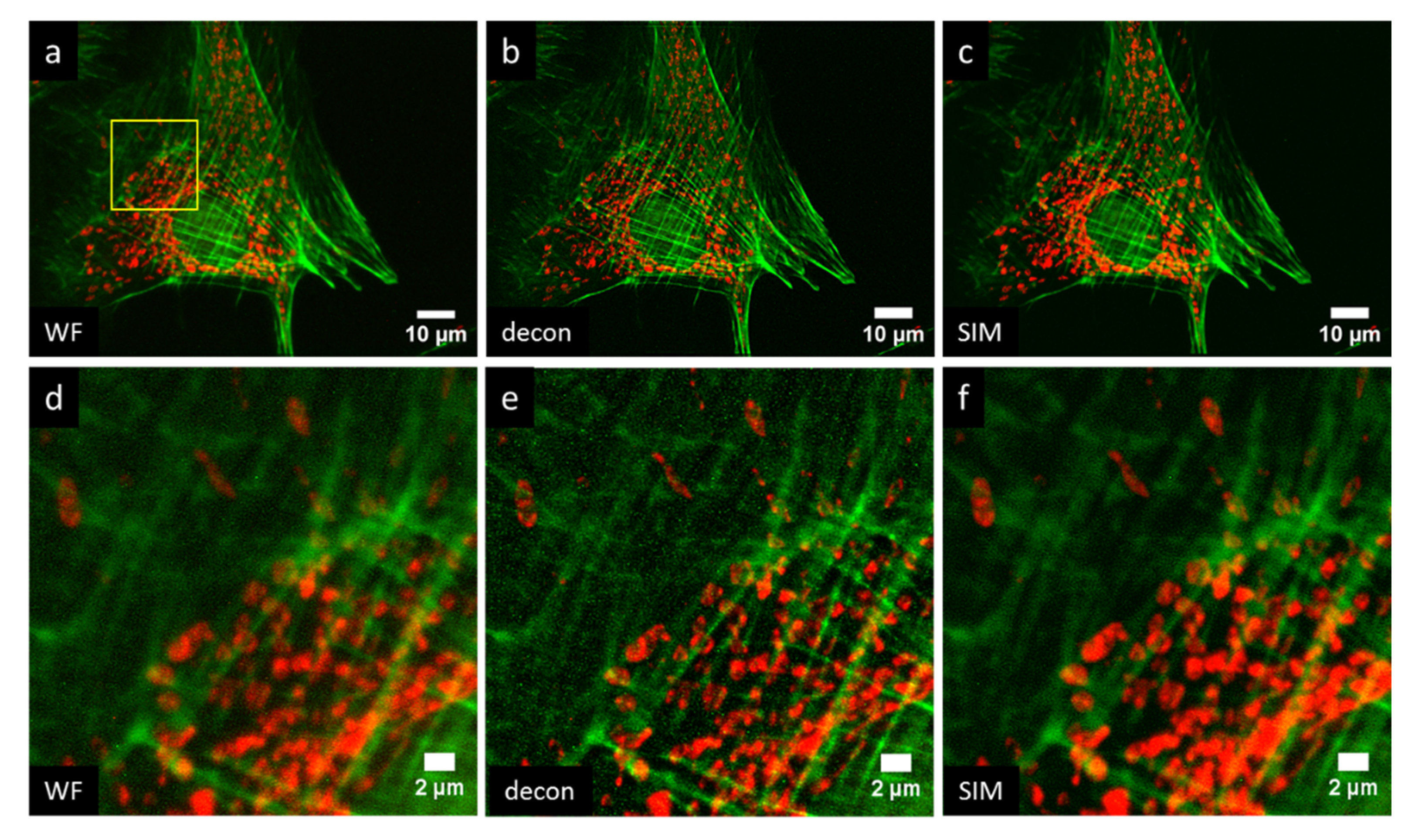
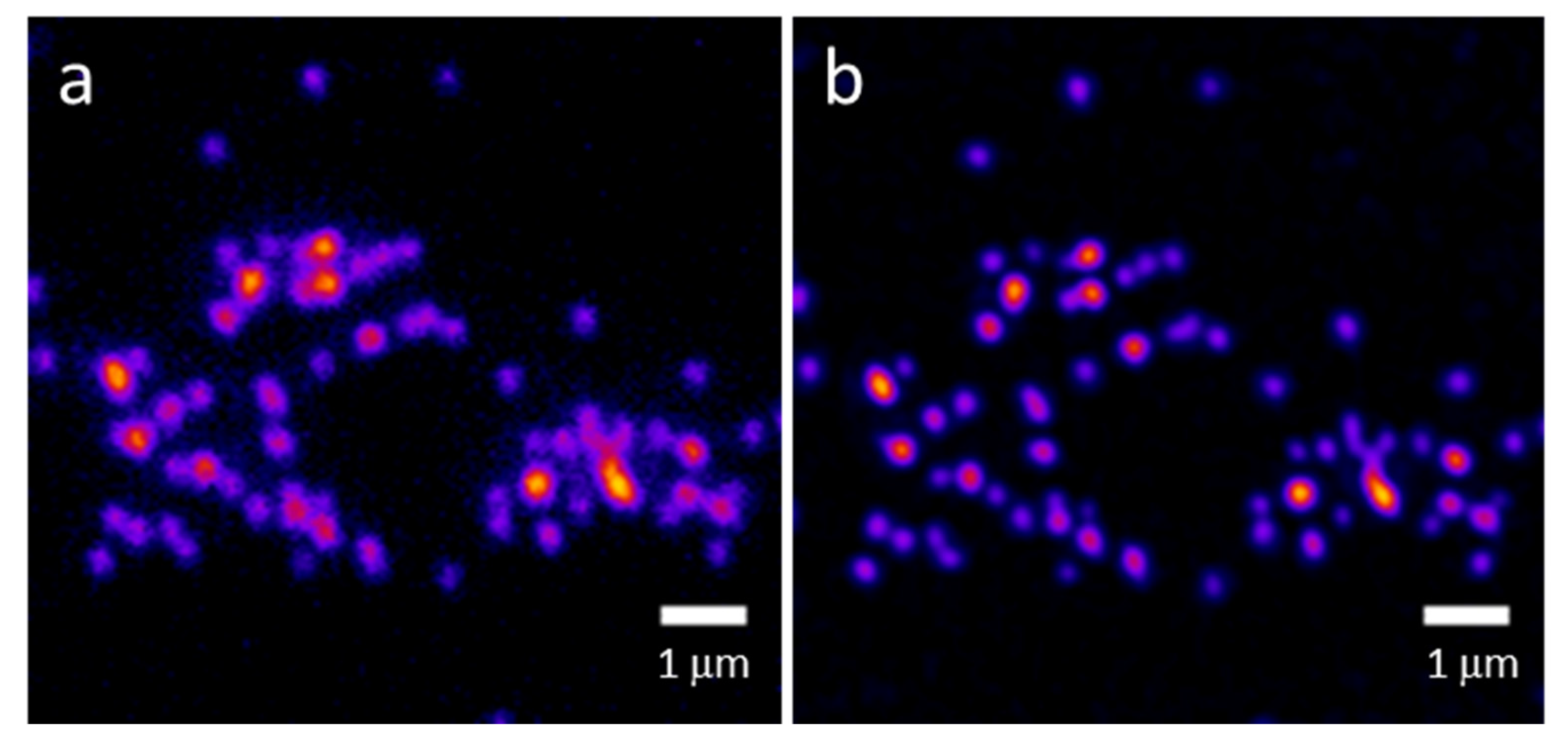
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gustafsson, M.G.L. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc. 2000, 198, 82–87. [Google Scholar] [CrossRef]
- Shao, L.; Kner, P.; Rego, E.H.; Gustafsson, M.G.L. Super-resolution 3D microscopy of live whole cells using structured illumination. Nat. Methods 2011, 8, 1044–1048. [Google Scholar] [CrossRef] [PubMed]
- Heintzmann, R.; Cremer, C. Laterally modulated excitation microscopy: Improvement of resolution by using a diffraction grating. Proc. SPIE 1998, 3568, 185–196. [Google Scholar]
- Kner, P.; Chhun, B.B.; Griffis, E.R.; Winoto, L.; Gustafsson, M.G.L. Super-resolution video microscopy of live cells by structured illumination. Nat. Methods 2009, 6, 339–342. [Google Scholar] [CrossRef]
- Schermelleh, L.; Carlton, P.M.; Haase, S.; Shao, L.; Winoto, L.; Kner, P.; Burke, B.; Cardoso, M.C.; Agard, D.A.; Gustafsson, M.G.L.; et al. Subdiffraction multicolor imaging of the nuclear periphery with 3D structured illumination microscopy. Science 2008, 320, 1332–1336. [Google Scholar] [CrossRef]
- Gustafsson, M.G.L.; Shao, L.; Carlton, P.M.; Wang, C.J.R.; Golubovskaya, I.N.; Cande, W.Z.; Agard, D.A.; Sedat, J.W. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J. 2008, 94, 4957–4970. [Google Scholar] [CrossRef]
- Neil, M.A.A.; Juškaitis, R.; Wilson, T. Method of obtaining optical sectioning by using structured light in a conventional microscope. Opt. Lett. 1997, 22, 1905. [Google Scholar] [CrossRef] [PubMed]
- Pilger, C.; Pospíšil, J.; Müller, M.; Ruoff, M.; Schütte, M.; Spiecker, H.; Huser, T. Super-resolution fluorescence microscopy by line-scanning with an unmodified two-photon microscope. Philos. Trans. R. Soc. A Math. Phys. Eng. Sci. 2021, 379, 20200300. [Google Scholar] [CrossRef]
- Liu, W.; Liu, Q.; Zhang, Z.; Han, Y.; Kuang, C.; Xu, L.; Yang, H.; Liu, X. Three-dimensional super-resolution imaging of live whole cells using galvanometer-based structured illumination microscopy. Opt. Express 2019, 27, 7237. [Google Scholar] [CrossRef] [PubMed]
- Brown, P.T.; Kruithoff, R.; Seedorf, G.J.; Shepherd, D.P. Multicolor structured illumination microscopy and quantitative control of polychromatic light with a digital micromirror device. Biomed. Opt. Express 2021, 12, 3700. [Google Scholar] [CrossRef]
- Chang, B.J.; Meza, V.D.P.; Stelzer, E.H.K. csiLSFM combines light-sheet fluorescence microscopy and coherent Structured illumination for a lateral resolution below 100 nm. Proc. Natl. Acad. Sci. USA 2017, 114, 4869–4874. [Google Scholar] [CrossRef] [PubMed]
- Křížek, P.; Raška, I.; Hagen, G.M. Flexible structured illumination microscope with a programmable illumination array. Opt. Express 2012, 20, 24585. [Google Scholar] [CrossRef] [PubMed]
- Rossberger, S.; Best, G.; Baddeley, D.; Heintzmann, R.; Birk, U.; Dithmar, S.; Cremer, C. Combination of structured illumination and single molecule localization microscopy in one setup. J. Opt. 2013, 15, 094003. [Google Scholar] [CrossRef]
- Young, L.J.; Ströhl, F.; Kaminski, C.F. A Guide to Structured Illumination TIRF Microscopy at High Speed with Multiple Colors. J. Vis. Exp. 2016, 111, e53988. [Google Scholar] [CrossRef] [PubMed]
- Poher, V.; Zhang, H.X.; Kennedy, G.T.; Griffin, C.; Oddos, S.; Gu, E.; Elson, D.S.; Girkin, M.; French, P.M.W.; Dawson, M.D.; et al. Optical sectioning microscope with no moving parts using a micro-stripe array light emitting diode. Opt. Express 2007, 15, 11196–11206. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, J.; Wiebusch, G.; Fliegel, K.; Klíma, M.; Huser, T. Highly compact and cost-effective 2-beam super-resolution structured illumination microscope based on all-fiber optic components. Opt. Express 2021, 29, 11833. [Google Scholar] [CrossRef] [PubMed]
- Hinsdale, T.A.; Stallinga, S.; Rieger, B. High-speed multicolor structured illumination microscopy using a hexagonal single mode fiber array. Biomed. Opt. Express 2021, 12, 1181. [Google Scholar] [CrossRef]
- Lukeš, T.; Křížek, P.; Švindrych, Z.; Benda, J.; Ovesný, M.; Fliegel, K.; Klíma, M.; Hagen, G.M. Three-dimensional super-resolution structured illumination microscopy with maximum a posteriori probability image estimation. Opt. Express 2014, 22, 29805–29817. [Google Scholar] [CrossRef]
- Orieux, F.; Sepulveda, E.; Loriette, V.; Dubertret, B.; Olivo-Marin, J.C. Bayesian estimation for optimized structured illumination microscopy. IEEE Trans. Image Process. 2012, 21, 601–614. [Google Scholar] [CrossRef]
- Huang, X.; Fan, J.; Li, L.; Liu, H.; Wu, R.; Wu, Y.; Wei, L.; Mao, H.; Lal, A.; Xi, P.; et al. Fast, long-term, super-resolution imaging with Hessian structured illumination microscopy. Nat. Biotechnol. 2018, 36, 451–459. [Google Scholar] [CrossRef]
- Lukeš, T.; Hagen, G.M.; Křížek, P.; Švindrych, Z.; Fliegel, K.; Klíma, M. Comparison of image reconstruction methods for structured illumination microscopy. Proc. SPIE 2014, 9129, 91293J. [Google Scholar]
- Chakrova, N.; Rieger, B.; Stallinga, S. Deconvolution methods for structured illumination microscopy. J. Opt. Soc. Am. A 2016, 33, B12. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, S.; Chen, J.; Hajj, B.; Stallinga, S.; Katsov, A.Y.; Wisniewski, J.; Mizuguchi, G.; Soule, P.; Mueller, F.; Darzacq, C.D.; et al. Fast multicolor 3D imaging using aberration-corrected multifocus microscopy. Nat. Methods 2013, 10, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsson, S.; Blom, H.; Agostinho, A.; Jans, D.C.; Jost, A.; Müller, M.; Nilsson, L.; Bernhem, K.; Lambert, T.J.; Heintzmann, R.; et al. Multifocus structured illumination microscopy for fast volumetric super-resolution imaging. Biomed. Opt. Express 2017, 8, 4135–4414. [Google Scholar] [CrossRef] [PubMed]
- Hajj, B.; Wisniewski, J.; el Beheiry, M.; Chen, J.; Revyakin, A.; Wu, C.; Dahan, M. Whole-cell, multicolor superresolution imaging using volumetric multifocus microscopy. Proc. Natl. Acad. Sci. USA 2014, 111, 17480–17485. [Google Scholar] [CrossRef]
- Hajj, B.; el Beheiry, M.; Dahan, M. PSF engineering in multifocus microscopy for increased depth volumetric imaging. Biomed. Opt. Express 2016, 7, 726. [Google Scholar] [CrossRef]
- Abrahamsson, S.; Ilic, R.; Wisniewski, J.; Mehl, B.; Yu, L.; Chen, L.; Davanco, M.; Oudjedi, L.; Fiche, J.-B.; Hajj, B.; et al. Multifocus microscopy with precise color multi-phase diffractive optics applied in functional neuronal imaging. Biomed. Opt. Express 2016, 7, 855. [Google Scholar] [CrossRef]
- Geissbuehler, S.; Sharipov, A.; Godinat, A.; Bocchio, N.L.; Sandoz, P.A.; Huss, A.; Jensen, N.A.; Jakobs, S.; Enderlein, J.; van der Goot, F.G.; et al. Live-cell multiplane three-dimensional super-resolution optical fluctuation imaging. Nat. Commun. 2014, 5, 5830. [Google Scholar] [CrossRef]
- Descloux, A.; Müller, M.; Navikas, V.; Markwirth, A.; van den Eynde, R.; Lukes, T.; Hübner, W.; Lasser, T.; Radenovic, A.; Dedecker, P.; et al. High-speed multiplane structured illumination microscopy of living cells using an image-splitting prism. Nanophotonics 2020, 9, 143–148. [Google Scholar] [CrossRef]
- Xiao, S.; Gritton, H.; Tseng, H.; Zemel, D.; Han, X.; Mertz, J. High-contrast multifocus microscopy with a single camera and z-splitter prism. Optica 2020, 7, 1477–1486. [Google Scholar] [CrossRef]
- Prabhat, P.; Ram, S.; Ward, E.S.; Ober, R.J. Simultaneous imaging of different focal planes in fluorescence microscopy for the study of cellular dynamics in three dimensions. IEEE Trans. Nanobiosci. 2004, 3, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Babcock, H.P. Multiplane and spectrally-resolved single molecule localization microscopy with industrial grade CMOS cameras. Sci. Rep. 2018, 8, 1726. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Sun, M.; Gumpper, K.; Chi, Y.; Ma, J. 3D multifocus astigmatism and compressed sensing (3D MACS) based superresolution reconstruction. Biomed. Opt. Express 2015, 6, 902–915. [Google Scholar] [CrossRef][Green Version]
- Křížek, P.; Lukeš, T.; Ovesný, M.; Fliegel, K.; Hagen, G.M. SIMToolbox: A MATLAB toolbox for structured illumination fluorescence microscopy. Bioinformatics 2015, 32, 318–320. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, J.; Lukeš, T.; Bendesky, J.; Fliegel, K.; Spendier, K.; Hagen, G.M. Imaging tissues and cells beyond the diffraction limit with structured illumination microscopy and Bayesian image reconstruction. Gigascience 2018, 8, giy126. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.A.; Hagen, G.M. Artifact-free whole-slide imaging with structured illumination microscopy and Bayesian image reconstruction. Gigascience 2021, 9, giaa035. [Google Scholar] [CrossRef]
- Fiolka, R.; Shao, L.; Rego, E.H.; Davidson, M.W.; Gustafsson, M.G.L. Time-lapse two-color 3D imaging of live cells with doubled resolution using structured illumination. Proc. Natl. Acad. Sci. USA 2012, 109, 5311–5315. [Google Scholar] [CrossRef]
- Hagen, G.M.; Caarls, W.; Lidke, K.A.; de Vries, A.H.B.; Fritsch, C.; Barisas, B.G.; Arndt-Jovin, D.J.; Jovin, T.M. Fluorescence recovery after photobleaching and photoconversion in multiple arbitrary regions of interest using a programmable array microscope. Microsc. Res. Tech. 2009, 72, 431–440. [Google Scholar] [CrossRef]
- Kantelhardt, S.R.; Caarls, W.; de Vries, A.H.B.; Hagen, G.M.; Jovin, T.M.; Schulz-Schaeffer, W.; Rohde, V.; Giese, A.; Arndt-Jovin, D.J. Specific visualization of glioma cells in living low-grade tumor tissue. PLoS ONE 2010, 5, e11323. [Google Scholar] [CrossRef]
- Chen, B.C.; Legant, W.R.; Wang, K.; Shao, L.; Milkie, D.E.; Davidson, M.W.; Janetopoulos, C.; Wu, X.S.; Hammer, J.A.; Liu, Z.; et al. Lattice light-sheet microscopy: Imaging molecules to embryos at high spatiotemporal resolution. Science 2014, 346, 1257998. [Google Scholar] [CrossRef] [PubMed]
- Diaspro, A.; Federici, F.; Robello, M. Influence of refractive-index mismatch in high-resolution three-dimensional confocal microscopy. Appl. Opt. 2002, 41, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Preibisch, S.; Saalfeld, S.; Tomancak, P. Globally optimal stitching of tiled 3D microscopic image acquisitions. Bioinformatics 2009, 25, 1463–1465. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, J.; Fliegel, K.; Klíma, M. Assessing resolution in live cell structured illumination microscopy. Proc. SPIE 2017, 10603, 70–76. [Google Scholar]
- Geissbuehler, M.; Lasser, T. How to display data by color schemes compatible with red-green color perception deficiencies. Opt. Express 2013, 21, 9862. [Google Scholar] [CrossRef]
- Sage, D.; Dona, L.; Soulez, F.; Fortun, D.; Schmit, G.; Seitz, A.; Guiet, R.; Vonesch, C.; Unsera, M. DeconvolutionLab2: An open-source software for deconvolution microscopy. Methods 2017, 115, 28–41. [Google Scholar] [CrossRef]
- Ovesný, M.; Křížek, P.; Borkovec, J.; Švindrych, Z.; Hagen, G.M. ThunderSTORM: A comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 2014, 30, 2389–2390. [Google Scholar] [CrossRef]
- Ram, S.; Prabhat, P.; Ward, E.S.; Ober, R.J. Improved single particle localization accuracy with dual objective multifocal plane microscopy. Opt. Express 2009, 17, 6881–6898. [Google Scholar] [CrossRef]
- Pospíšil, J. Hot Pixels Suppression in Structured Illumination Microscopy. In Proceedings of the 21th International Student Conference on Electrical Engineering, POSTER 2017, Prague, Czech Republic, 23 May 2017; pp. 1–7. [Google Scholar]
- Khademi, W.; Rao, S.; Minnerath, C.; Hagen, G.; Ventura, J. Self-Supervised Poisson-Gaussian Denoising. In Proceedings of the IEEE/CVF Winter Conference on Applications of Computer Vision (WACV), Virtual, 5–9 January 2021; pp. 2131–2139. [Google Scholar]
- Hagen, G.; Bendesky, J.; Machado, R.; Nguyen, T.A.; Kumar, T.; Ventura, J. Fluorescence microscopy datasets for training deep neural networks. Gigascience 2021, 10, giab032. [Google Scholar] [CrossRef]
- Niu, Y.; Wu, J.; Liu, W.; Guo, W.; Lau, R.W.H. HDR-GAN: HDR image reconstruction from multi-exposed ldr images with large motions. arXiv 2020. [Google Scholar] [CrossRef]
- Gryaditskaya, Y.; Pouli, T.; Reinhard, E.; Myszkowski, K.; Seidel, H.P. Motion Aware Exposure Bracketing for HDR Video. Comput. Graph. Forum 2015, 34, 119–130. [Google Scholar] [CrossRef]
- Aguerrebere, C.; Delon, J.; Gousseau, Y.; Musé, P. Best algorithms for HDR image generation. A study of performance bounds. SIAM J. Imaging Sci. 2014, 7, 1–34. [Google Scholar] [CrossRef]
- Wilburn, B.; Joshi, N.; Vaish, V.; Talvala, E.-V.; Antunez, E.; Barth, A.; Adams, A.; Horowitz, M.; Levoy, M. High Performance Imaging Using Large Camera Arrays. In ACM SIGGRAPH 2005 Papers on—SIGGRAPH ’05; Proceedings of the SIGGRAPH05: Special Interest Group on Computer Graphics and Interactive Techniques Conference, Los Angeles, CA, USA, 31 July–4 August 2005; ACM Press: New York, NY, USA, 2005. [Google Scholar]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Camera | FLIR Blackfly S | Andor Zyla 4.2+ |
|---|---|---|
| Part number | BFS-U3-31S4M | ZYLA-4.2P-CL10 |
| Sensor type | Sony IMX265 | Andor Zyla |
| Quantum efficiency | 62% at 525 nm | 82% at 555 nm |
| Read noise | 2.26 e− | 0.9 e− |
| Pixel size | 3.45 µm | 6.5 µm |
| Maximum frame rate | 55 FPS | 100 FPS |
| Readout method | global shutter | rolling shutter |
| Sensor size | 7.18 × 5.32 mm | 13.3 × 13.3 mm |
| Approximate price | $530 | $18,000 |
| 30×/1.05 NA | 60×/1.35 NA | |||
|---|---|---|---|---|
| WF | MAPSIM | WF | MAPSIM | |
| Cam 1 | 357.4 | 253.2 | 254.4 | 236.0 |
| Cam 2 | 354.6 | 250.1 | 262.4 | 236.9 |
| Cam 3 | 362.4 | 253.9 | 260.3 | 236.0 |
| Cam 4 | 456.0 | 301.9 | 276.7 | 236.9 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Johnson, K.A.; Noble, D.; Machado, R.; Paul, T.C.; Hagen, G.M. Flexible Multiplane Structured Illumination Microscope with a Four-Camera Detector. Photonics 2022, 9, 501. https://doi.org/10.3390/photonics9070501
Johnson KA, Noble D, Machado R, Paul TC, Hagen GM. Flexible Multiplane Structured Illumination Microscope with a Four-Camera Detector. Photonics. 2022; 9(7):501. https://doi.org/10.3390/photonics9070501
Chicago/Turabian StyleJohnson, Karl A., Daniel Noble, Rosa Machado, Tristan C. Paul, and Guy M. Hagen. 2022. "Flexible Multiplane Structured Illumination Microscope with a Four-Camera Detector" Photonics 9, no. 7: 501. https://doi.org/10.3390/photonics9070501
APA StyleJohnson, K. A., Noble, D., Machado, R., Paul, T. C., & Hagen, G. M. (2022). Flexible Multiplane Structured Illumination Microscope with a Four-Camera Detector. Photonics, 9(7), 501. https://doi.org/10.3390/photonics9070501