Photobiomodulation for Alzheimer’s Disease: Has the Light Dawned?


{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
1.1. Introduction to Photobiomodulation
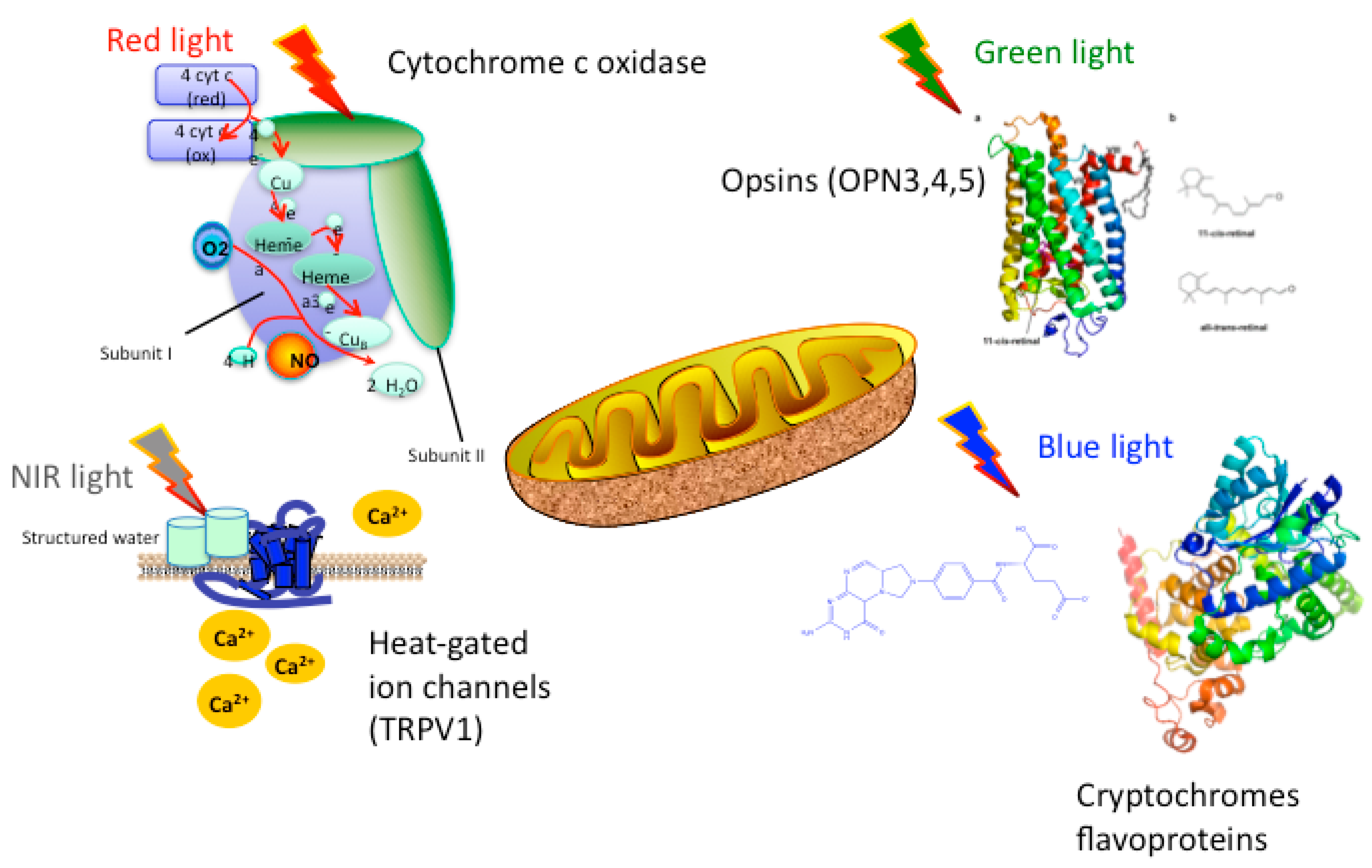
1.2. Mechanisms of PBM
2. Alzheimer’s Disease and Dementia
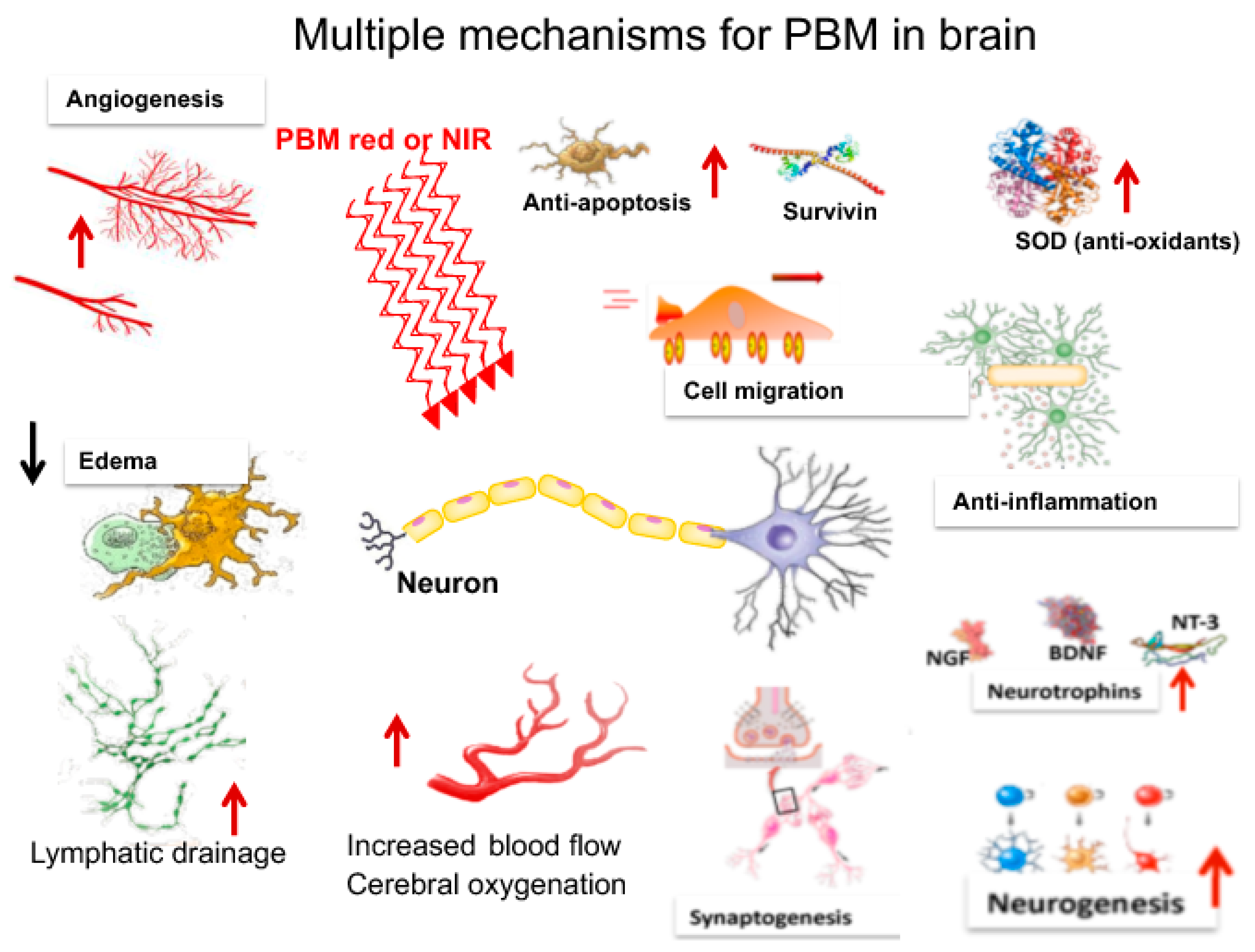
3. Mechanisms of PBM in the Brain
3.1. Metabolism
3.2. Blood Flow
3.3. Neuroprotection
3.4. Oxidative Stress
3.5. Anti-Inflammatory Effects
3.6. Neurogenesis
3.7. Synaptogenesis
3.8. Stem Cells
3.9. Gamma Rhythms
4. tPBM for AD in Animal Models
5. Clinical Trials of PBM in AD and Dementia
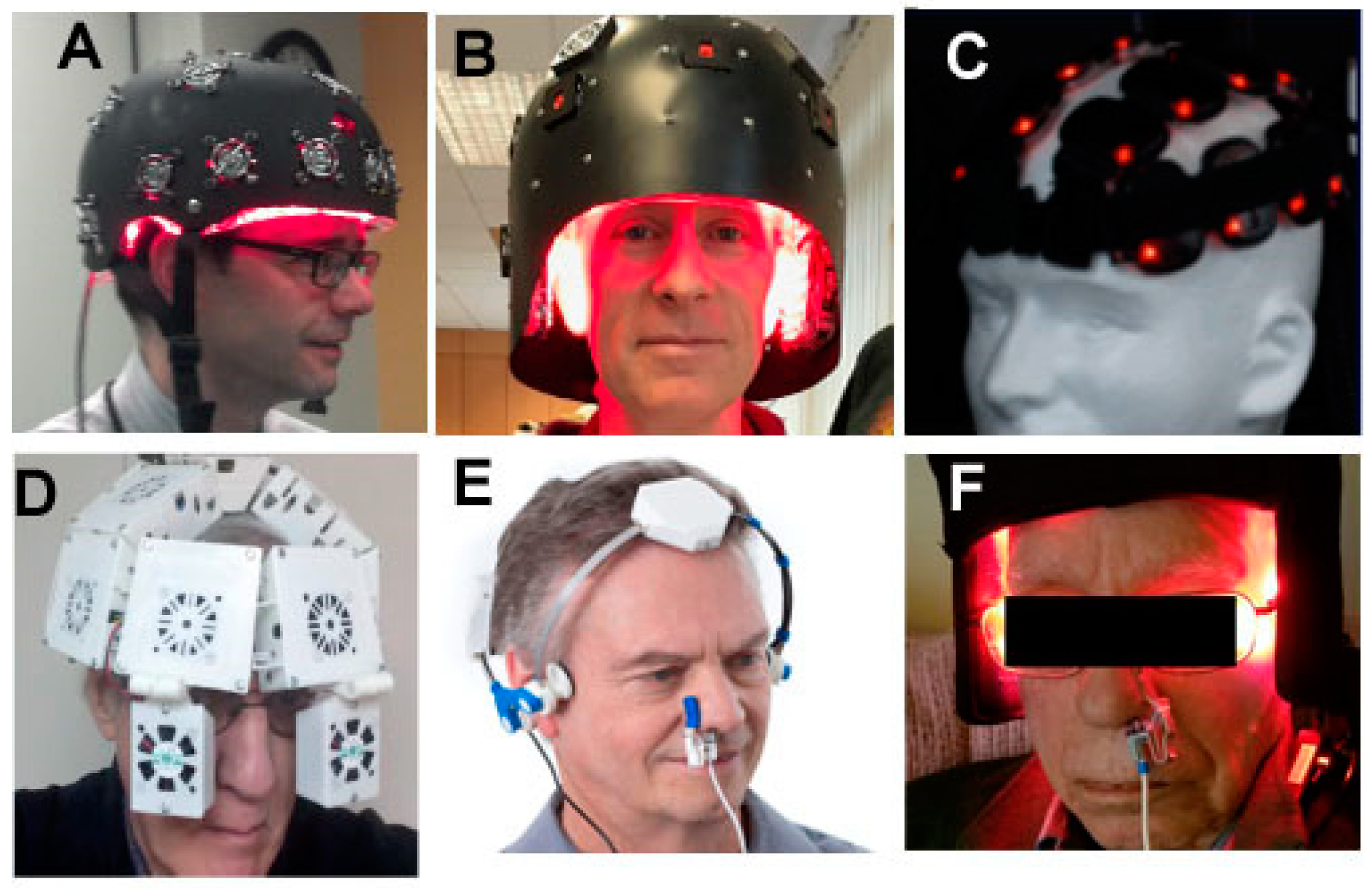
6. Devices and Parameters for Brain tPBM
7. Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Anders, J.J.; Arany, P.R.; Baxter, G.D.; Lanzafame, R.J. Light-Emitting Diode Therapy and Low-Level Light Therapy Are Photobiomodulation Therapy. Photobiomodul. Photomed. Laser Surg. 2019, 37, 63–65. [Google Scholar] [CrossRef] [PubMed]
- Anders, J.J.; Lanzafame, R.J.; Arany, P.R. Low-Level Light/Laser Therapy versus Photobiomodulation Therapy. Photomed. Laser Surg. 2015, 33, 183–184. [Google Scholar] [CrossRef] [PubMed]
- Mester, A.; Mester, A. The History of Photobiomodulation: Endre Mester (1903–1984). Photomed. Laser Surg. 2017, 35, 393–394. [Google Scholar] [CrossRef] [PubMed]
- Heiskanen, V.; Hamblin, M.R.; Hamblin, M.R. Photobiomodulation: Lasers vs. light emitting diodes? Photochem. Photobiol. Sci. 2018, 17, 1003–1017. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.E.; Cope, M.; Springett, R.; Amess, P.N.; Penrice, J.; Tyszczuk, L.; Punwani, S.; Ordidge, R.; Wyatt, J.; Delpy, D.T. Use of Mitochondrial Inhibitors to Demonstrate That Cytochrome Oxidase Near-Infrared Spectroscopy Can Measure Mitochondrial Dysfunction Noninvasively in the Brain. Br. J. Pharmacol. 1999, 19, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Karu, T.I. Multiple roles of cytochrome c oxidase in mammalian cells under action of red and IR-A radiation. IUBMB Life 2010, 62, 607–610. [Google Scholar] [CrossRef] [PubMed]
- Wong-Riley, M.T.; Liang, H.L.; Eells, J.T.; Chance, B.; Henry, M.M.; Buchmann, E.; Kane, M.; Whelan, H.T. Photobiomodulation directly benefits primary neurons functionally inactivated by toxins: Role of cytochrome c oxidase. J. Biol. Chem. 2005, 280, 4761–4771. [Google Scholar] [CrossRef]
- De Freitas, L.F.; Hamblin, M.R. Proposed Mechanisms of Photobiomodulation or Low-Level Light Therapy. IEEE J. Sel. Top. Quantum Electron. 2016, 22, 348–364. [Google Scholar] [CrossRef]
- Lima, P.L.; Pereira, C.V.; Nissanka, N.; Arguello, T.; Gavini, G.; Maranduba, C.M.D.C.; Diaz, F.; Moraes, C.T. Photobiomodulation enhancement of cell proliferation at 660 nm does not require cytochrome c oxidase. J. Photochem. Photobiol. B Boil. 2019, 194, 71–75. [Google Scholar] [CrossRef]
- Spitler, R.; Ho, H.; Norpetlian, F.; Kong, X.; Jiang, J.; Yokomori, K.; Andersen, B.; Boss, G.R.; Berns, M.W. Combination of low level light therapy and nitrosyl-cobinamide accelerates wound healing. J. Biomed. Opt. 2015, 20, 51022. [Google Scholar] [CrossRef]
- Núñez-Álvarez, C.; Del Olmo-Aguado, S.; Merayo-Lloves, J.; Osborne, N.N. Near infra-red light attenuates corneal endothelial cell dysfunction in situ and in vitro. Exp. Eye Res. 2017, 161, 106–115. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Zhou, F.; Wei, Y.; Chen, W.R.; Chen, Q.; Xing, D. Cancer Phototherapy via Selective Photoinactivation of Respiratory Chain Oxidase to Trigger a Fatal Superoxide Anion Burst. Antioxid. Redox Signal. 2014, 20, 733–746. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.P. Mitochondrial cytochrome c oxidase is not the primary acceptor for near infrared light—It is mitochondrial bound water: The principles of low-level light therapy. Ann. Transl. Med. 2019, 7, S13. [Google Scholar] [CrossRef] [PubMed]
- Karu, T.I. Mitochondrial Signaling in Mammalian Cells Activated by Red and Near-IR Radiation. Photochem. Photobiol. 2008, 84, 1091–1099. [Google Scholar] [CrossRef] [PubMed]
- Passarella, S.; Karu, T. Absorption of monochromatic and narrow band radiation in the visible and near IR by both mitochondrial and non-mitochondrial photoacceptors results in photobiomodulation. J. Photochem. Photobiol. B Boil. 2014, 140, 344–358. [Google Scholar] [CrossRef] [PubMed]
- Kamiński, M.M.; Roth, D.; Sass, S.; Sauer, S.W.; Krammer, P.H.; Gülow, K. Manganese superoxide dismutase: A regulator of T cell activation-induced oxidative signaling and cell death. Biochim. Biophys. Acta 2012, 1823, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Koyanagi, M.; Terakita, A. Diversity of animal opsin-based pigments and their optogenetic potential. Biochim. Biophys. Acta 2014, 1837, 710–716. [Google Scholar] [CrossRef]
- Losi, A.; Gärtner, W. Old Chromophores, New Photoactivation Paradigms, Trendy Applications: Flavins in Blue Light-Sensing Photoreceptors. Photochem. Photobiol. 2011, 87, 491–510. [Google Scholar] [CrossRef]
- Chaves, I.; Pokorný, R.; Byrdin, M.; Hoang, N.; Ritz, T.; Brettel, K.; Essen, L.-O.; Van Der Horst, G.T.J.; Batschauer, A.; Ahmad, M. The Cryptochromes: Blue Light Photoreceptors in Plants and Animals. Annu. Rev. Plant Boil. 2011, 62, 335–364. [Google Scholar] [CrossRef]
- Katz, B.; Payne, R.; Minke, B. TRP Channels in Vision. In Neurobiology of TRP Channels, 2nd ed.; Emir, T.L.R., Ed.; CRC Press: Boca Raton, FL, USA, 2017; pp. 27–63. [Google Scholar]
- Katz, B.; Minke, B. The Drosophila light-activated TRP and TRPL channels-Targets of the phosphoinositide signaling cascade. Prog. Retin. Eye Res. 2018, 66, 200–219. [Google Scholar] [CrossRef]
- Natasha, G.; Tan, A.; Farhatnia, Y.; Rajadas, J.; Hamblin, M.R.; Khaw, P.T.; Seifalian, A.M. Channelrhodopsins: Visual Regeneration & Neural Activation by a Light Switch. New Biotechnol. 2013, 30, 461–474. [Google Scholar]
- Yang, W.-Z.; Chen, J.-Y.; Yu, J.-T.; Zhou, L.-W. Effects of Low Power Laser Irradiation on Intracellular Calcium and Histamine Release in RBL-2H3 Mast Cells. Photochem. Photobiol. 2007, 83, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Ryu, J.-J.; Yoo, S.; Kim, K.; Park, J.-S.; Bang, S.; Lee, S.; Yang, T.-J.; Cho, H.; Hwang, S. Laser Modulation of Heat and Capsaicin Receptor TRPV1 Leads to Thermal Antinociception. J. Dent. Res. 2010, 89, 1455–1460. [Google Scholar] [CrossRef] [PubMed]
- Albert, E.S.; Bec, J.M.; Desmadryl, G.; Chekroud, K.; Travo, C.; Gaboyard, S.; Bardin, F.; Marc, I.; Dumas, M.; Lenaers, G.; et al. TRPV4 channels mediate the infrared laser-evoked response in sensory neurons. J. Neurophysiol. 2012, 107, 3227–3234. [Google Scholar] [CrossRef] [PubMed]
- Gu, Q.; Wang, L.; Huang, F.; Schwarz, W. Stimulation of TRPV1 by Green Laser Light. Evid. Based Complement. Altern. Med. 2012, 2012, 857123. [Google Scholar] [CrossRef]
- Wang, X.; Tian, F.; Reddy, D.D.; Nalawade, S.S.; Barrett, D.W.; Gonzalez-Lima, F.; Liu, H. Up-regulation of cerebral cytochrome-c-oxidase and hemodynamics by transcranial infrared laser stimulation: A broadband near-infrared spectroscopy study. Br. J. Pharmacol. 2017, 37, 3789–3802. [Google Scholar] [CrossRef] [PubMed]
- Blanco, N.J.; Maddox, W.T.; Gonzalez-Lima, F. Improving executive function using transcranial infrared laser stimulation. J. Neuropsychol. 2017, 11, 14–25. [Google Scholar] [CrossRef]
- Trevors, J.; Pollack, G. Origin of microbial life hypothesis: A gel cytoplasm lacking a bilayer membrane, with infrared radiation producing exclusion zone (EZ) water, hydrogen as an energy source and thermosynthesis for bioenergetics. Biochimie 2012, 94, 258–262. [Google Scholar] [CrossRef]
- Pollack, G.H.; Reitz, F.B. Phase transitions and molecular motion in the cell. Cell. Mol. Boil. 2001, 47, 885–900. [Google Scholar]
- Pollack, G.H. The role of aqueous interfaces in the cell. Adv. Colloid Interface Sci. 2003, 103, 173–196. [Google Scholar] [CrossRef]
- Ritchie, C.W.; Terrera, G.M.; Quinn, T.J. Dementia trials and dementia tribulations: Methodological and analytical challenges in dementia research. Alzheimer’s Res. Ther. 2015, 7, 31. [Google Scholar] [CrossRef] [PubMed]
- Day, G.; Tang-Wai, D.F. When dementia progresses quickly: A practical approach to the diagnosis and management of rapidly progressive dementia. Neurodegener. Dis. Manag. 2014, 4, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer’s Disease International. World Alzheimer Report 2015. Available online: https://www.alz.co.uk/research/WorldAlzheimerReport2015.pdf (accessed on 3 July 2019).
- Burns, A.; Iliffe, S. Dementia. BMJ 2009, 338, b75. [Google Scholar] [CrossRef] [PubMed]
- Möller, H.J.; Graeber, M.B. The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Eur. Arch. Psychiatry Clin. Neurosci. 1998, 248, 111–122. [Google Scholar] [PubMed]
- Ballard, C.; Gauthier, S.; Corbett, A.; Brayne, C.; Aarsland, D.; Jones, E. Alzheimer’s disease. Lancet 2011, 377, 1019–1031. [Google Scholar] [CrossRef]
- Amemori, T.; Jendelova, P.; Ruzicka, J.; Urdzíková, L.M.; Syková, E. Alzheimer’s Disease: Mechanism and Approach to Cell Therapy. Int. J. Mol. Sci. 2015, 16, 26417–26451. [Google Scholar] [CrossRef] [PubMed]
- Mullane, K.; Williams, M. Alzheimer’s disease (AD) therapeutics-1: Repeated clinical failures continue to question the amyloid hypothesis of AD and the current understanding of AD causality. Biochem. Pharmacol. 2018, 158, 359–375. [Google Scholar] [CrossRef] [PubMed]
- Mullane, K.; Williams, M. Alzheimer’s therapeutics: Continued clinical failures question the validity of the amyloid hypothesis—but what lies beyond? Biochem. Pharmacol. 2013, 85, 289–305. [Google Scholar] [CrossRef]
- Kametani, F.; Hasegawa, M. Reconsideration of Amyloid Hypothesis and Tau Hypothesis in Alzheimer’s Disease. Front. Mol. Neurosci. 2018, 12, 25. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, G.G. Tauopathies. Handb. Clin. Neurol. 2017, 145, 355–368. [Google Scholar] [PubMed]
- Regen, F.; Hellmann-Regen, J.; Costantini, E.; Reale, M.; Regen, J.H.-R.F. Neuroinflammation and Alzheimer’s Disease: Implications for Microglial Activation. Curr. Alzheimer’s Res. 2017, 14, 1140–1148. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Orehek, A.J. The Micron Stroke Hypothesis of Alzheimer’s Disease and Dementia. Med. Hypotheses 2012, 78, 562–570. [Google Scholar] [CrossRef] [PubMed]
- Cullen, K.M.; Kócsi, Z.; Stone, J. Pericapillary Haem-Rich Deposits: Evidence for Microhaemorrhages in Aging Human Cerebral Cortex. Br. J. Pharmacol. 2005, 25, 1656–1667. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Montagne, A.; Sagare, A.P.; Nation, D.A.; Schneider, L.S.; Chui, H.C.; Harrington, M.G.; Pa, J.; Law, M.; Wang, D.J.; et al. Vascular dysfunction—The disregarded partner of Alzheimer’s disease. Alzheimer’s Dement. 2019, 15, 158–167. [Google Scholar] [CrossRef]
- Cullen, K.M.; Kócsi, Z.; Stone, J. Microvascular pathology in the aging human brain: Evidence that senile plaques are sites of microhaemorrhages. Neurobiol. Aging 2006, 27, 1786–1796. [Google Scholar] [CrossRef] [PubMed]
- Nation, D.A.; Sweeney, M.D.; Montagne, A.; Sagare, A.P.; D’Orazio, L.M.; Pachicano, M.; Sepehrband, F.; Nelson, A.R.; Buennagel, D.P.; Harrington, M.G.; et al. Blood–brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat. Med. 2019, 25, 270–276. [Google Scholar] [CrossRef]
- Praticò, D. Oxidative stress hypothesis in Alzheimer’s disease: A reappraisal. Trends Pharmacol. Sci. 2008, 29, 609–615. [Google Scholar] [CrossRef]
- Persson, T.; Popescu, B.O.; Cedazo-Minguez, A. Oxidative Stress in Alzheimer’s Disease: Why did Antioxidant Therapy Fail? Oxidative Med. Cell. Longev. 2014, 2014, 427318. [Google Scholar] [CrossRef]
- Weidling, I.; Swerdlow, R.H. Mitochondrial Dysfunction and Stress Responses in Alzheimer’s Disease. Boilogy 2019, 8, 39. [Google Scholar] [CrossRef]
- Jiang, C.; Li, G.; Huang, P.; Liu, Z.; Zhao, B. The Gut Microbiota and Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 58, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Kowalski, K.; Mulak, A. Brain-Gut-Microbiota Axis in Alzheimer’s Disease. J. Neurogastroenterol. Motil. 2019, 25, 48–60. [Google Scholar] [CrossRef] [PubMed]
- Dominy, S.S.; Lynch, C.; Ermini, F.; Benedyk, M.; Marczyk, A.; Konradi, A.; Nguyen, M.; Haditsch, U.; Raha, D.; Griffin, C.; et al. Porphyromonas gingivalis in Alzheimer’s disease brains: Evidence for disease causation and treatment with small-molecule inhibitors. Sci. Adv. 2019, 5, eaau3333. [Google Scholar] [CrossRef] [PubMed]
- Fulop, T.; Itzhaki, R.F.; Balin, B.J.; Miklossy, J.; Barron, A.E. Role of Microbes in the Development of Alzheimer’s Disease: State of the Art—An International Symposium Presented at the 2017 IAGG Congress in San Francisco. Front. Genet. 2018, 9, 362. [Google Scholar] [CrossRef] [PubMed]
- Mintzopoulos, D.; Gillis, T.E.; Tedford, C.E.; Kaufman, M.J. Effects of Near-Infrared Light on Cerebral Bioenergetics Measured with Phosphorus Magnetic Resonance Spectroscopy. Photomed. Laser Surg. 2017, 35, 395–400. [Google Scholar] [CrossRef]
- Ando, T.; Xuan, W.; Xu, T.; Dai, T.; Sharma, S.K.; Kharkwal, G.B.; Huang, Y.-Y.; Wu, Q.; Whalen, M.J.; Sato, S.; et al. Comparison of Therapeutic Effects between Pulsed and Continuous Wave 810-nm Wavelength Laser Irradiation for Traumatic Brain Injury in Mice. PLoS ONE 2011, 6, e26212–e26220. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, F.; Ahmadian, N.; Rasta, S.H.; Farhoudi, M.; Karimi, P.; Sadigh-Eteghad, S. Transcranial low-level laser therapy improves brain mitochondrial function and cognitive impairment in D-galactose–induced aging mice. Neurobiol. Aging 2017, 58, 140–150. [Google Scholar] [CrossRef]
- Kann, O. The interneuron energy hypothesis: Implications for brain disease. Neurobiol. Dis. 2016, 90, 75–85. [Google Scholar] [CrossRef]
- Cao, X.; Li, L.-P.; Wang, Q.; Wu, Q.; Hu, H.-H.; Zhang, M.; Fang, Y.-Y.; Zhang, J.; Li, S.-J.; Xiong, W.-C.; et al. Astrocyte-derived ATP modulates depressive-like behaviors. Nat. Med. 2013, 19, 773–777. [Google Scholar] [CrossRef]
- Lyons, D.N.; Vekaria, H.; Macheda, T.; Bakshi, V.; Powell, D.K.; Gold, B.T.; Lin, A.-L.; Sullivan, P.G.; Bachstetter, A.D.; Sulllivan, P. A Mild Traumatic Brain Injury in Mice Produces Lasting Deficits in Brain Metabolism. J. Neurotrauma 2018, 35, 2435–2447. [Google Scholar] [CrossRef]
- Briston, T.; Hicks, A.R. Mitochondrial dysfunction and neurodegenerative proteinopathies: Mechanisms and prospects for therapeutic intervention. Biochem. Soc. Trans. 2018, 46, 829–842. [Google Scholar] [CrossRef] [PubMed]
- Swerdlow, R.H.; Burns, J.M.; Khan, S.M. The Alzheimer’s disease mitochondrial cascade hypothesis: Progress and perspectives. Biochim. Biophys. Acta 2014, 1842, 1219–1231. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Tian, F.; Soni, S.S.; Gonzalez-Lima, F.; Liu, H. Interplay between up-regulation of cytochrome-c-oxidase and hemoglobin oxygenation induced by near-infrared laser. Sci. Rep. 2016, 6, 30540. [Google Scholar] [CrossRef] [PubMed]
- Schiffer, F.; Johnston, A.L.; Ravichandran, C.; Polcari, A.; Teicher, M.H.; Webb, R.H.; Hamblin, M.R. Psychological benefits 2 and 4 weeks after a single treatment with near infrared light to the forehead: A pilot study of 10 patients with major depression and anxiety. Behav. Brain Funct. 2009, 5, 46. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.I.; Lee, S.-W.; Kim, S.Y.; Kim, N.G.; Park, K.-J.; Choi, B.T.; Shin, Y.-I.; Shin, H.K. Pretreatment with light-emitting diode therapy reduces ischemic brain injury in mice through endothelial nitric oxide synthase-dependent mechanisms. Biochem. Biophys. Res. Commun. 2017, 486, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Charriaut-Marlangue, C.; Bonnin, P.; Pham, H.; Loron, G.; Leger, P.-L.; Gressens, P.; Renolleau, S.; Baud, O.; Charriaut-Marlangue, C. Nitric oxide signaling in the brain: A new target for inhaled nitric oxide? Ann. Neurol. 2013, 73, 442–448. [Google Scholar] [CrossRef]
- Bragin, D.E.; Statom, G.L.; Hagberg, S.; Nemoto, E.M. Increases in microvascular perfusion and tissue oxygenation via pulsed electromagnetic fields in the healthy rat brain. J. Neurosurg. 2015, 122, 1239–1247. [Google Scholar] [CrossRef]
- Kisler, K.; Nelson, A.R.; Montagne, A.; Zlokovic, B.V. Cerebral blood flow regulation and neurovascular dysfunction in Alzheimer disease. Nat. Rev. Neurosci. 2017, 18, 419–434. [Google Scholar] [CrossRef]
- Liang, J.; Liu, L.; Xing, D. Photobiomodulation by low-power laser irradiation attenuates Abeta-induced cell apoptosis through the Akt/GSK3beta/beta-catenin pathway. Free Radic. Biol. Med. 2012, 53, 1459–1467. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, Y.; Xing, D. LPLI inhibits apoptosis upstream of Bax translocation via a GSK-3beta-inactivation mechanism. J. Cell. Physiol. 2010, 224, 218–228. [Google Scholar]
- Ling, Q.; Meng, C.; Chen, Q.; Xing, D. Activated ERK/FOXM1 pathway by low-power laser irradiation inhibits UVB-induced senescence through down-regulating p21 expression. J. Cell. Physiol. 2014, 229, 108–116. [Google Scholar] [CrossRef] [PubMed]
- Eells, J.T.; Gopalakrishnan, S.; Valter, K. Near-Infrared Photobiomodulation in Retinal Injury and Disease. Adv. Exp. Med. Biol. 2016, 854, 437–441. [Google Scholar] [PubMed]
- Eells, J.T.; Henry, M.M.; Summerfelt, P.; Wong-Riley, M.T.T.; Buchmann, E.V.; Kane, M.; Whelan, N.T.; Whelan, H.T. Therapeutic photobiomodulation for methanol-induced retinal toxicity. Proc. Natl. Acad. Sci. USA 2003, 100, 3439–3444. [Google Scholar] [CrossRef] [PubMed]
- Liang, H.; Whelan, H.; Eells, J.; Meng, H.; Büchmann, E.; Lerch-Gaggl, A.; Wong-Riley, M. Photobiomodulation partially rescues visual cortical neurons from cyanide-induced apoptosis. Neuroscience 2006, 139, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Nagata, K.; Tedford, C.E.; McCarthy, T.; Hamblin, M.R. Low-level laser therapy (LLLT) reduces oxidative stress in primary cortical neurons in vitro. J. Biophotonics 2013, 6, 829–838. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.K.; Kharkwal, G.B.; Sajo, M.; Huang, Y.-Y.; De Taboada, L.; McCarthy, T.; Hamblin, M.R. Dose Response Effects of 810 nm Laser Light on Mouse Primary Cortical Neurons. Lasers Surg. Med. 2011, 43, 851–859. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.Y.; Nagata, K.; Tedford, C.E.; Hamblin, M.R. Low-level laser therapy (810 nm) protects primary cortical neurons against excitotoxicity in vitro. J. Biophotonics 2014, 7, 656–664. [Google Scholar] [CrossRef]
- Habtemariam, S. Natural Products in Alzheimer’s Disease Therapy: Would Old Therapeutic Approaches Fix the Broken Promise of Modern Medicines? Molecules 2019, 24, 1519. [Google Scholar] [CrossRef]
- Roomruangwong, C.; Anderson, G.; Berk, M.; Stoyanov, D.; Carvalho, A.F.; Maes, M. A neuro-immune, neuro-oxidative and neuro-nitrosative model of prenatal and postpartum depression. Prog. Neuro-Psychopharmacol. Boil. Psychiatry 2018, 81, 262–274. [Google Scholar] [CrossRef]
- Rodríguez-Rodríguez, A.; Egea-Guerrero, J.J.; Murillo-Cabezas, F.; Carrillo-Vico, A. Oxidative Stress in Traumatic Brain Injury. Curr. Med. Chem. 2014, 21, 1201–1211. [Google Scholar] [CrossRef]
- Wu, J.; Xia, S.; Kalionis, B.; Wan, W.; Sun, T. The Role of Oxidative Stress and Inflammation in Cardiovascular Aging. BioMed Res. Int. 2014, 2014, 615312. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Wang, W.; Li, L.; Perry, G.; Lee, H.-G.; Zhu, X. Oxidative stress and mitochondrial dysfunction in Alzheimer’s disease. Biochim. Biophys. Acta 2014, 1842, 1240–1247. [Google Scholar] [CrossRef] [PubMed]
- Steinhubl, S.R. Why Have Antioxidants Failed in Clinical Trials? Am. J. Cardiol. 2008, 101, S14–S19. [Google Scholar] [CrossRef] [PubMed]
- Rahal, A.; Kumar, A.; Singh, V.; Yadav, B.; Tiwari, R.; Chakraborty, S.; Dhama, K. Oxidative Stress, Prooxidants, and Antioxidants: The Interplay. BioMed Res. Int. 2014, 2014, 761264. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K.; Oberbach, A.; Klöting, N.; Birringer, M.; Kiehntopf, M.; Stumvoll, M.; Kahn, C.R.; Blüher, M. Antioxidants prevent health-promoting effects of physical exercise in humans. Proc. Natl. Acad. Sci. USA 2009, 106, 8665–8670. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, F.; Farajdokht, F.; Erfani, M.; Sadigh-Eteghad, S.; Shotorbani, S.S.; Karimi, P.; Rasta, S.H.; Mahmoudi, J.; Hamblin, M.R. Transcranial near-infrared photobiomodulation attenuates memory impairment and hippocampal oxidative stress in sleep-deprived mice. Brain Res. 2018, 1682, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Bartos, A.; Grondin, Y.; Bortoni, M.E.; Ghelfi, E.; Sepúlveda, R.; Carroll, J.; Rogers, R.A. Pre-conditioning with near infrared photobiomodulation reduces inflammatory cytokines and markers of oxidative stress in cochlear hair cells. J. Biophotonics 2016, 9, 1125–1135. [Google Scholar] [CrossRef]
- Mungrue, I.N.; Husain, M.; Stewart, D.J. The Role of NOS in Heart Failure: Lessons from Murine Genetic Models. Heart Fail. Rev. 2002, 7, 407–422. [Google Scholar] [CrossRef]
- Ahmed, I.; Bose, S.K.; Pavese, N.; Ramlackhansingh, A.; Turkheimer, F.; Hotton, G.; Hammers, A.; Brooks, D.J. Glutamate NMDA receptor dysregulation in Parkinson’s disease with dyskinesias. Brain 2011, 134, 979–986. [Google Scholar] [CrossRef]
- Assis, L.; Moretti, A.I.; Abrahao, T.B.; de Souza, H.P.; Hamblin, M.R.; Parizotto, N.A. Low-level laser therapy (808 nm) contributes to muscle regeneration and prevents fibrosis in rat tibialis anterior muscle after cryolesion. Lasers Med. Sci. 2013, 28, 947–955. [Google Scholar] [CrossRef]
- Cury, V.; Moretti, A.I.; Assis, L.; Bossini, P.; Crusca, J.S.; Neto, C.B.; Fangel, R.; de Souza, H.P.; Hamblin, M.R.; Parizotto, N.A. Low level laser therapy increases angiogenesis in a model of ischemic skin flap in rats mediated by VEGF, HIF-1alpha and MMP-2. J. Photochem. Photobiol. B Boil. 2013, 125, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Walski, T.; Drohomirecka, A.; Bujok, J.; Czerski, A.; Wąż, G.; Trochanowska-Pauk, N.; Gorczykowski, M.; Cichon, R.; Komorowska, M. Low-Level Light Therapy Protects Red Blood Cells Against Oxidative Stress and Hemolysis during Extracorporeal Circulation. Front. Physiol. 2018, 9, 647. [Google Scholar] [CrossRef] [PubMed]
- Sarandol, A.; Sarandol, E.; Eker, S.S.; Erdinc, S.; Vatansever, E.; Kirli, S. Major depressive disorder is accompanied with oxidative stress: Short-term antidepressant treatment does not alter oxidative–antioxidative systems. Hum. Psychopharmacol. Clin. Exp. 2007, 22, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhong, C. Oxidative stress in Alzheimer’s disease. Neurosci. Bull. 2014, 30, 271–281. [Google Scholar] [CrossRef] [PubMed]
- Wadsworth, T.L.; Bishop, J.A.; Pappu, A.S.; Woltjer, R.L.; Quinn, J.F. Evaluation of Coenzyme Q as an Antioxidant Strategy for Alzheimer’s Disease. J. Alzheimers Dis. 2008, 14, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.; Kim, J.; Kim, S.; Karna, S.; Won, J.; Jeon, S.M.; Kim, S.Y.; Choi, Y.; Choi, H.; Kim, O. Modulation of lipopolysaccharide-induced NF-kappaB signaling pathway by 635 nm irradiation via heat shock protein 27 in human gingival fibroblast cells. Photochem. Photobiol. 2013, 89, 199–207. [Google Scholar] [CrossRef]
- Yang, Y.; Gao, L. Celecoxib Alleviates Memory Deficits by Downregulation of COX-2 Expression and Upregulation of the BDNF-TrkB Signaling Pathway in a Diabetic Rat Model. J. Mol. Neurosci. 2017, 62, 188–198. [Google Scholar] [CrossRef]
- Chen, A.C.-H.; Arany, P.R.; Huang, Y.-Y.; Tomkinson, E.M.; Sharma, S.K.; Kharkwal, G.B.; Saleem, T.; Mooney, D.; Yull, F.E.; Blackwell, T.S.; et al. Low-Level Laser Therapy Activates NF-kB via Generation of Reactive Oxygen Species in Mouse Embryonic Fibroblasts. PLoS ONE 2011, 6, e22453. [Google Scholar] [CrossRef]
- Chen, A.C.-H.; Huang, Y.-Y.; Sharma, S.K.; Hamblin, M.R. Effects of 810-nm Laser on Murine Bone-Marrow-Derived Dendritic Cells. Photomed. Laser Surg. 2011, 29, 383–389. [Google Scholar] [CrossRef]
- Yamaura, M.; Yao, M.; Yaroslavsky, I.; Cohen, R.; Smotrich, M.; Kochevar, I.E. Low level light effects on inflammatory cytokine production by rheumatoid arthritis synoviocytes. Lasers Surg. Med. 2009, 41, 282–290. [Google Scholar] [CrossRef]
- Filiano, A.J.; Gadani, S.P.; Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015, 1617, 18–27. [Google Scholar] [CrossRef] [PubMed]
- Dissing-Olesen, L.; Ladeby, R.; Nielsen, H.; Toft-Hansen, H.; Dalmau, I.; Finsen, B. Axonal lesion-induced microglial proliferation and microglial cluster formation in the mouse. Neuroscience 2007, 149, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Orihuela, R.; McPherson, C.A.; Harry, G.J. Microglial M1/M2 polarization and metabolic states. Br. J. Pharmacol. 2016, 173, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Haschemi, A.; Kosma, P.; Gille, L.; Evans, C.R.; Burant, C.F.; Starkl, P.; Knapp, B.; Haas, R.; Schmid, J.A.; Jandl, C.; et al. The Sedoheptulose Kinase CARKL Directs Macrophage Polarization through Control of Glucose Metabolism. Cell Metab. 2012, 15, 813–826. [Google Scholar] [CrossRef]
- Fernandes, K.P.S.; Souza, N.H.C.; Mesquita-Ferrari, R.A.; Silva, D.D.F.T.D.; Rocha, L.A.; Alves, A.N.; Sousa, K.D.B.; Bussadori, S.K.; Hamblin, M.R.; Nunes, F.D. Photobiomodulation with 660-nm and 780-nm laser on activated J774 macrophage-like cells: Effect on M1 inflammatory markers. J. Photochem. Photobiol. B Boil. 2015, 153, 344–351. [Google Scholar] [CrossRef]
- Singer, A.C.; Martorell, A.J.; Douglas, J.M.; Abdurrob, F.; Attokaren, M.K.; Tipton, J.; Mathys, H.; Adaikkan, C.; Tsai, L.-H. Noninvasive 40-Hz light flicker to recruit microglia and reduce amyloid beta load. Nat. Protoc. 2018, 13, 1850–1868. [Google Scholar] [CrossRef]
- Bergmann, O.; Spalding, K.L.; Frisen, J. Adult Neurogenesis in Humans. Cold Spring Harb. Perspect. Biol. 2015, 7, a018994. [Google Scholar] [CrossRef]
- Lepousez, G.; Nissant, A.; Lledo, P.-M. Adult Neurogenesis and the Future of the Rejuvenating Brain Circuits. Neuron 2015, 86, 387–401. [Google Scholar] [CrossRef]
- Kirschen, G.W.; Kery, R.; Ge, S. The Hippocampal Neuro-Glio-Vascular Network: Metabolic Vulnerability and Potential Neurogenic Regeneration in Disease. Brain Plast. 2018, 3, 129–144. [Google Scholar] [CrossRef]
- Zhang, L.; Li, H.; Zeng, S.; Chen, L.; Fang, Z.; Huang, Q. Long-term tracing of the BrdU label-retaining cells in adult rat brain. Neurosci. Lett. 2015, 591, 30–34. [Google Scholar] [CrossRef]
- Pozhilenkova, E.A.; Lopatina, O.L.; Komleva, Y.K.; Salmin, V.V.; Salmina, A.B. Blood-brain barrier-supported neurogenesis in healthy and diseased brain. Rev. Neurosci. 2017, 28, 397–415. [Google Scholar] [CrossRef] [PubMed]
- Hu, M.; Zhu, K.; Chen, X.-L.; Zhang, Y.-J.; Zhang, J.-S.; Xiao, X.-L.; Liu, J.-X.; Liu, Y. Newly generated neurons at 2 months post-status epilepticus are functionally integrated into neuronal circuitry in mouse hippocampus. Exp. Neurol. 2015, 273, 273–287. [Google Scholar] [CrossRef] [PubMed]
- Xavier, A.L.R.; Kress, B.T.; Goldman, S.A.; De Menezes, J.R.L.; Nedergaard, M.; Xavier, A.L.R. A Distinct Population of Microglia Supports Adult Neurogenesis in the Subventricular Zone. J. Neurosci. 2015, 35, 11848–11861. [Google Scholar] [CrossRef] [PubMed]
- Shen, S.-W.; Duan, C.-L.; Chen, X.-H.; Wang, Y.-Q.; Sun, X.; Zhang, Q.-W.; Cui, H.-R.; Sun, F.-Y. Neurogenic effect of VEGF is related to increase of astrocytes transdifferentiation into new mature neurons in rat brains after stroke. Neuropharmacology 2016, 108, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Oron, A.; Oron, U.; Chen, J.; Eilam, A.; Zhang, C.; Sadeh, M.; Lampl, Y.; Streeter, J.; DeTaboada, L.; Chopp, M.; et al. Low-Level Laser Therapy Applied Transcranially to Rats after Induction of Stroke Significantly Reduces Long-Term Neurological Deficits. Stroke 2006, 37, 2620–2624. [Google Scholar] [CrossRef]
- Xuan, W.; Vatansever, F.; Huang, L.; Hamblin, M.R. Transcranial low-level laser therapy enhances learning, memory, and neuroprogenitor cells after traumatic brain injury in mice. J. Biomed. Opt. 2014, 19, 108003. [Google Scholar] [CrossRef] [PubMed]
- Moreno-Jiménez, E.P.; Flor-García, M.; Terreros-Roncal, J.; Rábano, A.; Cafini, F.; Pallas-Bazarra, N.; Ávila, J.; Llorens-Martín, M. Adult hippocampal neurogenesis is abundant in neurologically healthy subjects and drops sharply in patients with Alzheimer’s disease. Nat. Med. 2019, 25, 554–560. [Google Scholar] [CrossRef]
- Bothwell, M. NGF, BDNF, NT3, and NT4. Handb. Exp. Pharmacol. 2014, 220, 3–15. [Google Scholar]
- Yang, J.; Harte-Hargrove, L.C.; Siao, C.-J.; Marinic, T.; Clarke, R.; Ma, Q.; Jing, D.; Lafrancois, J.J.; Bath, K.G.; Mark, W.; et al. ProBDNF negatively regulates neuronal remodeling, synaptic transmission and synaptic plasticity in hippocampus. Cell Rep. 2014, 7, 796–806. [Google Scholar] [CrossRef]
- Yan, X.; Liu, J.; Zhang, Z.; Li, W.; Sun, S.; Zhao, J.; Dong, X.; Qian, J.; Sun, H. Low-level laser irradiation modulates brain-derived neurotrophic factor mRNA transcription through calcium-dependent activation of the ERK/CREB pathway. Lasers Med. Sci. 2017, 32, 169–180. [Google Scholar] [CrossRef]
- Barbieri, R.; Contestabile, A.; Ciardo, M.G.; Forte, N.; Marte, A.; Baldelli, P.; Benfenati, F.; Onofri, F. Synapsin I and Synapsin II regulate neurogenesis in the dentate gyrus of adult mice. Oncotarget 2018, 9, 18760–18774. [Google Scholar] [CrossRef] [PubMed]
- Meng, C.; He, Z.; Xing, D. Low-Level Laser Therapy Rescues Dendrite Atrophy via Upregulating BDNF Expression: Implications for Alzheimer’s Disease. J. Neurosci. 2013, 33, 13505–13517. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Huang, Y.-Y.; Wang, Y.; Lyu, P.; Hamblin, M.R. Photobiomodulation of human adipose-derived stem cells using 810 nm and 980 nm lasers operates via different mechanisms of action. Biochim. Biophys. Acta 2016, 1861, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Forrest, M.P.; Parnell, E.; Penzes, P. Dendritic structural plasticity and neuropsychiatric disease. Nat. Rev. Neurosci. 2018, 19, 215–234. [Google Scholar] [CrossRef] [PubMed]
- Nahum, M.; Lee, H.; Merzenich, M.M. Principles of Neuroplasticity-Based Rehabilitation. Pathol. Situat. Dis. 2013, 207, 141–171. [Google Scholar]
- Kuhn, M.; Weidenauer, A.; Pezawas, L. Neuroplasticity and memory formation in major depressive disorder: An imaging genetics perspective on serotonin and BDNF. Restor. Neurol. Neurosci. 2014, 32, 25–49. [Google Scholar]
- Felling, R.J.; Song, H. Epigenetic mechanisms of neuroplasticity and the implications for stroke recovery. Exp. Neurol. 2015, 268, 37–45. [Google Scholar] [CrossRef] [PubMed]
- Tomaszczyk, J.C.; Green, N.L.; Frasca, D.; Colella, B.; Turner, G.R.; Christensen, B.K.; Green, R.E.A. Negative Neuroplasticity in Chronic Traumatic Brain Injury and Implications for Neurorehabilitation. Neuropsychol. Rev. 2014, 24, 409–427. [Google Scholar] [CrossRef]
- Creed, M.; Pascoli, V.J.; Luscher, C. Refining deep brain stimulation to emulate optogenetic treatment of synaptic pathology. Science 2015, 347, 659–664. [Google Scholar] [CrossRef]
- Arany, P.R. Photobiomodulation Therapy: Communicating with Stem Cells for Regeneration? Photomed. Laser Surg. 2016, 34, 497–499. [Google Scholar] [CrossRef]
- Abrahamse, H.; Hamblin, M.R. Photomedicine and Stem Cells; Morgan and Claypool Publishing: San Rafael, CA, USA, 2017; ISBN 978-1-6817-4321-9. [Google Scholar]
- Farfara, D.; Tuby, H.; Trudler, D.; Doron-Mandel, E.; Maltz, L.; Vassar, R.J.; Frenkel, D.; Oron, U. Low-level laser therapy ameliorates disease progression in a mouse model of Alzheimer’s disease. J. Mol. Neurosci. 2015, 55, 430–436. [Google Scholar] [CrossRef] [PubMed]
- Blatt, A.; Elbaz-Greener, G.A.; Tuby, H.; Maltz, L.; Siman-Tov, Y.; Ben-Aharon, G.; Copel, L.; Eisenberg, I.; Efrati, S.; Jonas, M.; et al. Low-Level Laser Therapy to the Bone Marrow Reduces Scarring and Improves Heart Function Post-Acute Myocardial Infarction in the Pig. Photomed. Laser Surg. 2016, 34, 516–524. [Google Scholar] [CrossRef] [PubMed]
- Tuby, H.; Maltz, L.; Oron, U. Induction of autologous mesenchymal stem cells in the bone marrow by low-level laser therapy has profound beneficial effects on the infarcted rat heart. Lasers Surg. Med. 2011, 43, 401–409. [Google Scholar] [CrossRef] [PubMed]
- Oron, U.; Tuby, H.; Maltz, L.; Sagi-Assif, O.; Abu-Hamed, R.; Yaakobi, T.; Doenyas-Barak, K.; Efrati, S. Autologous Bone-Marrow Stem Cells Stimulation Reverses Post-Ischemic-Reperfusion Kidney Injury in Rats. Am. J. Nephrol. 2014, 40, 425–433. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.M.; Yeon, B.K.; Cho, S.J.; Suh, Y.H. Stem Cell Therapy for Alzheimer’s Disease: A Review of Recent Clinical Trials. J. Alzheimer’s Dis. 2016, 54, 879–889. [Google Scholar] [CrossRef]
- Iaccarino, H.F.; Singer, A.C.; Martorell, A.J.; Rudenko, A.; Gao, F.; Gillingham, T.Z.; Mathys, H.; Seo, J.; Kritskiy, O.; Abdurrob, F.; et al. Gamma frequency entrainment attenuates amyloid load and modifies microglia. Nature 2016, 540, 230–235. [Google Scholar] [CrossRef]
- Adaikkan, C.; Middleton, S.J.; Marco, A.; Pao, P.-C.; Mathys, H.; Kim, D.N.-W.; Gao, F.; Young, J.Z.; Suk, H.-J.; Boyden, E.S.; et al. Gamma Entrainment Binds Higher-Order Brain Regions and Offers Neuroprotection. Neuron 2019, 102, 929–943. [Google Scholar] [CrossRef]
- Martorell, A.J.; Paulson, A.L.; Suk, H.-J.; Abdurrob, F.; Drummond, G.T.; Guan, W.; Young, J.Z.; Kim, D.N.-W.; Kritskiy, O.; Barker, S.J.; et al. Multi-sensory Gamma Stimulation Ameliorates Alzheimer’s-Associated Pathology and Improves Cognition. Cell 2019, 177, 256–271. [Google Scholar] [CrossRef]
- De Taboada, L.; Yu, J.; El-Amouri, S.; Gattoni-Celli, S.; Richieri, S.; McCarthy, T.; Jackson, S.; Mark, K.S. Transcranial laser therapy attenuates amyloid-beta peptide neuropathology in amyloid-beta protein precursor transgenic mice. J. Alzheimer’s Dis. 2011, 23, 521–535. [Google Scholar] [CrossRef]
- Purushothuman, S.; Johnstone, D.M.; Nandasena, C.; Mitrofanis, J.; Stone, J. Photobiomodulation with near infrared light mitigates Alzheimer’s disease-related pathology in cerebral cortex-evidence from two transgenic mouse models. Alzheimer’s Res. Ther. 2014, 6, 2. [Google Scholar] [CrossRef]
- Purushothuman, S.; Johnstone, D.M.; Nandasena, C.; van Eersel, J.; Ittner, L.M.; Mitrofanis, J.; Stone, J. Near infrared light mitigates cerebellar pathology in transgenic mouse models of dementia. Neurosci. Lett. 2015, 591, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Wang, R.; Dong, Y.; Tucker, D.; Zhao, N.; Ahmed, M.E.; Zhu, L.; Liu, T.C.-Y.; Cohen, R.M.; Zhang, Q. Low-level laser therapy for beta amyloid toxicity in rat hippocampus. Neurobiol. Aging 2017, 49, 165–182. [Google Scholar] [CrossRef] [PubMed]
- Da Luz Eltchechem, C.; Salgado, A.S.I.; Zangaro, R.A.; da Silva Pereira, M.C.; Kerppers, I.I.; da Silva, L.A.; Parreira, R.B. Transcranial LED therapy on amyloid-beta toxin 25–35 in the hippocampal region of rats. Lasers Med. Sci. 2017, 32, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Blivet, G.; Meunier, J.; Roman, F.J.; Touchon, J. Neuroprotective effect of a new photobiomodulation technique against Aβ25-35 peptide-induced toxicity in mice: Novel hypothesis for therapeutic approach of Alzheimer’s disease suggested. Alzheimer’s Dement. 2018, 4, 54–63. [Google Scholar] [CrossRef]
- Saltmarche, A.E.; Naeser, M.A.; Ho, K.F.; Hamblin, M.R.; Lim, L. Significant Improvement in Cognition in Mild to Moderately Severe Dementia Cases Treated with Transcranial Plus Intranasal Photobiomodulation: Case Series Report. Photomed. Laser Surg. 2017, 35, 432–441. [Google Scholar] [CrossRef]
- Chao, L.L. Effects of Home Photobiomodulation Treatments on Cognitive and Behavioral Function, Cerebral Perfusion, and Resting-State Functional Connectivity in Patients with Dementia: A Pilot Trial. Photobiomodul. Photomed. Laser Surg. 2019, 37, 133–141. [Google Scholar] [CrossRef]
- Berman, M.H.; Halper, J.P.; Nichols, T.W.; Jarrett, H.; Lundy, A.; Huang, J.H. Photobiomodulation with Near Infrared Light Helmet in a Pilot, Placebo Controlled Clinical Trial in Dementia Patients Testing Memory and Cognition. J. Neurol. Neurosci. 2017, 8, 176. [Google Scholar] [CrossRef]
- Salehpour, F.; Hamblin, M.R.; DiDuro, J.O. Rapid Reversal of Cognitive Decline, Olfactory Dysfunction, and Quality of Life Using Multi-Modality Photobiomodulation Therapy: Case Report. Photobiomodul. Photomed. Laser Surg. 2019, 37, 159–167. [Google Scholar] [CrossRef]
- Maksimovich, I.V. Laser Technologies as a New Direction in Transcatheter Interventions. Photobiomodul. Photomed. Laser Surg. 2019. [Google Scholar] [CrossRef]
- Maksimovich, I.V. Endovascular Application of Low-Energy Laser in the Treatment of Dyscirculatory Angiopathy of Alzheimer’s Type. J. Behav. Brain Sci. 2012, 2, 67–81. [Google Scholar] [CrossRef][Green Version]
- Jameie, S.B.; Masoumipoor, M.; Janzadeh, A.; Nasirinezhad, F.; Kerdari, M.; Soleimani, M. Combined therapeutic effects of low power laser (980 nm) and CoQ10 on Neuropathic Pain in adult male rat. Med. J. Islam. Repub. Iran 2014, 28, 58. [Google Scholar] [PubMed]
- Salehpour, F.; Farajdokht, F.; Cassano, P.; Sadigh-Eteghad, S.; Erfani, M.; Hamblin, M.R.; Salimi, M.M.; Karimi, P.; Rasta, S.H.; Mahmoudi, J. Near-infrared photobiomodulation combined with coenzyme Q10 for depression in a mouse model of restraint stress: Reduction in oxidative stress, neuroinflammation, and apoptosis. Brain Res. Bull. 2019, 144, 213–222. [Google Scholar] [CrossRef] [PubMed]
- Salehpour, F.; Farajdokht, F.; Mahmoudi, J.; Erfani, M.; Farhoudi, M.; Karimi, P.; Rasta, S.H.; Sadigh-Eteghad, S.; Hamblin, M.R.; Gjedde, A. Photobiomodulation and Coenzyme Q10 Treatments Attenuate Cognitive Impairment Associated with Model of Transient Global Brain Ischemia in Artificially Aged Mice. Front. Cell. Neurosci. 2019, 13, 74. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, Y.; Xu, H.; Luo, X.; Yu, J.; Liu, J.; Chang, R.C.-C. Neuroprotection of Coenzyme Q10 in Neurodegenerative Diseases. Curr. Top. Med. Chem. 2016, 16, 858–866. [Google Scholar] [CrossRef]



© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hamblin, M.R. Photobiomodulation for Alzheimer’s Disease: Has the Light Dawned? Photonics 2019, 6, 77. https://doi.org/10.3390/photonics6030077
Hamblin MR. Photobiomodulation for Alzheimer’s Disease: Has the Light Dawned? Photonics. 2019; 6(3):77. https://doi.org/10.3390/photonics6030077
Chicago/Turabian StyleHamblin, Michael R. 2019. "Photobiomodulation for Alzheimer’s Disease: Has the Light Dawned?" Photonics 6, no. 3: 77. https://doi.org/10.3390/photonics6030077
APA StyleHamblin, M. R. (2019). Photobiomodulation for Alzheimer’s Disease: Has the Light Dawned? Photonics, 6(3), 77. https://doi.org/10.3390/photonics6030077