1. Introduction
The mammalian brain is highly interconnected [
1,
2], comprised of specialized neuronal sub-circuits that link together into a larger, functional network [
2]. Information transmission and processing within the nervous system occurs through simultaneous, parallel neuronal activity across a multitude of different brain areas. Mapping neuronal activation with high spatio-temporal resolution across major portions of the intact brain, including deep and hard-to-access areas, has been one of the long-term goals of neuroscience, and could help discern its fundamental operating principles. To this end, an array of imaging techniques is in development to allow non-invasive investigation of the brain’s circuitry and network functionality.
Among the established methods of functional whole-brain imaging, blood oxygen-level dependent functional magnetic resonance imaging (BOLD fMRI) is the most well-known. This approach is able to detect blood oxygenation changes and blood flow in the brain [
3]. Cerebral hemodynamic changes are similarly linked to neural activity via neurovascular coupling [
4] and thus BOLD fMRI has been used to make a number of observations regarding brain activity. The biggest drawback of this method is that it can only indirectly access neuronal function. An important variant, Pharmacological MRI [
5] allows the study of vasoactive neurotransmitter dynamics by targeting specific receptors or neurotransmitters chemically, but similarly lacks temporal resolution to resolve fast neuronal events, providing only an indirect link between observed signal changes and the cellular and physiological mechanisms underlying them [
6]. Electroencephalography (EEG), another well-established method, is able to measure voltage fluctuations induced by ionic currents within neurons at millisecond-range temporal resolutions [
7,
8]. However, EEG suffers from poor spatial resolution in its ability to infer underlying neural sources by solving an ill-posed inverse problem [
9]. In similar fashion, magnetoencephalography (MEG) can record the magnetic induction generated by electrochemical currents between and within neurons, combining the high temporal resolution of EEG with an improved spatial resolution and less distortion by modeling approximations [
10]. The high acquisition and maintenance cost of MEG systems, especially the need for magnetically shielded rooms to eliminate electromagnetic noise sources, together with challenges in signal extraction and interpretation, make the method less suitable for small animal neuroimaging.
A multitude of optical imaging methods have also emerged over the years as alternative approaches for observing large neuronal populations [
11], from macroscopic optical imaging techniques like diffuse optical tomography [
12] to several fluorescence microscopy methods based on voltage and calcium indicators for direct neuronal activity mapping [
13]. Recent two-photon microscopy systems can visualize a large number of neurons across several cortical layers [
14]. However, only cortical layers can typically be imaged by those methods due to strong photon scattering by brain tissues. Newly introduced three-photon systems can capture both cortical layers and the Stratum pyramidale (SP) layer of the CA1 hippocampal region by utilizing infrared excitation wavelengths, but strongly compromise their acquisition pixel rate and struggle to resolve structures beyond 1.1 mm of depth [
15]. Several optical imaging strategies have been introduced to visualize deep brain structures beyond this limit, such as the removal of overlying cortical tissues [
16] and the insertion of fiber bundles [
17], graded index lenses [
18], or minimally invasive multimode fibers [
19]. However, the effective field of view (FOV) remains well below 1 mm
3 when attempting to resolve fast neuronal activity at high volumetric frame rates [
15,
20]. Another caveat of optical brain microscopy approaches is the general requirement for invasive procedures such as scalp and/or skull removal, the insertion of fibers, or the implantation of lenses. Additionally, the installment of cranial windows is often necessary.
In recent years, optoacoustic (OA) tomography has proven to be a powerful alternative for imaging optical absorption in highly scattering tissues such as the brain. In general, snapshots of large tissue volumes can be recorded instantaneously as just a single laser pulse suffices to induce an OA response in the entire volume of interest [
21]. By relying on ultrasound detection of light-induced signals, the OA approach overcomes the limitations of pure optical imaging systems in terms of penetration depth. In this way, non-invasive, highly-specific molecular imaging is possible at depths where conventional optical methods fail, up to a few centimeters in biological tissues [
21,
22]. Through the use of high-end data acquisition systems and graphic-processing-unit (GPU)-based image rendering, non-invasive acquisition, processing and visualization of five-dimensional OA data in real time has recently become possible [
21,
23]. Functional brain imaging can be achieved by utilizing real-time volumetric spectrally enriched tomography recordings [
24]. Due to the strong and spectrally distinctive OA contrast provided by deoxygenated and oxygenated hemoglobin, OA brain imaging has mainly focused on blood oxygenation variations and hemodynamics [
25], e.g., in order to observe stimulus-induced brain function [
26] and seizure activity in mice [
23]. However, changes in hemodynamics only reflect neuronal activity indirectly. In contrast, genetically encoded calcium indicators (GECIs) are a potent tool for the direct observation of rapid neuronal activity of large brain networks. Using GECIs from the GCaMP-family [
27], we have recently shown that functional OA neuro-tomography (FONT) can image fast calcium dynamics in the brains of zebra fish [
21] and mice [
28]. Yet, since GCaMP excitation is performed at its peak absorption wavelength of 488 nm, the strong absorption of light in this spectral range by blood precludes visualization beyond the cortical areas in mammalian brains.
To demonstrate the fundamental capacity for calcium imaging in deep regions of the rodent brain using FONT, we developed and validated here a GCaMP6f-expressing in situ mouse brain model intracardially perfused with artificial cerebrospinal fluid that overcomes the limitations of low penetration at visible wavelengths. This preparation is shown to resemble in vivo conditions, demonstrating high viability and functional activity for more than 45 minutes while preserving realistic indicator responses. Following the removal of highly absorbing blood background, large-scale OA recording from mouse brains through skull and skin is implemented at penetration depths and spatio-temporal resolution scales not possible with other existing neuroimaging techniques.
2. Materials and Methods
2.1. Perfused Mouse Model Preparation
A total of 8 mice ranging in age from eight- to twelve-weeks-old C57BL/6J-Tg (Thy1-GCaMP6f) GP5.5Dkim/J (The Jackson Laboratory, Maine, ME, USA; stock number 024276) were used for these experiments in full compliance with the institutional guidelines of the Institute for Biological and Medical Imaging along with approval from the Government District of Upper Bavaria.
For cardiac perfusion, freshly prepared artificial cerebrospinal fluid (ACSF) [
29] was continuously supplied with Carbogen (Linde Group, Munich, Germany) and remained in a water bath (ThermoFisher Scientific, Waltham, MA, USA) at 42 °C. Using a perfusion pump (Cole-Parmer, Vernon Hills, IL, USA), ACSF was circulated at a rate of 6–8 ml/min through a glass heating coil (Radnoti LTD, Dublin, Ireland). The flow continued through a bubble trap and a physiological pressure transducer (AD Instruments, Sydney, Australia) connected to a pressure transducer Simulator/Tester (Utah Medical Products, Midvale, UT, USA) and a 25G butterfly infusion set. ACSF was pumped through the setup for at least 2 min to ensure no bubbles remained.
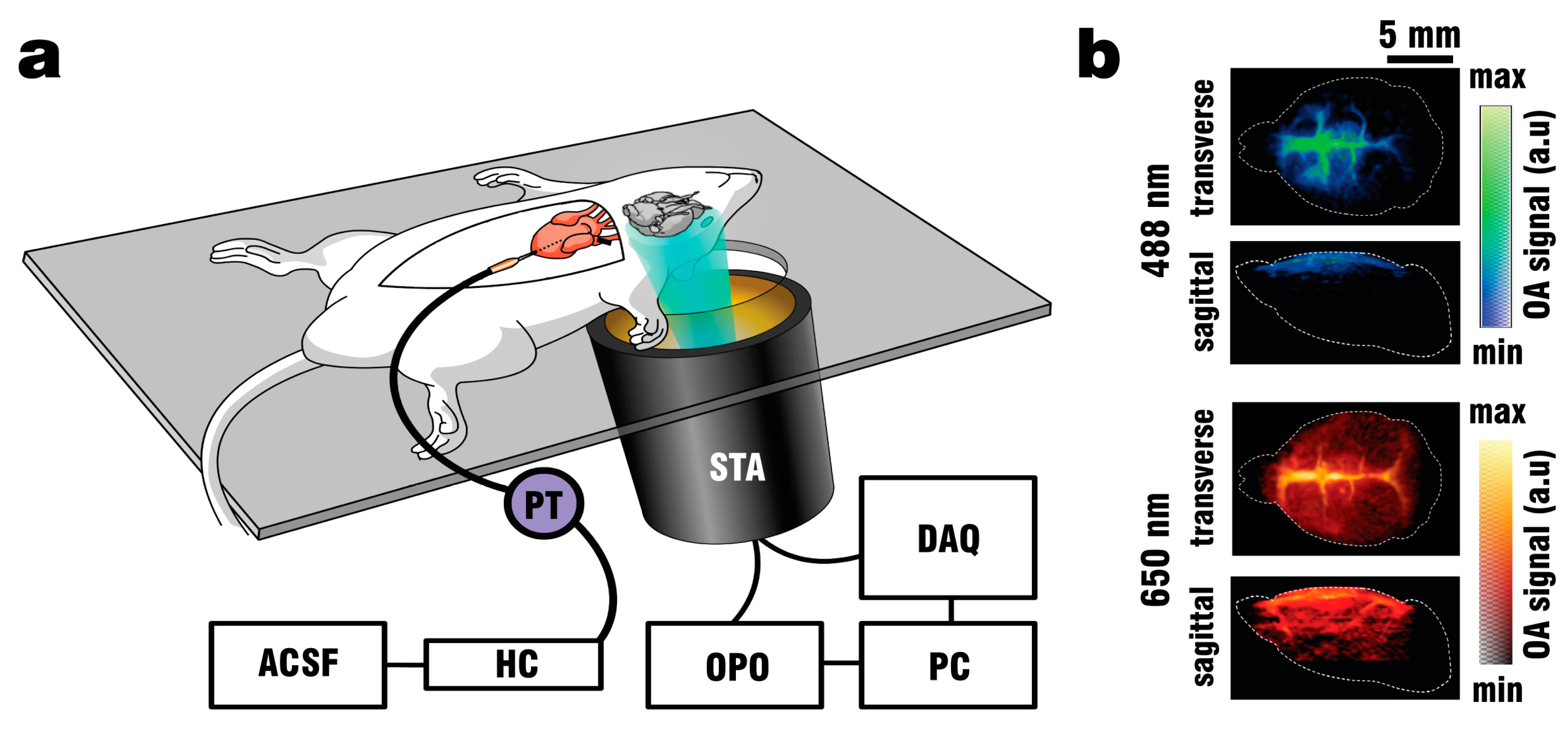
Mice were anaesthetized with a mixture of 87.5 mg/kg Ketamine (Bremer Pharma, Wabing, Germany) and 12.5 mg/kg Xylazine (Bela-pharm, Vechta, Germany) and additionally given 75 U of Heparin (Ratiofarm GbmH, Ulm, Germany) diluted in 100 µl 0.9% NaCl Solution (Braun AG, Melsungen, Germany), both administered via intraperitoneal (IP) injection. Once the mice reached a stable plane of deep anesthesia as determined by the absence of a toe-pitch reflex at both hind paws, the fur on top of the mouse head was removed using hair-removal cream. Animals were placed onto a 3.8 mm thick silicon sheet as seen in
Figure 1a, with the head positioned above an opening covering the field of view (FOV) of the FONT imaging system.
The silicon sheet was attached to a 4°-sloped holder that additionally supported the spherical transducer array used for capturing the FONT data. To avoid motion artefacts during all experiments, the mouse head was fixed into a custom designed stereotactic mouse head holder (Narishige International Limited, London, UK). At this stage, mice were injected with a second, lethal dose of 87.5 mg/kg Ketamine and 12.5 mg/kg Xylazine. After a second toe-pinch reflex test of the hind paws, all extremities were secured with 25G needles by pinning onto the silicon sheet. Surgery began with an incision from the mid abdomen to the sternum followed by removing the ventral portion of the ribcage to allow unhindered access to the heart. Once the mouse heart stopped beating, intracardiac perfusion with ACSF was performed by inserting the 25G butterfly needle into the left ventricle of the heart followed by an incision into the right atrium. Pump speed was adjusted to ensure the pressure recorded by the transducer did not exceed the physiological maximum of 100 mmHg. Fresh, oxygenated ACSF was supplied during the entire duration of the experiment to preserve brain functionality and serve as a vehicle for delivering the epileptic drug Pentylenetetrazol (PTZ). This convulsant significantly blocks GABA
A receptor function [
30] and can induce seizures and thus widespread brain activity when given in sufficient concentration [
31,
32]. PTZ (100 mg/ml in saline) could be added to the ACSF flow through a stopcock (Braun, Melsungen, Germany) connected to an Aladdin programmable syringe pump (World Precision instruments, Sarasota, FL, USA).
A schematic of the perfusion and imaging setup is depicted in
Figure 1a. Note that intracardiac perfusion was selected as the vehicle of ACSF to provide consistent clearing results. Other methods of ACSF delivery, such as perfusion through the tail vein with an incision of the external jugular vein to serve as an outlet were explored, but deemed too unreliable for proper visualization of the brain. By pumping oxygenated ACSF at 37.5 °C (when delivered through the heart) physiological conditions for the blood-free brain could be established. Fixating the animals onto the silicon pad in combination with the use of a head holder was necessary to avoid motion artifacts during recording, especially random spasms induced by the addition of PTZ to the perfusion mix. The spherical array transducer holder was designed with a 4° incline to allow easier collection of blood and ACSF flowing through the incision in the right atrium and reduce the chance of contaminating the coupling medium. The use of a pressure transducer improved the clearing efficiency during perfusion and reduced the amount of vessels retaining blood when pressures did not exceed the physiological limit of 100 mmHg.
2.2. Electroencephalography Recordings
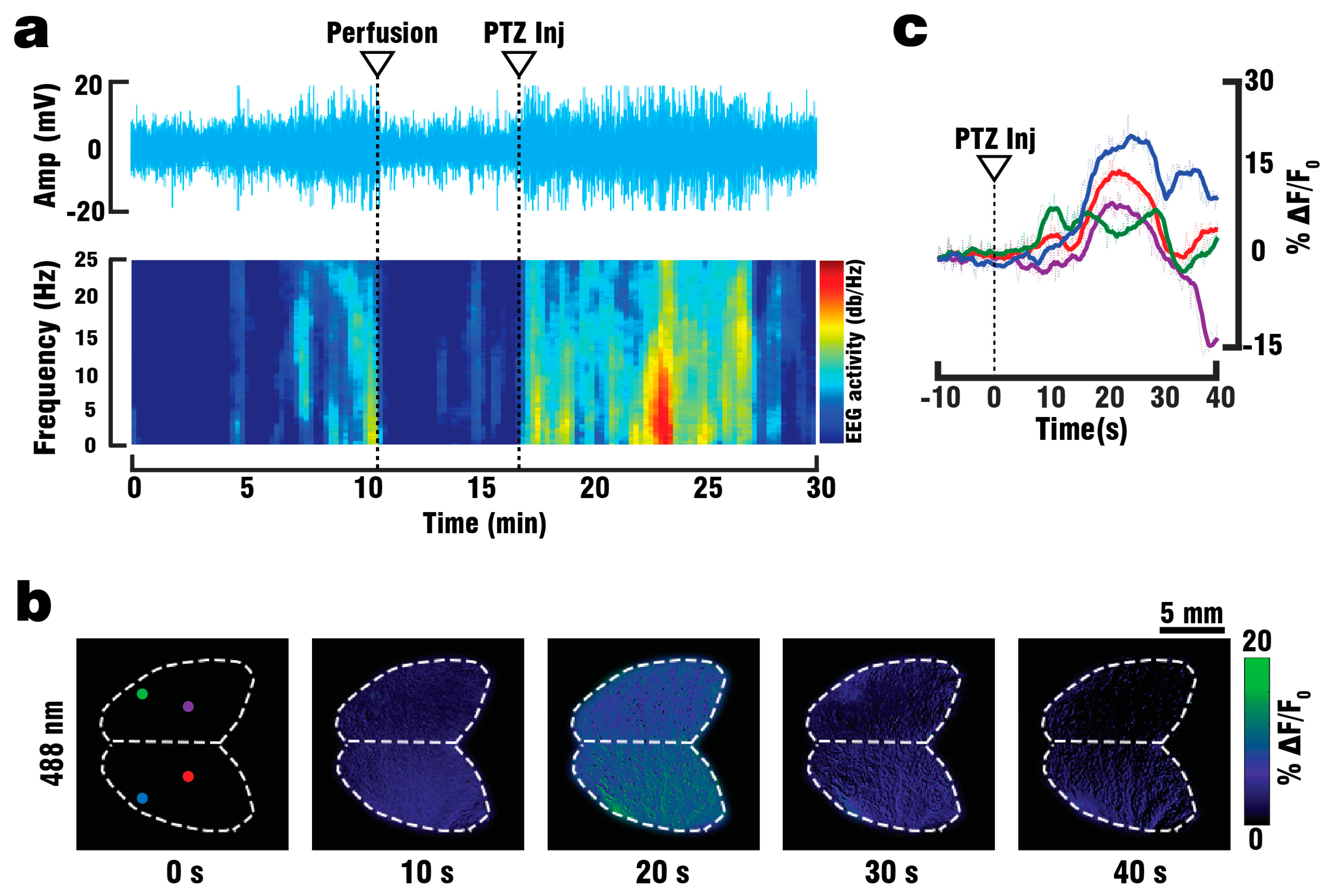
For electroencephalography (EEG) recordings, a GCaMP6f mouse (n = 1) was anaesthetized with a Ketamine/Xylazine-mixture and administered Heparin as described above. Once the mouse reached a stable level of anesthesia, the scalp was removed and a droplet of Xylonest 2% topical anesthetic (AstraZeneca, Cambridge, UK) was applied to prevent excessive bleeding. Two small openings, one in each hemisphere and set 5 mm apart, were cut into the mouse skull using a micro drill system (CircuitMedic, Haverhill, MA, USA). The animal was then placed on the STR holder and fixed in place using 25G needles. The head of the mouse was secured via a head holder. EEG-signals were recorded via two custom-made needle electrodes fixed 5 mm apart to a micromanipulator (Narishige International Limited, London, UK) and inserted into the openings drilled on the skull. Both electrodes were connected to a to a DP-311 differential amplifier (Warner Instruments, Hamden, CT, USA) set to a high pass of 10 Hz, a low pass of 100 Hz and a gain of 100. Amplified signals were digitized via a PowerLab26T data acquisition module (AD Instruments, Sydney, Australia) controlled through a host PC running the Labchart 8 software (AD Instruments, Sydney, Australia). A grounding electrode connected to the Powerlab DAQ was inserted into the skin around the neck of the mouse. After 10 min of baseline recording, a lethal dose of Ketamine/Xylazine-mixture was administered via IP injection and intracardiac perfusion was started as described above. To validate the responsiveness of the brain, 3 mL of PTZ was injected into the perfusion setup over the course of 60 s. The recorded EEG signals were processed using MatLab (Math Works, Natick, MA, USA) to compare the anesthetized and perfused states and to identify periods of elevated brain activity following PTZ injection. For this, the EEG spectrogram was calculated as the short-time Fourier transform with a window of 20 s, sufficient for detecting higher frequency components in the 10–20 Hz range corresponding to seizure-like activity caused by PTZ.
2.3. Fluorescence Recordings
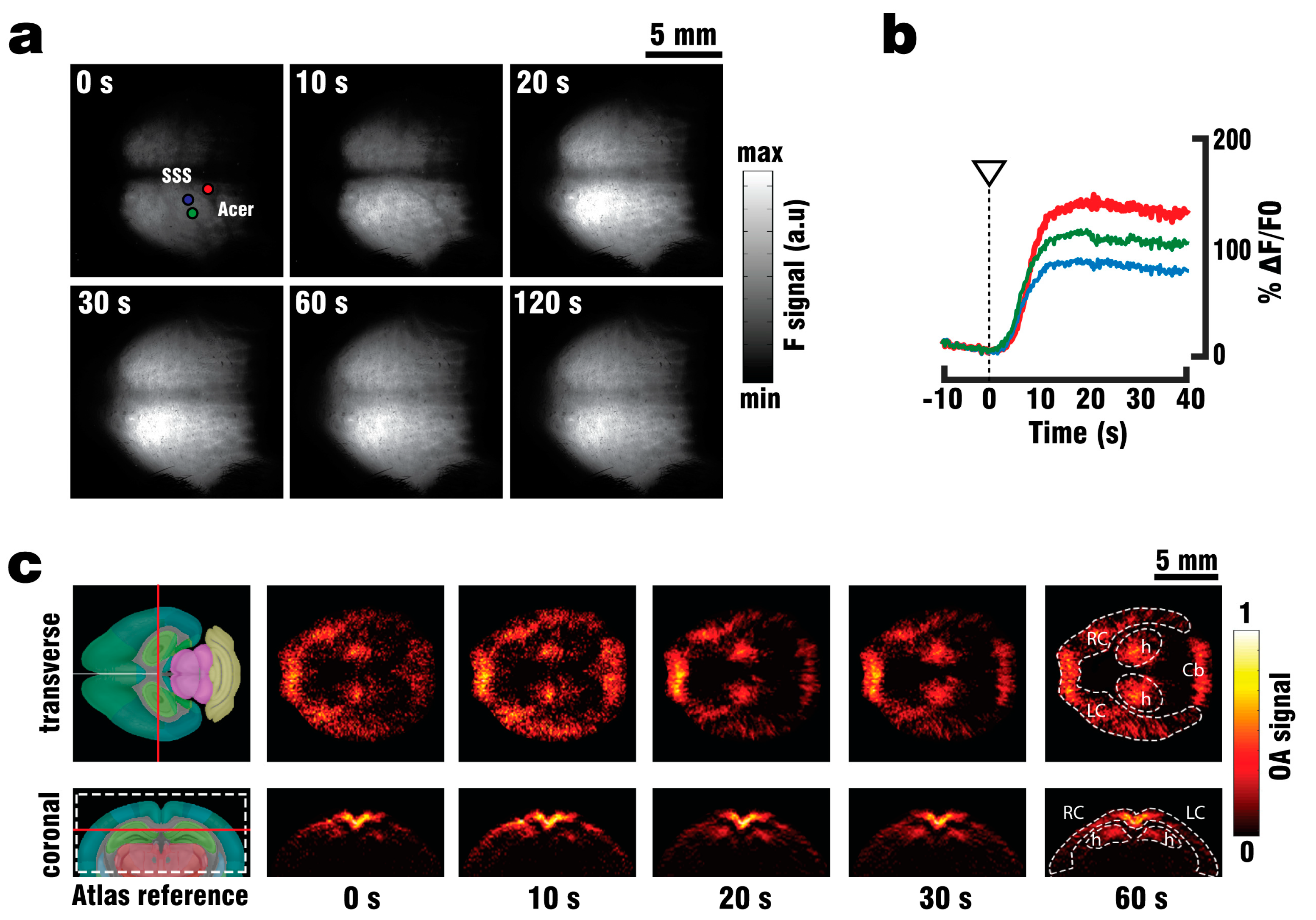
Wide-field surface imaging of GCaMP6f fluorescence were performed during perfusion and PTZ stimulation by means of a Luca R 604 high speed EMCCD camera (Andor Technology, Belfast, UK). The camera was equipped with a 105 mm Nikon F mount objective (Nikon, Chiyoda, Tokio, Japan) and a one-inch emission filter with 525 nm center wavelength and 39 nm bandwidth (MF525-39, Thorlabs Inc., Newton, MA, USA). The illumination source was an optical parametric oscillator (OPO)-based laser (EVO I OPO 355nm broadband, Innolas GmbH, Krailling, Germany) providing short (<10 ns) pulses at 25 Hz with output wavelength set to 488 nm. The acquisition time of the camera was set to 200 ms, corresponding to the integration of five laser pulses.
A GCaMP6f mouse (n = 1) was anaesthetized, shaved and fixed in place on the silicon layer as described above. The camera position was manually adjusted until the mouse head was in focus. A single custom-made silica fused-end fiber bundle (CeramOptics GmbH, Bonn, Germany), positioned below the mouse head and outside of the FOV of the camera, was used to illuminate the imaged area from a single direction. The perfusion procedure was then continued, with the camera recording both the clearing of blood and the subsequent injection of 3 ml PTZ over the course of 60 s. The relative fluorescence signal changes (ΔF/F0) were calculated from the recorded images as changes in signal intensity following PTZ injection. These changes correspond to calcium responses evoked by neural activity. The baseline fluorescence signal level F0 was calculated as the average of frames over 10 s preceding the injection.
2.4. Functional Optoacoustic Neuro-Tomography (FONT) Recordings
The FONT imaging set-up consisted of a custom-made matrix transducer array (Imasonic SaS, Voray, France) having 512 individual piezocomposite elements densely distributed over a 40 mm radius spherical aperture covering an angle of 140° (1.31p solid angle). Each element has 2.5 mm diameter, 5 MHz central frequency and ~100% -6dB bandwidth, thus the array provides an almost isotropic resolution of 150–250 µm over an effective FOV of ~2 cm
3 [
33]. The 512 OA signals detected by the array elements were simultaneously digitized with a custom-made data acquisition unit (DAQ, Falkenstein Microsysteme, Taufkirchen, Germany) triggered with the Q-switch output of the tunable OPO laser used for the excitation of OA responses. The digitized signals were transferred to a PC via 1 Gbit/s Ethernet for further processing. The spherical matrix array transducer was placed into the holder at a distance of ~1 cm from the bottom of the silicon sheet as seen in
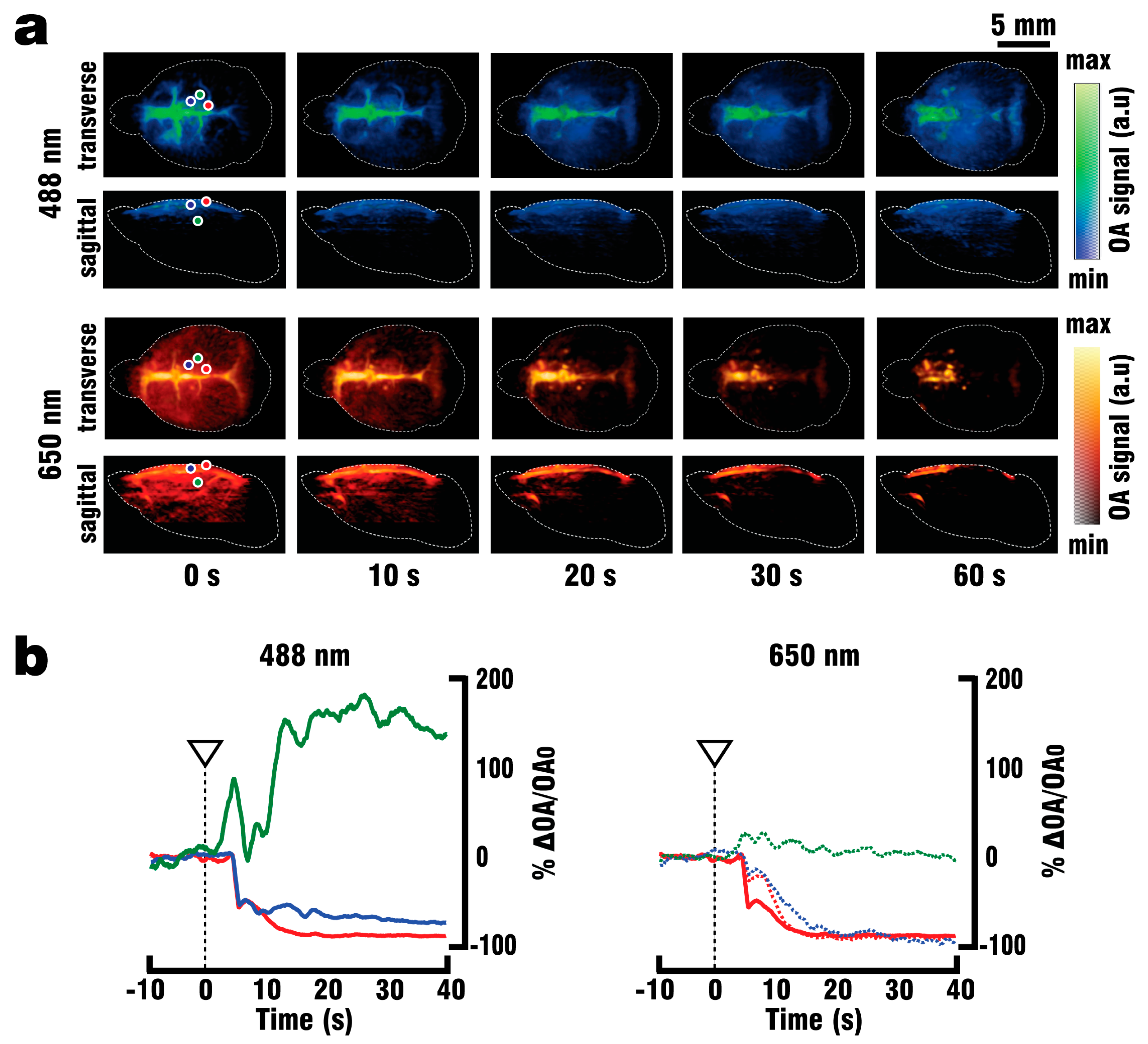
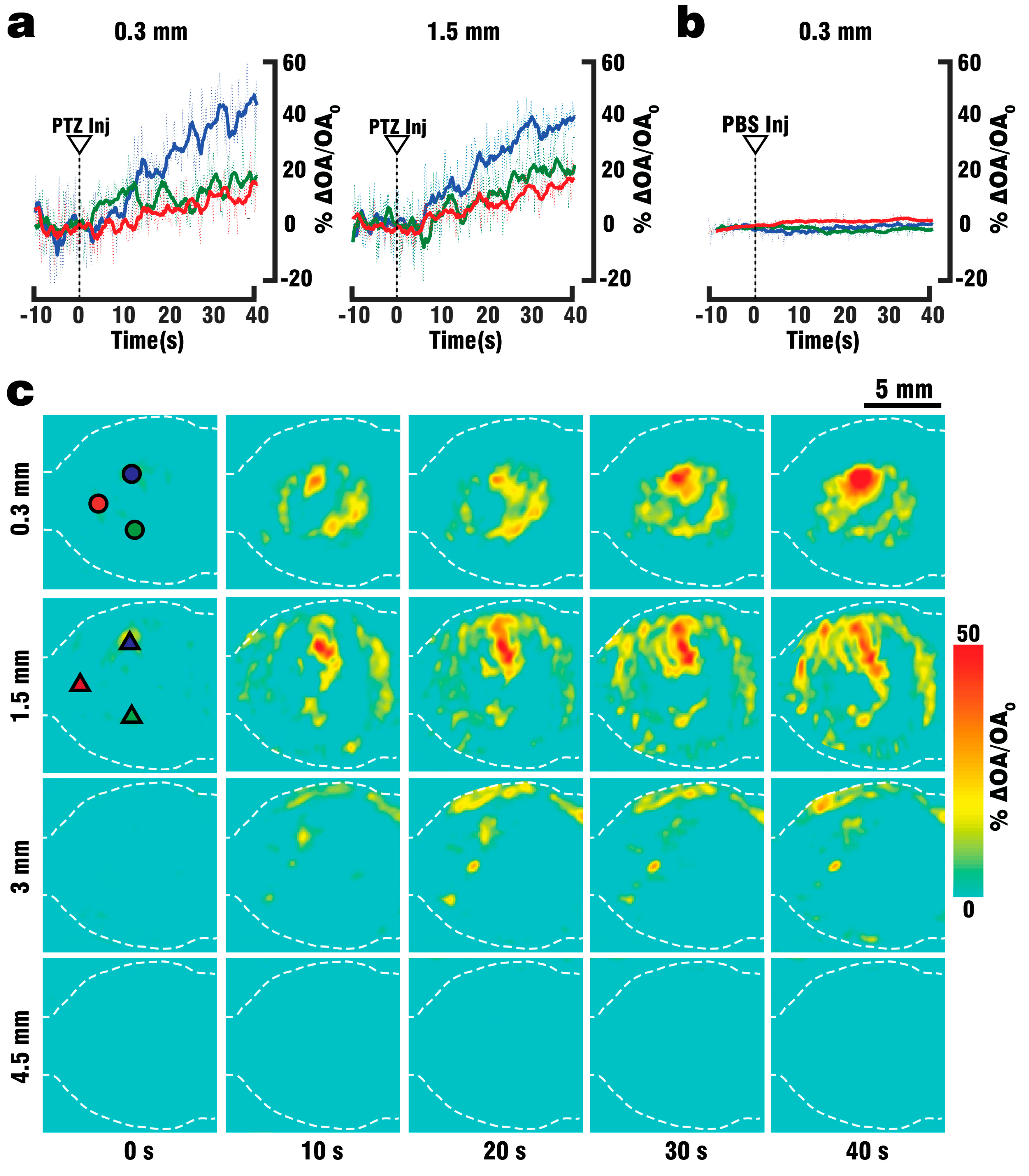
Figure 1a, where a fiber bundle guided the illumination beam through a central cylindrical aperture. The bundle was connected to the output of the OPO laser, which was set to emit alternating pulses of 488 nm/650 nm light at 20 Hz repetition rate. GCaMP excitation was performed at 488 nm wavelength, while 650 nm served as a control wavelength for which hemoglobin has significantly less optical absorption with GCaMP having no residual absorption. The outer rim of the array transducer was sealed with 1.5% Agar (Sigma-Aldrich, Taufkirchen, Germany) to prevent leakage, and the space between the silicone sheet and the transducer’s active surface was filled with water to provide acoustic coupling for the optoacoustically-generated ultrasound waves. GCaMP6f mice (n = 3) were perfused as described above, and OA data was collected both during the blood clearing procedure and the PTZ-induced neural activation which commenced after 2 min of clearing.
Volumetric (3D) FONT images were reconstructed from the acquired signals at each wavelength using GPU-based implementation of a back-projection formula [
34]. The signals were deconvolved with the impulse response of the transducer array elements before reconstruction. In addition, a band-pass filter with cut-off frequencies of 0.1 and 7.5 MHz was applied. Reconstruction of the volumetric image data was performed on a grid of 150 × 150 × 100 voxels
3 (equivalent to 15 × 15 × 10 mm
3) to match system’s spatial resolution. An example reconstruction for the data acquired before perfusion started is seen in
Figure 1b.
4. Discussion and Conclusions
Optoacoustic imaging has recently been demonstrated as a valid method for functional, non-invasive observations of transient calcium fluxes in cortical structures of zebrafish [
21] and an in-vivo mouse model expressing GCaMP6-type indicators [
28]. By combining an effective field of view covering the entire mouse brain with spatio-temporal scales not achievable by other neuroimaging modalities and deeper tissue penetration than conventional optical approaches, FONT has proven to be a viable new tool for the observation of whole-brain neuronal network interactions across long distances. However, strong absorption of light by hemoglobin at the GCaMP6 excitation wavelength of 488 nm makes direct visualization of calcium activity through GCaMP6 signal changes in deeper brain regions highly challenging in-vivo. The development of near-infrared calcium indicators with absorption peaks above 650 nm, such as NIR-GECO1 [
37], is underway, and adapting them to OA imaging would allow even higher depth penetration due to a significantly lower blood absorption background at these wavelengths. Unfortunately, this indicator will require further rounds of improvement before its performance in OA mode is comparable to GCaMP.
A possible solution to acquiring FONT signals from subcortical brain areas with existing calcium indicators is the use of a blood-free brain model. In a previous study, we have established a functional blood-free excised brain preparation superfused with oxygenated ACSF [
28], for which in-vivo-like neuronal functionality could be observed during ~30 min after brain extraction. However, the brain extraction method has major caveats. The reliance on passive diffusion complicates delivery of oxygen and nutrients to deep brain regions past the 30 min mark, increasing the chance of hypoxia and cell death that adversely affects the model. In addition, the complexity of the preparation raised the chance of failure and the absence of anatomical references (e.g., Bregma) made targeting specific regions for stimulation at specific depths more challenging. Moreover, small volumes of highly concentrated PTZ had to be injected directly into the brain and depending on the glass micro-capillary position some of its contents would leak into the superfusion chamber, adding more variability to the experimental results. Blood-free studies in larger rodents, such as isolated guinea-pig brains [
38] and isolated rat brains [
39], utilized perfusion through the cortical vasculature to mimic in vivo conditions in the entire brain and keep it responsive to electric stimuli for up to 8 h post excision. Such approaches are however surgically challenging when applied to the much smaller mouse model, for which the suggested intracardiac perfusion was found to be suitable.
The presented results showcase a strategy for eliminating the highly absorbing blood background from a functional murine whole brain model, enabling direct tracking of calcium dynamics associated with neuronal activity at depths previously inaccessible with FONT and extending the time window in which a response to stimuli can be triggered. Furthermore, it is now possible to observe and evaluate the transition between an in-vivo brain model (prior to perfusion) and a blood free in-situ preparation (after perfusion), providing a platform to further explore the relation between epifluorescence and optoacoustic signals for GCaMP6 indicators. A simpler, quicker surgical procedure in comparison to the isolated brain model [
40] increased the success rate of performed experiments, thus reducing the number of animals required for the study. By utilizing the vascular system we were able to fully replace the blood with ACSF, and thereby double the maximum depth from which GCaMP6f signals could be collected. Furthermore, the whole brain could be efficiently supplied with nutrients and oxygen for more than 30 min following blood clearance, which in combination with physiological pressure, temperature and glucose levels should allow for closer mimicry of in-vivo conditions. Brain viability was validated by EEG recordings indicating the presence of neuronal activity for at least 45 min following the start of perfusion, while the stimulus response to PTZ injection was observed in EEG, planar fluorescence, and FONT recordings. The FONT imaging enabled a clear detection of calcium fluxes as true high-resolution 3D-information not affected by intense light scattering in the brain, while epi-fluorescence recordings failed to provide high resolution maps of depth-resolved calcium dynamics. Since the total duration of the stimulation experiments including the full removal of blood was below 10 min, the data acquired can be attributed to in-vivo-like neuronal functionality.
The presented model is not without limitations and multiple aspects can be further improved. Simultaneous acquisition of epifluorescence and optoacoustic data at sufficient depth through the use of a fibroscope [
41] was not achievable at the time of the study, complicating direct comparisons of specific regions in both modes beyond a proof-of-concept viewpoint between animals. This is now becoming possible thanks to recent advancements in fibroscope design. In addition, due to the use of very high anesthesia doses and general setup layout, common stimulation paradigms such as paw or whisker stimulation could not be applied to our mouse model, necessitating the use of a pharmacological compound to induce neuronal activity. Excitation with PTZ invoked robust, albeit widespread changes in calcium dynamics across large areas of the brain. To adapt this preparation for use in functional mapping of network connectivity, the use of drugs targeting specific neuronal populations must be explored in future studies. However, this methodology is not limited to pharmacological stimulation paradigms. The use of widely tunable lasers with 0.1-1 kHz repetition rates for excitation in our technology could in theory allow the interrogation of specific circuits through optogenetics [
42], while the spherical matrix transducer array used for data acquisition could be modified for concurrent use with transcranial ultrasound stimulation to excite small volumes of brain tissue [
43,
44]. Another caveat is the mesoscopic resolution of 150 µm which can be scaled down to ~35 µm using high-frequency matrix array probes at the cost of a smaller FOV [
45]. The use of GCaMP mouse models with sparse expression [
46] in areas of interest such as the hippocampus or specific cortex layers could theoretically enable access to normal and dysregulated neuronal network interactions at a whole-brain level within previously homogeneous non-sparse cell populations with high temporal resolution. Yet the most glaring issue is the limited viability of the preparation (45 min) and the impossibility of longitudinal studies due to the invasive and final cardiac perfusion procedure. Survivability of the brain after blood clearing could be prolonged through the improvement of the blood replacement mixture by adjusting ion concentrations, inclusion of cytoprotective agents [
47] and the addition of a dedicated, perfluorocarbon-based oxygen carrier [
48] lacking strong absorption in OA mode. Furthermore, adaptation of a “bloodless” mouse model akin to existing rat protocols [
49,
50] could allow long-term studies over the span of days or weeks until animals replace missing hematologic components, but great care must be taken to ensure adherence to current ethics regulations and animal welfare protocols.
In conclusion, we have developed a novel intracardiac perfusion in situ brain model to optoacoustically monitor deep brain calcium dynamics under blood-free conditions at unprecedented depths and spatio-temporal resolutions not attainable with other optical microscopy modalities. The model provides a framework to further explore the relation between fluorescence and optoacoustic signals for existing or emerging calcium/voltage indicators. It can be further adapted for the functional observation and/or mapping of network connectivity between specific cortical and subcortical structures in the murine model.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




