Ultrafast Hyperspectral Transient Absorption Spectroscopy: Application to Single Layer Graphene


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
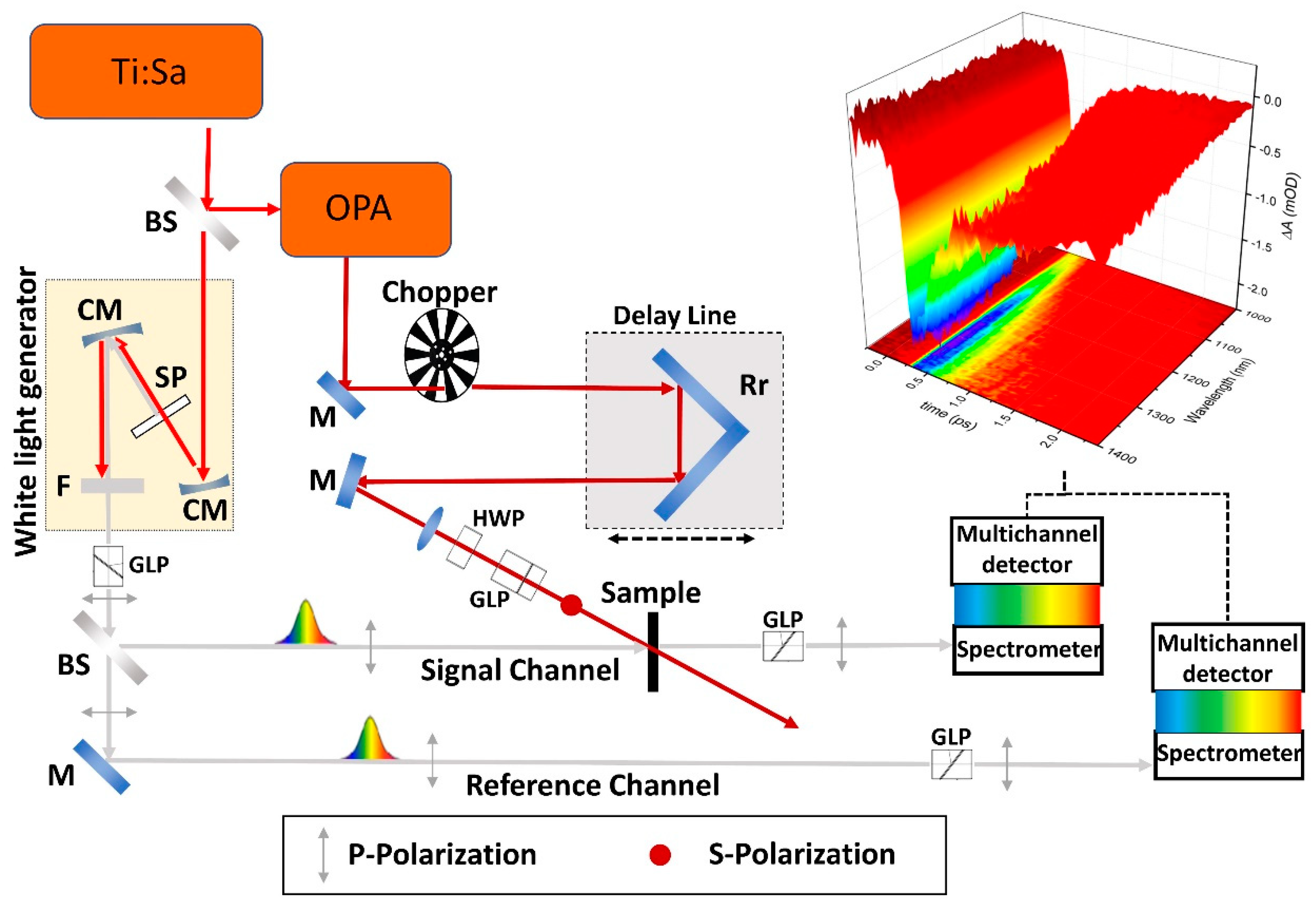
2. Experimental Setup
3. Results
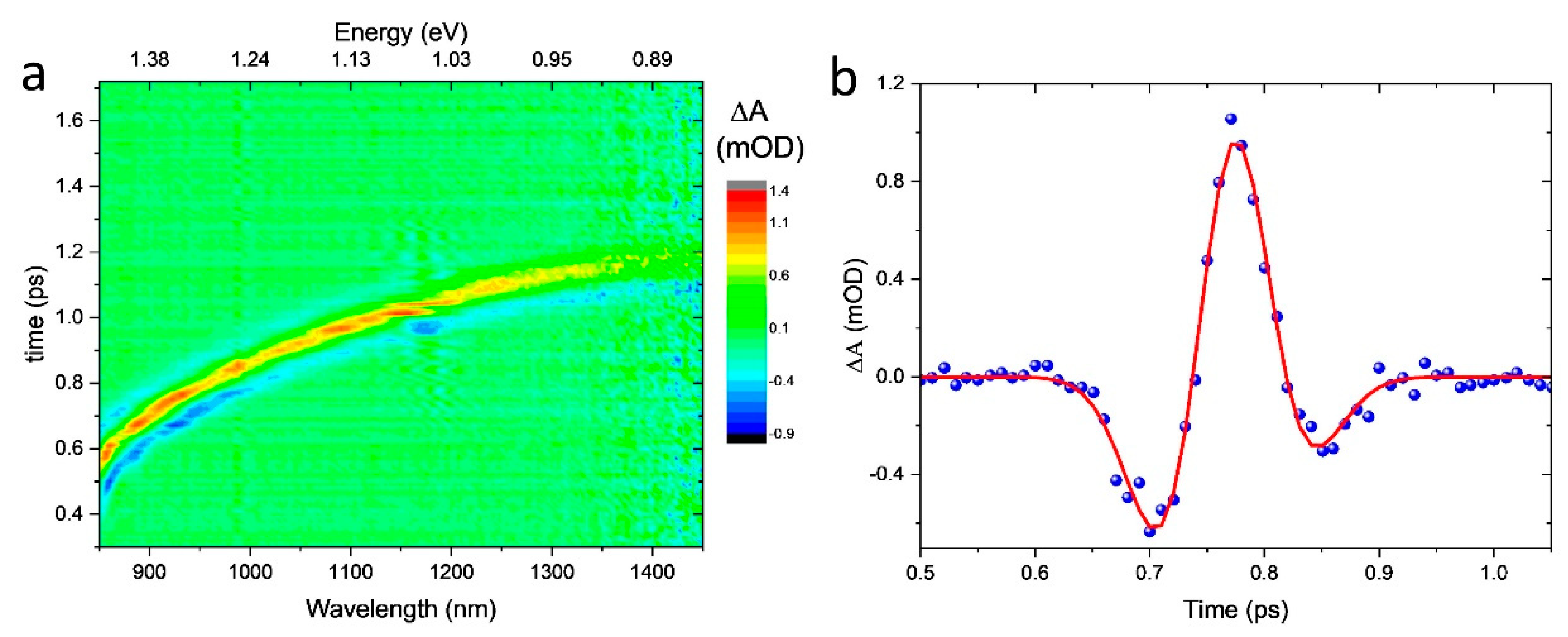
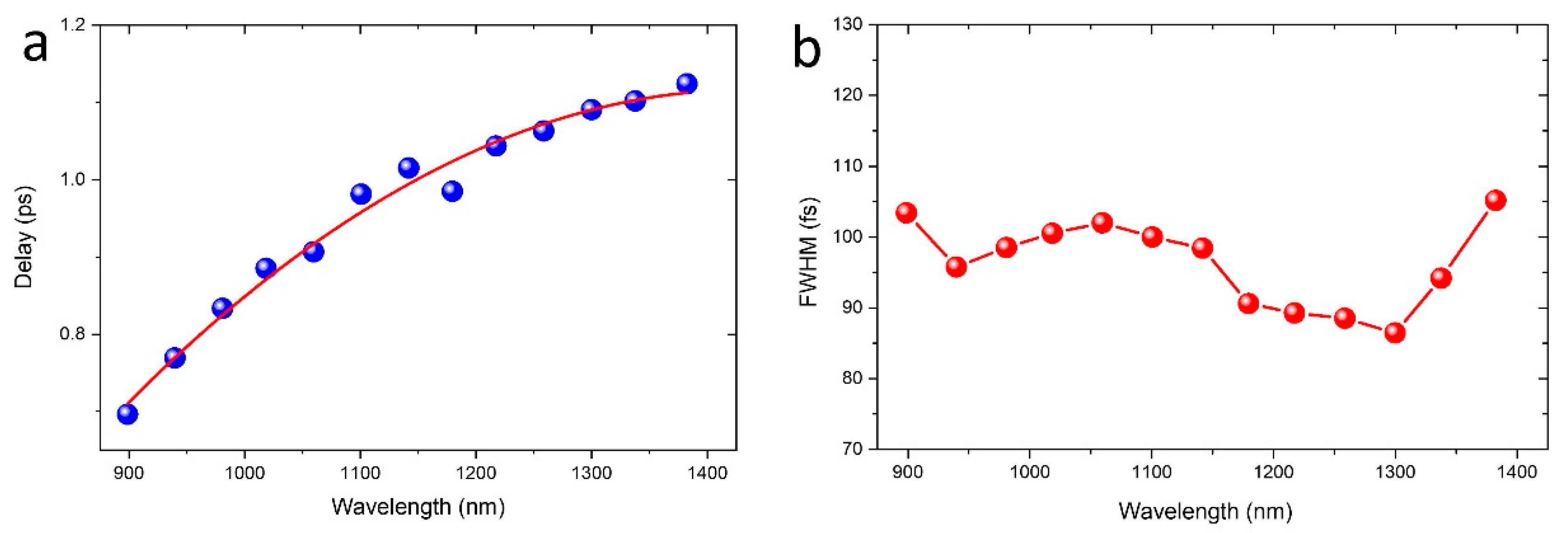
3.1. Chirp Correction and Time Resolution
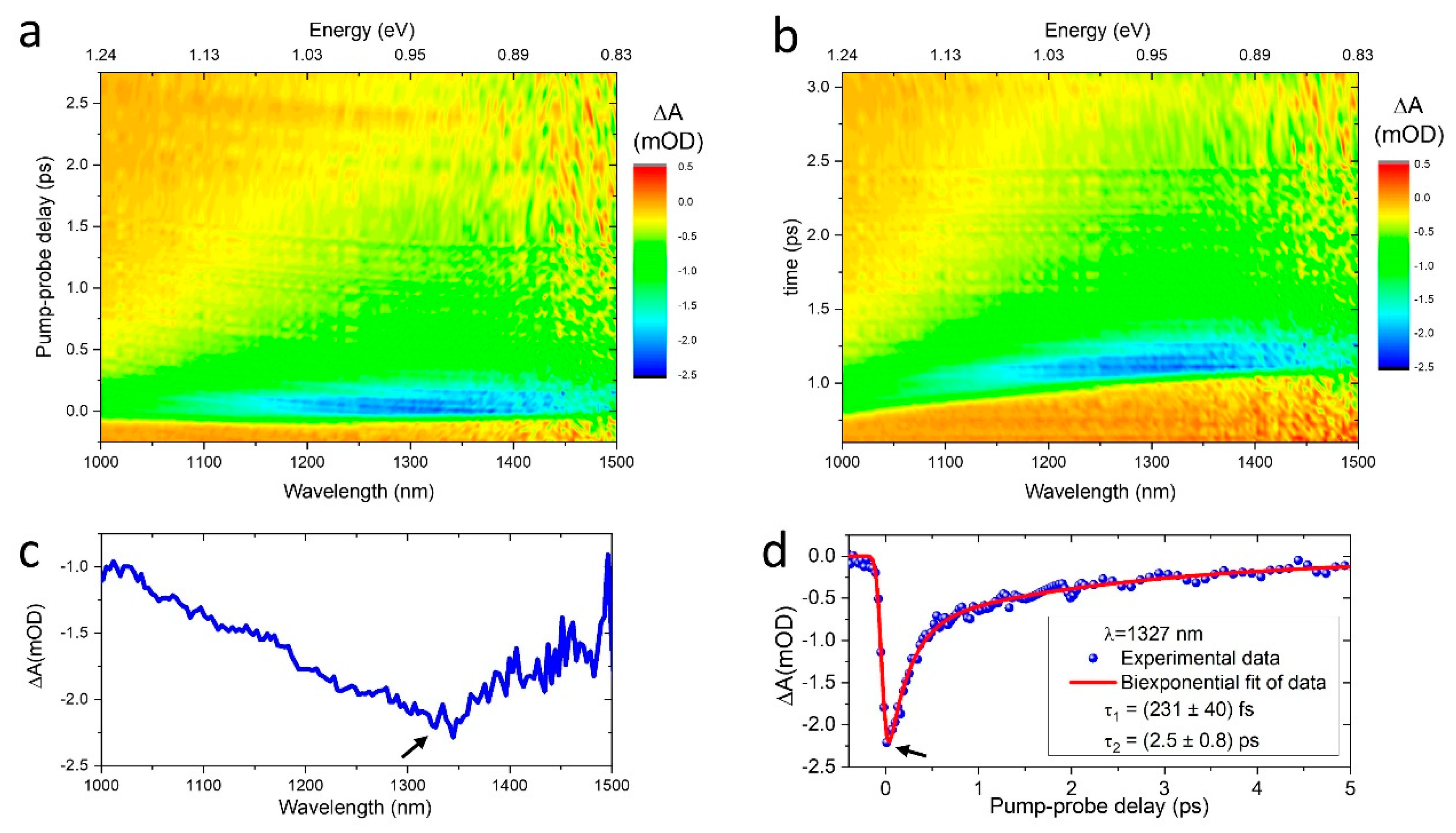
3.2. Ultrafast Carrier Dynamics in 2D Graphene
4. Discussion and Conclusions
Funding
Conflicts of Interest
References
- Kambhampati, P. Unraveling the Structure and Dynamics of Excitons in Semiconductor Quantum Dots. Acc. Chem. Res. 2011, 44, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sfeir, M.Y.; Qian, H.; Nobusada, K.; Jin, R. Ultrafast Relaxation Dynamics of Rod-Shaped 25-Atom Gold Nanoclusters. J. Phys. Chem. C 2011, 115, 6200–6207. [Google Scholar] [CrossRef]
- Gesuele, F.; Sfeir, M.Y.; Koh, W.-K.; Murray, C.B.; Heinz, T.F.; Wong, C.W. Ultrafast Supercontinuum Spectroscopy of Carrier Multiplication and Biexcitonic Effects in Excited States of PbS Quantum Dots. Nano Lett. 2012, 12, 2658–2664. [Google Scholar] [CrossRef] [PubMed]
- Berera, R.; van Grondelle, R.; Kennis, J.T.M. Ultrafast transient absorption spectroscopy: Principles and application to photosynthetic systems. Photosynth. Res. 2009, 101, 105–118. [Google Scholar] [CrossRef] [PubMed]
- Zewail, A.H. Femtochemistry: Atomic-Scale Dynamics of the Chemical Bond. J. Phys. Chem. A 2000, 104, 5660–5694. [Google Scholar] [CrossRef]
- Itri, F.; Monti, D.M.; Della Ventura, B.; Vinciguerra, R.; Chino, M.; Gesuele, F.; Lombardi, A.; Velotta, R.; Altucci, C.; Birolo, L.; et al. Femtosecond UV-laser pulses to unveil protein–protein interactions in living cells. Cell. Mol. Life Sci. 2016, 73, 637–648. [Google Scholar] [CrossRef] [PubMed]
- Auböck, G.; Consani, C.; van Mourik, F.; Chergui, M. Ultrabroadband femtosecond two-dimensional ultraviolet transient absorption. Opt. Lett. 2012, 37, 2337. [Google Scholar] [CrossRef] [PubMed]
- Alfano, R.R. The Supercontinuum Laser Source; Springer: New York, NY, USA, 2016; ISBN 978-1-4939-3324-2. [Google Scholar]
- Vulto, S.I.E.; Kennis, J.T.M.; Streltsov, A.M.; Amesz, J.; Aartsma, T.J. Energy Relaxation within the B850 Absorption Band of the Isolated Light-Harvesting Complex LH2 from Rhodopseudomonas acidophila at Low Temperature. J. Phys. Chem. B 1999, 103, 878–883. [Google Scholar] [CrossRef]
- Savikhin, S.; van Amerongen, H.; Kwa, S.L.; van Grondelle, R.; Struve, W.S. Low-temperature energy transfer in LHC-II trimers from the Chl a/b light-harvesting antenna of photosystem II. Biophys. J. 1994, 66, 1597–1603. [Google Scholar] [CrossRef][Green Version]
- Tkachenko, N.V. Optical Spectroscopy: Methods and Instrumentations; Elsevier: Amsterdam, The Netherlands, 2006; p. 307. ISBN 9780444521262. [Google Scholar]
- Auböck, G.; Consani, C.; Monni, R.; Cannizzo, A.; van Mourik, F.; Chergui, M. Femtosecond pumpsupercontinuum-probe setup with 20 kHz repetition rate. Rev. Sci. Instrum. 2012, 83, 2010–2015. [Google Scholar] [CrossRef]
- Moon, J.A. Optimization of signal-to-noise ratios in pump-probe spectroscopy. Rev. Sci. Instrum. 1993, 64, 1775–1778. [Google Scholar] [CrossRef]
- Dobryakov, A.L.; Kovalenko, S.A.; Weigel, A.; Pérez-Lustres, J.L.; Lange, J.; Müller, A.; Ernsting, N.P. Femtosecond pump/supercontinuum-probe spectroscopy: Optimized setup and signal analysis for single-shot spectral referencing. Rev. Sci. Instrum. 2010, 81, 113106. [Google Scholar] [CrossRef] [PubMed]
- Megerle, U.; Pugliesi, I.; Schriever, C.; Sailer, C.F.; Riedle, E. Sub-50 fs broadband absorption spectroscopy with tunable excitation: Putting the analysis of ultrafast molecular dynamics on solid ground. Appl. Phys. B Lasers Opt. 2009, 96, 215–231. [Google Scholar] [CrossRef]
- Klimov, V.I.; McBranch, D.W. Femtosecond high-sensitivity, chirp-free transient absorption spectroscopy using kilohertz lasers. Opt. Lett. 1998, 23, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Lorenc, M.; Ziolek, M.; Naskrecki, R.; Karolczak, J.; Kubicki, J.; Maciejewski, A. Artifacts in femtosecond transient absorption spectroscopy. Appl. Phys. B Lasers Opt. 2002, 74, 19–27. [Google Scholar] [CrossRef]
- Chachisvilis, M.; Fidder, H.; Sundström, V. Electronic coherence in pseudo two-colour pump-probe spectroscopy. Chem. Phys. Lett. 1995. [Google Scholar] [CrossRef]
- Kang, I.; Smolorz, S.; Krauss, T.; Wise, F.; Aitken, B.; Borrelli, N. Time-domain observation of nuclear contributions to the optical nonlinearities of glasses. Phys. Rev. B Condens. Matter Mater. Phys. 1996, 54, R12641–R12644. [Google Scholar] [CrossRef]
- Valadan, M.; D’Ambrosio, D.; Gesuele, F.; Velotta, R.; Altucci, C. Temporal and spectral characterization of femtosecond deep-UV chirped pulses. Laser Phys. Lett. 2015, 12, 025302. [Google Scholar] [CrossRef]
- Hellwarth, R.; Cherlow, J.; Yang, T.T. Origin and frequency dependence of nonlinear optical susceptibilities of glasses. Phys. Rev. B 1975, 11, 964–967. [Google Scholar] [CrossRef]
- Kovalenko, S.A.; Dobryakov, A.L.; Ruthmann, J.; Ernsting, N.P. Femtosecond spectroscopy of condensed phases with chirped supercontinuum probing. Phys. Rev. A 1999, 59, 2369–2384. [Google Scholar] [CrossRef]
- Trebino, R. Frequency-Resolved Optical Gating: The Measurement of Ultrashort Laser Pulses; Springer US: Boston, MA, USA, 2000; ISBN 978-1-4613-5432-1. [Google Scholar]
- Rasmusson, M.; Tarnovsky, A.N.; Åkesson, E.; Sundström, V. On the use of two-photon absorption for determination of femtosecond pump-probe cross-correlation functions. Chem. Phys. Lett. 2001. [Google Scholar] [CrossRef]
- Dobryakov, A.L.; Pérez Lustres, J.L.; Kovalenko, S.A.; Ernsting, N.P. Femtosecond transient absorption with chirped pump and supercontinuum probe: Perturbative calculation of transient spectra with general lineshape functions, and simplifications. Chem. Phys. 2008, 347, 127–138. [Google Scholar] [CrossRef]
- Descrovi, E.; Ricciardi, C.; Giorgis, F.; Lérondel, G.; Blaize, S.; Pang, C.X.; Bachelot, R.; Royer, P.; Lettieri, S.; Gesuele, F.; et al. Field localization and enhanced Second-Harmonic Generation in silicon-based microcavities. Opt. Express 2007, 15, 4159. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Gesuele, F.; Lettieri, S.; Maddalena, P.; Liscidini, M.; Andreani, L.C.; Ricciardi, C.; Ballarini, V.; Giorgis, F. Band-edge and cavity second harmonic conversion in doubly resonant microcavity. J. Phys. B At. Mol. Opt. Phys. 2007, 40, 727–734. [Google Scholar] [CrossRef]
- Bonaccorso, F.; Colombo, L.; Yu, G.; Stoller, M.; Tozzini, V.; Ferrari, A.C.; Ruoff, R.S.; Pellegrini, V. 2D materials. Graphene, related two-dimensional crystals, and hybrid systems for energy conversion and storage. Science 2015, 347, 1246501. [Google Scholar] [CrossRef] [PubMed]
- Xia, F.; Wang, H.; Xiao, D.; Dubey, M.; Ramasubramaniam, A. Two-dimensional material nanophotonics. Nat. Photonics 2014, 8, 899–907. [Google Scholar] [CrossRef]
- Bhimanapati, G.R.; Lin, Z.; Meunier, V.; Jung, Y.; Cha, J.; Das, S.; Xiao, D.; Son, Y.; Strano, M.S.; Cooper, V.R.; et al. Recent Advances in Two-Dimensional Materials beyond Graphene. ACS Nano 2015, 9, 11509–11539. [Google Scholar] [CrossRef]
- Nivas, J.J.J.; Gesuele, F.; Allahyari, E.; Oscurato, S.L.; Fittipaldi, R.; Vecchione, A.; Bruzzese, R.; Amoruso, S. Effects of ambient air pressure on surface structures produced by ultrashort laser pulse irradiation. Opt. Lett. 2017, 42, 2710. [Google Scholar] [CrossRef] [PubMed]
- Basov, D.N.; Fogler, M.M.; Garcia de Abajo, F.J. Polaritons in van der Waals materials. Science 2016, 354, aag1992. [Google Scholar] [CrossRef]
- Tran, T.T.; Bray, K.; Ford, M.J.; Toth, M.; Aharonovich, I. Quantum emission from hexagonal boron nitride monolayers. Nat. Nanotechnol. 2016, 11, 37–41. [Google Scholar] [CrossRef]
- Geim, A.K.; Novoselov, K.S. The rise of graphene. Nat. Mater. 2007, 6, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Ajayi, O.A.; Anderson, N.C.; Cotlet, M.; Petrone, N.; Gu, T.; Wolcott, A.; Gesuele, F.; Hone, J.; Owen, J.S.; Wong, C.W. Time-resolved energy transfer from single chloride-terminated nanocrystals to graphene. Appl. Phys. Lett. 2014, 104, 171101. [Google Scholar] [CrossRef]
- Geim, A.K.; Grigorieva, I.V. Van der Waals heterostructures. Nature 2013, 499, 419–425. [Google Scholar] [CrossRef]
- Attanzio, A.; Sapelkin, A.; Gesuele, F.; van der Zande, A.; Gillin, W.P.; Zheng, M.; Palma, M. Carbon Nanotube-Quantum Dot Nanohybrids: Coupling with Single-Particle Control in Aqueous Solution. Small 2017, 13, 1603042. [Google Scholar] [CrossRef]
- Hong, X.; Kim, J.; Shi, S.-F.; Zhang, Y.; Jin, C.; Sun, Y.; Tongay, S.; Wu, J.; Zhang, Y.; Wang, F. Ultrafast charge transfer in atomically thin MoS2/WS2 heterostructures. Nat. Nanotechnol. 2014, 9, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Freeley, M.; Attanzio, A.; Cecconello, A.; Amoroso, G.; Clement, P.; Fernandez, G.; Gesuele, F.; Palma, M. Tuning the Coupling in Single-Molecule Heterostructures: DNA-Programmed and Reconfigurable Carbon Nanotube-Based Nanohybrids. Adv. Sci. 2018, 5, 1800596. [Google Scholar] [CrossRef]
- Butler, S.Z.; Hollen, S.M.; Cao, L.; Cui, Y.; Gupta, J.A.; Gutiérrez, H.R.; Heinz, T.F.; Hong, S.S.; Huang, J.; Ismach, A.F. Opportunities in Two-Dimensional Materials Beyond Graphene. ACS Nano 2013, 7, 2898–2926. [Google Scholar] [CrossRef]
- Frisenda, R.; Castellanos-Gomez, A. Robotic assembly of artificial nanomaterials. Nat. Nanotechnol. 2018, 13, 441–442. [Google Scholar] [CrossRef]
- Novoselov, K.S.; Jiang, D.; Schedin, F.; Booth, T.J.; Khotkevich, V.V.; Morozov, S.V.; Geim, A.K. Two-dimensional atomic crystals. Proc. Natl. Acad. Sci. USA 2005, 102, 10451–10453. [Google Scholar] [CrossRef]
- Paton, K.R.; Varrla, E.; Backes, C.; Smith, R.J.; Khan, U.; O’Neill, A.; Boland, C.; Lotya, M.; Istrate, O.M.; King, P.; et al. Scalable production of large quantities of defect-free few-layer graphene by shear exfoliation in liquids. Nat. Mater. 2014, 13, 624–630. [Google Scholar] [CrossRef]
- Kaur, J.; Gravagnuolo, A.M.; Maddalena, P.; Altucci, C.; Giardina, P.; Gesuele, F. Green synthesis of luminescent and defect-free bio-nanosheets of MoS2: Interfacing two dimensional crystals with hydrophobins. RSC Adv. 2017, 7, 22400–22408. [Google Scholar] [CrossRef]
- Li, X.; Cai, W.; An, J.; Kim, S.; Nah, J.; Yang, D.; Piner, R.; Velamakanni, A.; Jung, I.; Tutuc, E.; et al. Large-area synthesis of high-quality and uniform graphene films on copper foils. Science 2009, 324, 1312–1314. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Magnuson, C.W.; Venugopal, A.; An, J.; Suk, J.W.; Han, B.; Borysiak, M.; Cai, W.; Velamakanni, A.; Zhu, Y.; et al. Graphene Films with Large Domain Size by a Two-Step Chemical Vapor Deposition Process. Nano Lett. 2010, 10, 4328–4334. [Google Scholar] [CrossRef] [PubMed]
- Lettieri, S.; Pallotti, D.K.; Gesuele, F.; Maddalena, P. Unconventional ratiometric-enhanced optical sensing of oxygen by mixed-phase TiO2. Appl. Phys. Lett. 2016, 109, 031905. [Google Scholar] [CrossRef]
- Pallotti, D.K.; Passoni, L.; Gesuele, F.; Maddalena, P.; Di Fonzo, F.; Lettieri, S. Giant O2-Induced Photoluminescence Modulation in Hierarchical Titanium Dioxide Nanostructures. ACS Sens. 2017, 2, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Coscia, U.; Ambrosone, G.; Gesuele, F.; Grossi, V.; Parisi, V.; Schutzmann, S.; Basa, D.K. Laser annealing study of PECVD deposited hydrogenated amorphous silicon carbon alloy films. Appl. Surf. Sci. 2007, 254, 984–988. [Google Scholar] [CrossRef]
- Gesuele, F.; Nivas, J.J.J.; Fittipaldi, R.; Altucci, C.; Bruzzese, R.; Maddalena, P.; Amoruso, S. Analysis of nascent silicon phase-change gratings induced by femtosecond laser irradiation in vacuum. Sci. Rep. 2018, 8, 12498. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.C.; Meyer, J.C.; Scardaci, V.; Casiraghi, C.; Lazzeri, M.; Mauri, F.; Piscanec, S.; Jiang, D.; Novoselov, K.S.; Roth, S.; et al. Raman Spectrum of Graphene and Graphene Layers. Phys. Rev. Lett. 2006, 97, 187401. [Google Scholar] [CrossRef]
- Ferrari, A.; Basko, D. Raman spectroscopy as a versatile tool for studying the properties of graphene. Nat. Nanotechnol. 2013, 8, 235–246. [Google Scholar] [CrossRef]
- Gesuele, F.; Pang, C.X.; Leblond, G.; Blaize, S.; Bruyant, A.; Royer, P.; Deturche, R.; Maddalena, P.; Lerondel, G. Towards routine near-field optical characterization of silicon-based photonic structures: An optical mode analysis in integrated waveguides by transmission AFM-based SNOM. Phys. E Low Dimens. Syst. Nanostruct. 2009, 41, 1130–1134. [Google Scholar] [CrossRef]
- Blaize, S.; Gesuele, F.; Stefanon, I.; Bruyant, A.; Lérondel, G.; Royer, P.; Martin, B.; Morand, A.; Benech, P.; Fedeli, J.-M. Real-space observation of spectral degeneracy breaking in a waveguide-coupled disk microresonator. Opt. Lett. 2010, 35, 3168. [Google Scholar] [CrossRef] [PubMed]
- Dawlaty, J.M.; Shivaraman, S.; Chandrashekhar, M.; Rana, F.; Spencer, M.G. Measurement of ultrafast carrier dynamics in epitaxial graphene. Appl. Phys. Lett. 2008, 92, 1–3. [Google Scholar] [CrossRef]
- Obraztsov, P.A.; Rybin, M.G.; Tyurnina, A.V.; Garnov, S.V.; Obraztsova, E.D.; Obraztsov, A.N.; Svirko, Y.P. Broadband light-induced absorbance change in multilayer graphene. Nano Lett. 2011, 11, 1540–1545. [Google Scholar] [CrossRef] [PubMed]
- Winnerl, S.; Mittendorff, M.; König-Otto, J.C.; Schneider, H.; Helm, M.; Winzer, T.; Knorr, A.; Malic, E. Ultrafast Processes in Graphene: From Fundamental Manybody Interactions to Device Applications. Ann. Phys. 2017, 529, 1–12. [Google Scholar] [CrossRef]
- Malic, E.; Winzer, T.; Bobkin, E.; Knorr, A. Microscopic theory of absorption and ultrafast many-particle kinetics in graphene. Phys. Rev. B Condens. Matter Mater. Phys. 2011, 84. [Google Scholar] [CrossRef]
- König-Otto, J.C.; Mittendorff, M.; Winzer, T.; Kadi, F.; Malic, E.; Knorr, A.; Berger, C.; De Heer, W.A.; Pashkin, A.; Schneider, H.; et al. Slow Noncollinear Coulomb Scattering in the Vicinity of the Dirac Point in Graphene. Phys. Rev. Lett. 2016, 117, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Mittendorff, M.; Winzer, T.; Malic, E.; Knorr, A.; Berger, C.; De Heer, W.A.; Schneider, H.; Helm, M.; Winnerl, S. Anisotropy of excitation and relaxation of photogenerated charge carriers in graphene. Nano Lett. 2014, 14, 1504–1507. [Google Scholar] [CrossRef]
- George, P.A.; Strait, J.; Dawlaty, J.; Shivaraman, S.; Chandrashekhar, M.; Rana, F.; Spencer, M.G. Ultrafast Optical-Pump Terahertz-Probe Spectroscopy of the Carrier Relaxation and Recombination Dynamics in Epitaxial Graphene. Nano Lett. 2008, 8, 4248–4251. [Google Scholar] [CrossRef]
- Sun, D.; Wu, Z.K.; Divin, C.; Li, X.; Berger, C.; De Heer, W.A.; First, P.N.; Norris, T.B. Ultrafast relaxation of excited dirac fermions in epitaxial graphene using optical differential transmission spectroscopy. Phys. Rev. Lett. 2008, 101, 1–4. [Google Scholar] [CrossRef]
- Brida, D.; Tomadin, A.; Manzoni, C.; Kim, Y.J.; Lombardo, A.; Milana, S.; Nair, R.R.; Novoselov, K.S.; Ferrari, A.C.; Cerullo, G.; et al. Ultrafast collinear scattering and carrier multiplication in graphene. Nat. Commun. 2013, 4, 1987. [Google Scholar] [CrossRef]
- Hwang, E.H.; Hu, B.Y.K.; Das Sarma, S. Inelastic carrier lifetime in graphene. Phys. Rev. B Condens. Matter Mater. Phys. 2007, 76, 1–6. [Google Scholar] [CrossRef]
- Polini, M.; Asgari, R.; Borghi, G.; Barlas, Y.; Pereg-Barnea, T.; MacDonald, A.H. Plasmons and the spectral function of graphene. Phys. Rev. B Condens. Matter Mater. Phys. 2008, 77, 3–6. [Google Scholar] [CrossRef]
- Chirayath, V.A.; Callewaert, V.; Fairchild, A.J.; Chrysler, M.D.; Gladen, R.W.; McDonald, A.D.; Imam, S.K.; Shastry, K.; Koymen, A.R.; Saniz, R.; et al. Auger electron emission initiated by the creation of valence-band holes in graphene by positron annihilation. Nat. Commun. 2017, 8, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Winzer, T.; Malić, E. Impact of Auger processes on carrier dynamics in graphene. Phys. Rev. B Condens. Matter Mater. Phys. 2012, 85, 1–5. [Google Scholar] [CrossRef]
- Winzer, T.; Knorr, A.; Malic, E. Carrier multiplication in graphene. Nano Lett. 2010, 10, 4839–4843. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gesuele, F. Ultrafast Hyperspectral Transient Absorption Spectroscopy: Application to Single Layer Graphene. Photonics 2019, 6, 95. https://doi.org/10.3390/photonics6030095
Gesuele F. Ultrafast Hyperspectral Transient Absorption Spectroscopy: Application to Single Layer Graphene. Photonics. 2019; 6(3):95. https://doi.org/10.3390/photonics6030095
Chicago/Turabian StyleGesuele, Felice. 2019. "Ultrafast Hyperspectral Transient Absorption Spectroscopy: Application to Single Layer Graphene" Photonics 6, no. 3: 95. https://doi.org/10.3390/photonics6030095
APA StyleGesuele, F. (2019). Ultrafast Hyperspectral Transient Absorption Spectroscopy: Application to Single Layer Graphene. Photonics, 6(3), 95. https://doi.org/10.3390/photonics6030095