Abstract
Lensless fluorescence microscopy (LLFM) has emerged as a promising approach for biological imaging, offering a simplified, high-throughput, portable, and cost-effective substitute for conventional microscopy techniques by removing lenses in favor of directly recording fluorescent light on a digital sensor. However, there are several obstacles that this novel approach must overcome, such as restrictions on the resolution, field-of-view (FOV), signal-to-noise ratio (SNR), and multicolor-imaging capabilities. This review looks at the most current developments aimed at addressing these challenges and enhancing the performance of LLFM systems. To address these issues, computational techniques, such as deconvolution and compressive sensing, hardware modifications and structured illumination, customized filters, and the utilization of fiber-optic plates, have been implemented. Finally, this review emphasizes the numerous applications of LLFM in tissue analysis, pathogen detection, and cellular imaging, highlighting its adaptability and potential influence in a range of biomedical research and clinical diagnostic areas.
1. Introduction
Fluorescence microscopy is a fundamental tool in current biological research, providing the precise and sensitive imaging of cellular and molecular processes [1,2,3]. This approach uses fluorescent markers that attach to certain cellular components to provide precise information about organelles [4,5,6]. Its ability to reveal the dynamic interactions within cells and between molecular constituents is invaluable, providing critical insights into biological functions and pathologies [7,8,9,10,11], and it is crucial in several different fields, including drug discovery, disease diagnostics, genetic research, oncology, and neuroscience, amongst others.
There is a growing need for high-throughput capabilities in the fields of scientific research and diagnostics. For thorough analysis and effective screening to produce statistically significant outcomes, the ability to monitor dynamic characteristics across a broad scale is crucial. Simultaneously, there has been a notable increase in the demand for portable microscopy solutions, especially for situations requiring in-field diagnostics or in environments with limited resources. Furthermore, in clinical and environmental applications, portability might be essential for quick decision making since it allows for direct on-site investigation. Also, bringing down the cost of microscopy technology is essential to increasing the accessibility to this effective diagnostic tool and making it reasonably priced for global application in a variety of clinical, educational, and research contexts.
One of the strategies utilized to overcome the aforementioned obstacles is the development and implementation of lensless fluorescence microscopy (LLFM), which has emerged as a potential tool for addressing these diverse demands in biomedical science. This novel imaging technology disregards standard lenses and optical components in favor of capturing fluorescent light directly on a digital sensor, followed by computer reconstruction to provide detailed images. This method streamlines the microscopy setup while also drastically lowering the cost and size of the imaging equipment. Aside from fluorescence, lensless imaging has been widely used in digital holography [12,13] for various purposes, such as the detection of biomolecules, nanoparticles [14,15,16], and pathogens [16,17], and for studying cells and tissues [18,19], amongst others.
LLFM is inherently more compact and lightweight compared to traditional microscopes, facilitating easy transportation and use in varied settings. The absence of complex optical components reduces the cost dramatically while allowing for the scalability of the technology. Additionally, LLFM offers a significantly larger field of view (FOV), often spanning several centimeters. This wide FOV allows for the rapid screening and analysis of sample regions without the need for translation, which is beneficial for high-throughput applications.
Although the advantages of LLFM are manifold, it also presents several challenges. The resolution often does not match those of its conventional lens-based counterparts, and the requirement for a simple design limits its flexibility (e.g., lower sensitivity and challenges in achieving multicolor imaging, which are critical for detailed biological analyses).
This review will explore the different aspects of LLFM in depth. The first part will delve into the challenges with the image resolution and the various innovations designed to improve its performance. Following this, the discussion will shift to the methods developed to expand and optimize the field of view, enhancing the signal-to-noise ratio (or sensitivity), and the multicolor imaging and lifetime imaging capabilities within lensless systems. Finally, the practical applications of LLFM in imaging cells, tissues, and pathogens will be comprehensively discussed, underscoring the technique’s versatility and potential across different domains of biomedical research and clinical diagnostics. Through this structured examination, this review aims to provide a thorough understanding of LLFM’s current capabilities, its ongoing development, and its future potential.
2. Resolution Enhancement
Resolution is a fundamental aspect of any microscopy technique, defining its ability to clearly distinguish two closely positioned points as separate entities. In the context of microscopy, a higher resolution translates to the capacity to visualize finer details within a sample, which is crucial for accurate scientific analysis and diagnosis. In this light, resolution is an important parameter in both conventional and lensless microscopy. Conventional optical microscopes utilize lenses to focus light and magnify the image of the sample. The resolution in these systems is inherently limited by the numerical aperture of the lenses and the wavelength of the light, as described by the Abbe limit [20]. This is defined by the following equation:
where d is the space between two adjacent particles (still allowing the particles to be perceived as separate), λ is the wavelength of illumination, and N.A. is the numerical aperture of the objective.
Improvements in the resolution for optical microscopes often involve using higher numerical aperture lenses, shorter wavelength light, and techniques such as oil immersion for refractive index matching. For example, a 10× objective with an NA of ~0.25–0.45 enables a resolution of ~1 µm or lower, whereas a 100× objective with an NA of 1.3–1.4 enables a resolution of about ~0.2 µm. In contrast, LLFM captures light directly on a digital sensor and uses computational methods to reconstruct images recorded by the sensor from the light that is emitted by the sample. The resolution in lensless systems is primarily determined by factors such as the sensor’s pixel size, the wavelength of the light, the distance between the sample and the sensor, and the sophistication of the computational algorithms used to reconstruct the image. With each sensor pixel acting as an individual detector, light from the sample is captured without the intermediation of lenses. The pixel size therefore limits the smallest point that the system can resolve. Larger pixel sizes in sensors inherently restrict the system’s resolution, making it difficult to observe fine details or closely spaced objects within the sample. One of the most common approaches to improve the resolution beyond the limitations imposed by the pixel size is using computational techniques such as digital deconvolution, compressive sensing, etc., to reconstruct the image captured by the sensor. Deconvolution is a mathematical and computational approach used in digital imaging to reduce out-of-focus, blurry images by transforming the image information. Deconvolution greatly relies on the point spread function (PSF) of the image to improve the image quality. Basically, the raw image captured by the image sensor can be said to be the object structure convolved with the PSF:
The image is reconstructed by solving an inverse problem. This is accomplished by first computing the Fourier transform of the captured image, which transforms the image parameters from the spatial domain to the frequency domain. The PSF is transformed into an Optical Transfer function after the Fourier transform is computed. Deconvolution is then achieved by taking the inverse Fourier transform to recover the higher spatial frequencies of the object that contains the finer details:
Overall, image reconstruction using computational techniques is an indispensable part of imaging when using LLFM along with different lensless imaging techniques [21,22,23]. For example, using digital deconvolution, Coskun et al. [24] were able to enhance the resolving power of LLFM by approximately 5-fold, resulting in a spatial resolution of around 40–50 µm across the entire sensor field of view of 2.5 cm × 3.5 cm. In this approach, the sample is put directly onto a decapped CMOS sensor whilst the sample-to-sensor distance is kept at ~0.5–1 mm. The sample is excited through a prism interface, where the excitation light is blocked due to the total internal reflection (TIR) at the bottom surface of the glass substrate. The emitted fluorescent signal from the sample, which does not undergo TIR, is captured directly by the image sensor over its full area (FOV). The image captured by the camera is then reconstructed using the digital deconvolution algorithm to obtain a higher-resolution image.
The compressive-sampling-based algorithm is another technique to increase the resolution for the imaging of sparse objects. Compressive sampling, also known as compressive sensing, is an innovative signal-processing technique that reconstructs a signal from far fewer samples or experiments than traditionally required [25,26]. It leverages the fact that many signals have sparse representation, meaning that most of their data points are not significant and can be omitted without losing crucial information. By applying mathematical algorithms, compressive sampling reconstructs the original signal accurately using only these essential samples.
The compressive decoding process can be formalized as an l1-regularized least-square problem (LSP), such that
where β > 0 is a regularization parameter; Idet is the detected raw fluorescence image at the sensor array (in the vector form); Mconv represents the 2D convolution matrix based on the incoherent point spread function of the system; is the fluorescent source distribution that creates the lensfree image at the detector plane; and represents the lp norm of vector .
Compressive sampling has been used along with TIR-based LED-illuminated LLFM to decode the blurring lens-free fluorescence patterns captured on the chip, yielding a resolution of 10 μm throughout the wide FOV of 2–8 cm2 [27,28]. By incorporating a faceplate into the same approach as discussed above, the efficiency of the fluorescent emission collection was increased, and by using the compressive-sampling approach, a resolution of <4 μm over an FOV of >0.6–8 cm2 has been achieved [29]. Another computational approach that can circumvent this issue includes pixel super-resolution [30], which involves capturing multiple sub-pixel shifted images to synthesize a higher-resolution image, but they have yet to be implemented for lensless fluorescence microscopes.
Another significant innovation is the development of a novel phase mask presented by Adams et al. tailored for low-contrast imaging environments typical in biological samples [31]. This phase mask enhances the contrast and localizes the sparse point spread function (PSF), capturing the essential spatial frequencies of biological tissues. To create this PSF, the study employed a unique method combining Perlin noise generation with Canny edge detection, which contributes significantly to improving the image reconstruction for dense, low-contrast samples. The Bio-FlatScope, a prototype developed in this study, leverages a near-field phase retrieval algorithm, demonstrating improved image reconstruction capabilities, particularly in dense, low-contrast biological samples. The Bio-FlatScope, leveraging computational imaging algorithms co-designed with optics and sensors, achieved a <9 μm lateral resolution, characterized by imaging United States Air Force (USAF) resolution targets, and a large field of view (FOV) of about 16 mm². Additionally, for axial resolution, the Bio-FlatScope achieved resolutions of approximately 50 μm in non-scattering media and 80 μm in scattering media. This approach enables 3D imaging capabilities. In another study from the same group, they employed a novel technique that replaces traditional lenses with an optimized amplitude mask positioned a few hundred micrometers above the sensor, coupled with an efficient computational algorithm for high-resolution 3D image reconstruction from a single frame of captured data. By using a separable mask pattern, the FlatScope simplifies both the calibration and image reconstruction, significantly reducing the computational complexity. The resulting device achieved a micrometer-scale lateral resolution and covers an imaging volume of several cubic millimeters. It can dynamically focus on different depths from a single captured frame, allowing efficient 3D reconstruction without mechanical scanning and achieving a resolution and speed comparable to those of confocal microscopes but at a fraction of the data acquisition time [32].
Moreover, designing efficient sample illumination can also lead to improved image resolution. Han et al. utilized a unique illumination design based on the Talbot effect to create an imaging platform with high resolution [22]. The fluorescence Talbot microscopy employed a microlens grid to convert the incident coherent plane wave with the wavelength (λ) into a grid of tightly focused light spots at the focal distance of the lenses. The distance between adjacent focal spots corresponds to the microlens grid pitch (d). The light grid then propagates, and regenerates focused light spots at a distance of z = nZT, where n is an integer and ZT = 2d2/λ is defined as the Talbot length. This self-imaging phenomenon is referred to as the Talbot effect. The tightly focused laser spots generated by a lenslet grid can scan across the sample by tilting the input light field using an MEMS mirror. The system setup involved a 488 nm laser light source, an MEMS mirror for the angular adjustment of the input light, and a lenslet grid to create a focused-light grid through the Talbot self-imaging effect. The Talbot focal spot is shown in Figure 1. The imaging platform achieved a resolution of 1.2 μm and a wide FOV of 13 mm2. Furthermore, in another study, the development and implementation of a fluorescence optofluidic microscope (FOFM) using an array of Fresnel zone plates (FZPs) was investigated. The array of FZPs in the FOFM was created using electron-beam lithography to pattern a 300 nm thick chrome layer deposited on a quartz substrate. This method defined concentric rings with a minimum line width of 400 nm, alternating between transparent quartz and opaque chrome. The patterned substrate was then etched to remove the chrome in designated areas, creating the FZP structure. Finally, the FZP array was aligned and bonded to the microfluidic channel made of polydimethylsiloxane (PDMS) and integrated with the CMOS sensor. The FZPs created focused light spots within a microfluidic channel, which locally excited fluorophores as the sample flew through. The fluorescence emissions were captured by a CMOS sensor coated with a filter, achieving a resolution limited by the focused-light-spot size, experimentally measured as 0.60 µm for a 405 nm excitation and 0.65 µm for a 488 nm excitation. The overall system resolution was established at 1.0 µm due to the Nyquist sampling criterion [33].
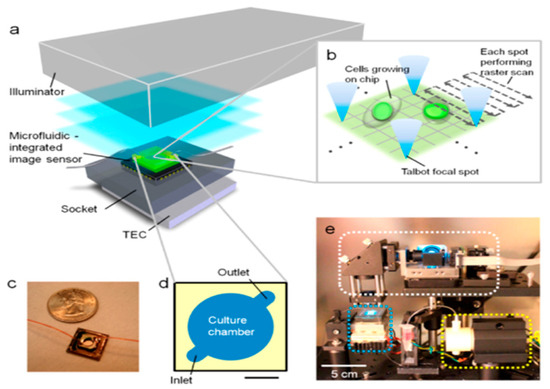
Figure 1.
System setup of on-chip Talbot fluorescence microscopy. (a) The system comprises an illuminator, a microfluidic-integrated image sensor, a socket, and a thermoelectric cooler (TEC). A laser with a wavelength of 488 nm was used to create a grid-focused laser spot through a lenslet grid, and an MEMS mirror was used to scan across the sample. (b) Depiction of the raster scan process; the Talbot grid spots are shifted laterally by tilting the input light field using the MEMS mirror, covering the entire sample area by scanning over a 30 μm × 30 μm area for each focal spot. (c) The assembled sensing platform consisted of a CMOS sensor chip with a precoated absorption fluorescence filter and a microfluidic component. (d) Detailed design of the microfluidic component consisting of the cell culture chamber, which includes two channels for the media inlet and outlet, and its ceiling is a glass coverslip. (e) The complete system including the illuminator (white rectangle), microfluidic-integrated image sensor, socket and TEC (blue rectangle), and micropump (yellow rectangle) placed inside the incubator. Scale bar in (d) represents 2 mm. Reproduced with permission from Ref. [22]. Copyright 2013, American Chemical Society.
3. Increasing Throughput
Throughput, which is determined by the imaging speed and FOV, is critical for high-efficiency biological imaging [34,35,36,37]. The FOV of a single snapshot depends on the size of the sensor chip (active area) and the N.A. of the objective lens. For example, using standard CMOS/CCD cameras, the FOV is typically extended to a few square centimeters, while using a 10 × 0.25 N.A. objective decreases it to ~3 mm2, and using a 100 × 1.4 N.A. objective decreases the FOV by 100-fold to ~0.03 mm2. A high-throughput system enables the quick acquisition of images across a wide field of view, allowing for the rapid examination of multiple samples, which is especially useful for large-scale biological investigations and medical diagnostics [38]. Techniques that enhance the imaging speed allow for the real-time capturing of dynamic biological processes, while a larger field of view allows for a thorough inspection of wide sample regions without the need for relocation. This section will look at the technological developments that have allowed for increased throughput in LLFM.
In a particular study, researchers effectively converted a traditional flatbed scanner into a fluorescence-imaging system capable of screening across an unprecedentedly wide field of view (FOV) of approximately 532 cm², which corresponds to dimensions of 19 cm × 28 cm [39]. The optical resolution of this platform was measured as 7.8 µm along the sensor direction and as 12.4 µm along the scanning direction. This extensive FOV was achieved by modifications to the hardware and software of a standard flatbed scanner, the incorporation of a specially designed absorbing fluorescent filter, and the addition of a two-dimensional array of external light sources to facilitate computer-controlled, high-angle fluorescent excitation. The scanner’s driver was reprogrammed to enable full control over the scanner hardware, optimizing the exposure time, gain, and sensitivity essential for the detection of fluorescent micro-objects through the gradient index self-focusing lens array positioned before the scanner sensor chip.
Furthermore, the high-throughput screening of large sample volumes was enabled using structured illumination [21]. The system employed a microfluidic device with an area of 7–18 cm², containing 0.3–0.7 mL of undiluted whole blood. The device was placed onto a wide-field opto-electronic sensor array. To create structured illumination, a diode laser beam (473 nm, 113 mW) was expanded by a telescopic-lens system to fill the active area of a Spatial Light Modulator (SLM), which then generated a 2D array of Gaussian-shaped excitation spots. These spots were projected onto the sample through two imaging lenses, relaying the SLM output to the sample plane with approximately 1.66 magnification. The structured illumination pattern was laterally scanned across the sample in small steps (90 μm at the sample plane), capturing 144 frames for the entire sample area. The fluorescent emissions excited by this pattern were collected directly by the sensor array and formed fluorescence images. These 144 images were digitally merged using a Maximum-Intensity-Projection (MIP) algorithm, which selects the highest intensity value for each pixel across all frames, significantly boosting the signal-to-noise ratio (SNR) and enhancing the contrast of the fluorescent objects. This method effectively detects fluorescent particles that would otherwise be hidden under uniform illumination, providing high sensitivity and specificity for the rapid and accurate screening of large sample volumes like undiluted whole blood.
4. Increasing Signal-to-Noise Ratio
The signal-to-noise ratio (SNR) is another importance factor in fluorescence microscopy, as it governs the clarity and quality of the acquired images. A high SNR is always desired, as it allows the precise visualization of the specimen’s features and helps identify and analyze the structures and processes within the sample. The SNR is highly influenced by the N.A. of the objective lens in conventional microscopes. A higher N.A. allows for better light collection from the fluorescent sample due to the larger angle coverage. Moreover, the SNR is strongly dependent on the type of camera used and the optical design of the microscopes. Highly sensitive cameras such as Electron-Multiplying Charge-Coupled Devices (EMCCDs) or scientific complementary-metal–oxide-semiconductor (sCMOS) cameras, coupled with high N.A. objectives, are regularly used for dynamic single-molecule fluorescence studies. However, due to the large cost and complexity, EMCCDs, sCMOS, and other high-end cameras are not suitable for LLFM, and the absence of high N.A. objectives necessitate innovative designs to improve the SNR. In this section, different methods used to increase the SNR of LLFM will be discussed.
Achieving a high SNR is crucial for the accurate computational reconstruction of the images captured by the sensor [40,41]. A low SNR can lead to errors in the reconstruction and the failure to detect low-abundance targets, which is essential for diagnostics and rare-cellular-event research. One important component that plays a critical role is the optical filters, which selectively allow fluorescence emissions while blocking excitation light and other unwanted wavelengths, reducing the background noise [42,43,44]. Integrated onto the sensor or imaging setup, filters enable clearer, more detailed images and the effective interpretation of diffraction patterns for reconstruction algorithms.
To enhance the SNR, Mudraboyina et al. deposited an emission filter directly onto the CMOS sensor array [23]. They created an emission filter with a thickness of 20 μm and spin-coated it onto the CMOS image sensor surface. This filter was built on top of a commercially available, packaged CMOS sensor that was already installed on a Field-Programmable Gate Array (FPGA) board. This manufacturing process is innovative in comparison to earlier research, which normally produced the filter separately and then mounted it onto the sensor, resulting in a higher filter thickness and decreased light-collecting efficiency. The ability to produce the filter directly on the sensor after the packaging and integration with other components enables an ideal filter thinness, resulting in a higher SNR. Furthermore, this filter can be readily removed by dissolving it in methanol, allowing the CMOS sensor to be reused while lowering the total system costs.
Another approach included the development of a chip-scale fluorescence microscope leveraging a unique silo-filter (SF) complementary-metal–oxide-semiconductor (CMOS) image sensor to increase the sensitivity of the imaging platform [45]. The SF design employed metal walls to isolate each pixel, channeling fluorescent light through the thick absorptive filter layer to the corresponding photodiode of a single pixel. Therefore, the researchers could increase the SNR significantly. The signal-to-noise ratio (SNR) calculation evaluates the fluorescence image quality. For additional examination, images with signal-to-noise ratios (SNRs) of more than 30 dB are usually classified as “good”. The evaluated SNRs of the fluorescent images were 9.5 dB for nucleic acid stain, 32 dB for red-fluorescence protein (RFP), and 35 dB for fluorescent beads. The high background from the unbound fluorophores resulted in low SNR values for the images with nucleic acid stains. In the study conducted by Sasagawa et al., a highly sensitive lensfree fluorescence-imaging device enabled by a complementary combination of interference and absorption filters was used to increase the SNR. In this setup, the interference filter is applied at the side of the fiber-optic plate (FOP) facing the sample and the absorption filter is applied at the other side facing the sensor. The interference filter transmits only scattered light from the target sample and rejects almost all other excitation light by reflection in its rejection band. Since interference filters do not absorb light, they emit no autofluorescence. The transmitted light from the interference filter is then relayed through the FOP and absorbed by the absorption filter. At this point, the autofluorescence intensity is greatly reduced due to reflection of the incident light by the interference filter; thus, the absorption filter transmits only the fluorescence signal from the sample with very little or no autofluorescence. In this regard, the proposed hybrid filter attains ultra-high selectivity for excitation light and fluorescence. This technique achieved a high-excitation rejection of approximately 108:1 at a wavelength of 450 nm without the use of a prism. This high-excitation rejection enables a high SNR, which enables the detection of more fluorescence emissions [46].
To increase the SNR, a hybrid bandpass emission filter was incorporated into an imaging system to enhance the fluorescence-imaging capabilities [47]. This was accomplished by employing a combination of different filters. To reflect the majority of the normal incoming excitation light, a longpass interference filter is placed on top. Because the FOP generates autofluorescence, which may reduce the performance, this filter is positioned at the top. Additionally, the observation target is exposed to another round of excitation light reflection, roughly doubling the fluorescence intensity. A shortpass interference filter is placed on the bottom of the FOP to reflect the autofluorescence of the target, which is substantially weaker than the excitation light. The third filter is an absorption filter. In a lensless imaging device, the excitation light scattered by the observation target hits the filter at different angles. Due to the shift in the transmission spectrum of an interference filter with varying angles of incidence, achieving high-excitation light rejection, as seen in lens-based fluorescence microscopy, is not feasible. Consequently, the hybrid filter uses an absorption filter to reject the scattered-light component. The structure of the proposed filter is shown in Figure 2.
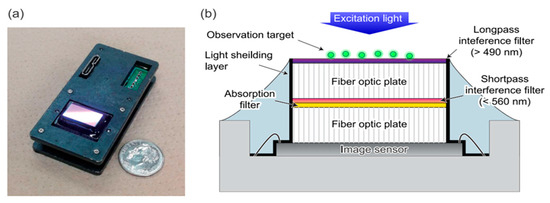
Figure 2.
Lensless fluorescence-imaging device with hybrid bandpass filter. (a) The fabricated lensless fluorescence-imaging device with dimensions of 60 mm × 30 mm × 17 mm. The commercially available image sensor (CMV2000, AMS, Wien, Austria) was used with a pixel count of 2048 × 1088 and a pixel size of 5.5 µm. The sensor’s glass lid was removed to add bandpass filters, enhancing its fluorescence detection capabilities. (b) The schematic of the device cross section depicts the integration of the sensor chip, electrodes, and filter components. A fiber-optic plate (FOP) was used to transmit images through bundled optical fibers. The hybrid filter consists of three parts: a longpass interference filter on top to reflect the normal incident excitation light, a shortpass interference filter on the bottom to block autofluorescence from the target, and an absorption filter to reject scattered excitation light at various angles. In this configuration, stray light from the sidewalls of the FOP or the image sensor causes artifacts. A layer of black paint (CS-37, Canon Chemical, Tokyo, Japan) was applied to avoid them. Reproduced with permission from Ref. [47]. Copyright 2019, AIP Publishing.
Moreover, Rustami et al. used plasma etching to transfer the thin and scalable hybrid emission filters, comprising an interference filter and an absorption filter, onto the image sensor to increase the SNR [48]. In their setup, the interference filter is first deposited on a silicon wafer substrate, and the absorption filter layer is then spin-coated on top of the interference filter. The image sensor is then bonded to this filter structure using a transparent epoxy resin. The entire assembly is then placed in a reactive-ion-etching (RIE) chamber, where the silicon substrate is chemically etched away using SF6 gas. This leaves the hybrid filter firmly attached to the image sensor surface, without the need for any of the fragile handling or laser-based lift-off methods used in previous works. This hybrid approach allowed them to fabricate large-area filters that can be uniformly integrated onto image sensor arrays, enabling the fluorescence imaging of optically dense biological samples due to the increased SNR. The spatial resolution of the device, calculated from microbead emissions, is 12.11 μm.
5. Multicolor Imaging
Multicolor imaging is critical in biology for observing various markers simultaneously within a single sample [49,50]. In conventional fluorescence microscopy, multicolor imaging is achieved by using different excitation and emission filters corresponding to the fluorophores that absorb and emit light at specific wavelengths. This process can be performed sequentially, switching filters to capture each color individually, or simultaneously, using multiple detectors with their own filters. Using motorized filter wheels to switch between excitation and emission filters enables multicolor imaging with a high frame rate. Although advanced systems with Acousto-Optic Tunable Filters (AOTFs) and Liquid Crystal Tunable Filters (LCTFs) can achieve faster switching speeds in the range of a few milliseconds, allowing for rapid multicolor imaging, they require more complex and expensive equipment. Nevertheless, their application in LLFM is challenging due to the need for separate filters to manage the excitation light for each dye. This is because the emission filter is usually sandwiched between the sample on the CMOS sensor, unlike traditional microscopy where the optical filters for color separation are installed on movable mounts. Lensless setups, lacking these opto-mechanical components, must adopt different methods for spectral separation. For instance, Shuo Pang et al. developed a Talbot grid-based microscopy system to illuminate the sample with a light-spot grid. They enabled multicolor imaging by inserting and exchanging optical filters between the relay lenses, blocking the specific excitation wavelength and collecting emission wavelengths of different fluorophores. The microscopy achieved an FOV of 12 mm × 10 mm with a resolution of 1.2 μm [51].
In one study, Shin and et al. presented a miniaturized multicolor fluorescence-imaging system by designing a polydimethylsiloxane (PDMS) light-guide plate (LGP) to guide the excitation light from multicolor LEDs to the fluorescent samples via total internal reflection [52]. This innovative approach overcame the limitations of the existing miniaturized imaging systems that face physical constraints with the integration of multiple filters for multicolor imaging within a compact structure. The LGP incorporated spherical-light out-couplers, which are 10 μm high and 15 μm in diameter, with a 45 μm pitch, to alter the incidence angle of the light, allowing it to exit and illuminate the sample uniformly. The LEDs are positioned 5.5 mm away from the CMOS image sensor to minimize the light leakage and maintain a high image resolution. The LGP efficiently directs light from the LEDs to the samples on its surface, ensuring uniform illumination and high-excitation light rejection, regardless of the wavelength. This innovative design allows the system to switch between blue and green LEDs for multicolor imaging, capturing high-resolution images of fluorescently labeled cells with a spatial resolution of about 10 μm.
6. Fluorescence Lifetime Imaging (FLIM)
The upperstate lifetime of the excited electrons in the fluorophores can be quantified by measuring the decay time of the fluorescence signal from a fluorophore after excitation. The fluorescence lifetime provides a host of information, including the chemical environment (oxygen, pH, etc.), redox state, molecular proximity, and more [53,54,55,56]. FLIM also results in high-SNR images, as the different fluorophores, particularly the background autofluorescence, can be easily separated based on their lifetimes. Therefore, FLIM is a powerful tool for gaining detailed, quantitative insights into the molecular and cellular mechanisms underlying various biological processes.
However, conventional FLIM requires expensive components, such as femtosecond lasers, highly sensitive detectors (e.g., PMTs), and timing electronics (e.g., Time-Correlated Single-Photon-Counting Modules), which are not viable for LLFMs. However, the combination of single-photon avalanche diodes (SPADs) and complementary-metal–oxide-semiconductor (CMOS) technology offers a compact alternative to conventional FLIM systems.
For instance, Lee et al., [57] present an innovative fluorescence lifetime imaging microscopy (FLIM) system that eliminates the need for traditional bulky lenses and optical filters. The core of this system is a 72 × 60 array of angle-sensitive single-photon avalanche diodes (A-SPADs). The array was fabricated using standard 180 nm CMOS technology and it integrates pixel-level counters to manage the data output rates efficiently and achieve accurate timing control. Each pixel is designed to capture both the spatial and angular information of the incoming light. The A-SPAD pixels are equipped with integrated diffraction gratings to detect incident light angles and SPADs to measure the photon arrival times precisely. In this technique, two fluorescent microspheres are suspended above the A-SPAD array integrated onto a CMOS chip using a micromanipulator, allowing fluorescent emissions to be detected from below. A UV laser diode excites the fluorophores in the sample with pulsed light, projected parallel to the SPAD array to minimize direct illumination. After the UV excitation pulse ends, the SPADs operate in a time-gated mode, driven into Geiger mode (ON state) by a controlled excess voltage. This timing is precisely managed by a digital-to-time converter (DTC), enabling the detection of photons from the fluorescent emissions. Each photon detection event triggers a voltage drop processed by an in-pixel comparator and counted by a 10-bit ripple counter. The inclusion of pixel-level counters allows for the accumulation of photon detection events over multiple excitation cycles, reducing the required data bandwidth for off-chip processing. The A-SPAD pixels, equipped with integrated diffraction gratings, determine the incident angles of the incoming light, combining this angular information with temporal data for the 3D localization of fluorescent sources with different lifetimes in 3D space down to the micrometer scale. This lensless and filterless approach can be utilized in various applications in which a small size and weight are crucial, such as surgical and endoscopic instruments, cost-sensitive field deployments, and high-throughput assays.
7. Biological Application of LLFM
LLMF has been used for various biological applications, including for imaging cells, tissues, and pathogens. Cell imaging is a cornerstone of biological and medical research, offering the real-time visualization of cellular processes like division, migration, and interaction to understand how cells function and respond to stimuli (e.g., drugs), as well as to diagnose diseases through the identification of morphological changes [58,59,60]. LLFM has been used to image different kinds of cells, such as the multicolor imaging of double-stained human breast cancer SK-BR-3, revealing detailed cellular structures such as membranes and nuclei [51]. Moreover, Hela cells [33] and blood cells (red and white blood cells) [21,24,39] have also been imaged using LLFM. Additionally, LLFM has been used for in situ live-cell imaging within incubators, which highlights the portability and lightweight nature of LLFM. For example, Hela cells expressing H2B-tagged eGFP (H2B-eGFP) were imaged using the Talbot Fluorescence ePetri system to observe dynamic cellular processes. The cells were cultured on the image sensor surface and a 3.7 mm × 3.5 mm FOV was imaged. The scan step size was set at 0.6 μm. It took 85 s to scan the entire FOV. Detailed time-lapse images of HeLa cells allow for the distinction of cells in various stages of the cell cycle (G1, G2, metaphase, and anaphase) and enable the visualization of nucleoli and dynamic cell behavior analysis within a standard incubator environment. Furthermore, in a quantitative study on camptothecin (CPT) treatment, the Hela cell division and migration behaviors in response to the anticancer drug CPT were studied and are shown in Figure 3. The study compared two groups: a control group and a CPT treatment group, each observed over a period of 21.5 h. At the start of the experiment, the control group had 1158 cells and the CPT group had 1297 cells. After 21.5 h, the control group exhibited a 52.4% increase in the cell count, whereas the CPT group showed only a 10.5% increase, indicating that CPT significantly inhibits cell proliferation. Additionally, the cell migration was assessed by tracking 373 cells in the control group and 422 cells in the CPT group. The control group had an average migration distance of 246.0 ± 59.2 μm, while the CPT group had a significantly lower average migration distance of 210.3 ± 68.5 μm (p < 0.001). These data demonstrate that CPT not only reduces cell proliferation but also impairs cell migration. The representative images in Figure 3c taken at 0, 6, 12, 18, and 21 h visually support these findings, highlighting the reduced cell division and movement in the CPT-treated cells compared to the control group [22]. HEK 293 cells expressing mCherry red fluorescent protein (RFP), MCF-7 cells stained with SYTO-64 red nucleic acid stain, and NIH 3T3 fibroblast cells are other kinds of cell lines imaged inside the incubator, allowing for the monitoring of dynamic cellular characteristics, such as spreading and proliferation [45,46,52]. For example, HEK 293-mCherry cells and MCF-7 cells stained with SYTO-64 red nucleic acid stain were cultured on an ePetri system for the long-term monitoring of the cells using a unique silo-filter (SF) CMOS image sensor. Fluorescence images were obtained at 20 min intervals for 20 h with a 450 ms exposure time. The time-sequence images indicate the movement and growth of the cells [45]. In addition, the real-time monitoring of green- and red-labeled NIH 3T3 fibroblast cells using the miniaturized multicolor fluorescence-imaging system integrated with a PDMS light-guide plate (LGP) has been performed. The migration of these labeled cells was monitored over time within an incubator. The system used blue LEDs (applied power of 0.26 W, 2.6 V, 0.1 A) to excite the green fluorescent samples and green LEDs (applied power of 0.26 W, 2.6 V, 0.1 A) to excite the red fluorescent samples. The fluorescence emissions from the cells were captured every 4 min, allowing for the observation of the cell movement and other dynamic characteristics.
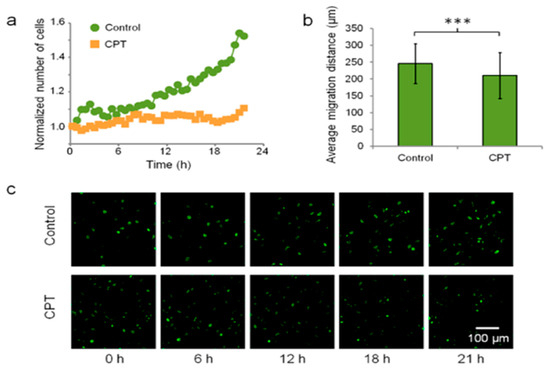
Figure 3.
Effect of camptothecin (CPT) on HeLa cells monitored by Talbot fluorescence-imaging system. (a) Cell count over time for the control group and the CPT-treated group. The CPT group showed significantly lower proliferation compared to the control group, demonstrating the inhibitory effect of CPT on cell division. (b) Average cell migration distances for the control group and the CPT-treated group over 21.5 h. Cells in the control group exhibited greater migration distances than those in the CPT-treated group, highlighting the impact of CPT on cell motility (*** p < 0.001). (c) Representative time-lapse images of HeLa cells at 0, 6, 12, 18, and 21 h for both the control and CPT-treated groups. The images reveal the differences in the cell densities and morphologies between the two groups, with the CPT-treated group showing reduced cell proliferation and altered migration patterns. The Talbot Fluorescence ePetri system enables the continuous, high-resolution monitoring of cellular responses to drug treatment within an incubator environment. Reproduced with permission from Ref. [22]. Copyright 2013, American Chemical Society.
The monitoring of the intracellular calcium dynamics in live Lymnaea stagnalis neurons is another application of LLFM. Calcium imaging was performed using Fura-2-loaded Lymnaea stagnalis neurons, excited at wavelengths of 340 nm and 380 nm, with fluorescence emissions detected at 510 nm. The system’s performance was validated by measuring the fluorescence changes induced by adding 50 μL of 1 M KCl stimulation, which depolarizes the neurons and triggers the action potentials, resulting in a rise in the intracellular calcium levels. A total of nine neurons were tested, and the fluorescence was recorded continuously. Furthermore, the wide field of view of the contact imaging system allowed for the monitoring of multiple neurons simultaneously [23]. In fact, monitoring the fluctuations in the calcium levels through fluorescence-imaging techniques is tremendously valuable, as it offers critical insights into the activity patterns and interconnectivity of neurons. Gaining this understanding is pivotal for advancing our comprehension of the brain function and for unraveling the complexities underlying neurological diseases and disorders.
In addition to cell imaging, tissue imaging has been performed using LLFM. For instance, fluorescently tagged transgenic Caenorhabditis elegans (C. elegans) were successfully imaged using the LLFM [28]. C. elegans were selected because they are widely used in studies related to neuroscience [61], oncology [62], and genetics [63]. The C elegans were genetically altered to express fluorescent markers, which allowed for close examination using the LLFM. Moreover, the Bio-FlatScope, a lensless microscopy incorporated with a designed phase mask, was used to monitor the neural activity in the motor cortexes of live mice. This was achieved by expressing the calcium indicator GCaMP6s in the neurons. The imaging system captured the fluorescence changes associated with neural activity, which were then reconstructed to provide insights into the calcium dynamics. The mice were free to move during the imaging process, and the Bio-FlatScope tracked their locomotion. The data revealed that the peak fluorescence signals during periods of movement were, on average, three times higher than during periods of inactivity. This significant increase in the fluorescence during locomotion was consistent with the neural-activity patterns observed using conventional epifluorescence microscopy, validating the Bio-FlatScope’s efficacy [31]. In the same study, The Bio-FlatScope was also utilized to image the calcium activity in Hydra vulgaris. Hydra vulgaris were genetically modified to express the calcium indicator GCaMP7b, which fluoresces in response to calcium ion binding, indicating cellular activity. The Bio-FlatScope was able to capture detailed images of the calcium dynamics in the freely moving Hydra, providing a unique perspective on the organism’s physiological processes in a natural, unrestrained state. The high-contrast contours and broad spatial frequency spectrum generated by the phase mask facilitated the clear visualization of the calcium signals, even in the presence of movement. For human applications, the Bio-FlatScope has been employed to image the microvasculature of the oral mucosa. This application is particularly significant for clinical diagnostics, as the ability to resolve microvessels less than 20 μm in diameter can aid in the early detection of conditions such as sepsis and cancer. The imaging was conducted in vivo, demonstrating the device’s potential for non-invasive medical diagnostics. The high-resolution images provided detailed views of the blood vessel structures, highlighting the fine capillaries and overall vascular network within the mucosal tissue.
In other studies, conducted using LLFM, a 100 µm thick brain slice from a green fluorescent protein (GFP) transgenic mouse was imaged to detect the fluorescent cell bodies within the brain slice. The slice was then placed directly on the device and imaged, and almost the whole slice was observed in a single shot [47,48]. Figure 4 compares images obtained using three different imaging setups: a lensless device with a hybrid longpass filter, a lensless device with a hybrid bandpass filter, and a conventional lens-based fluorescence microscope. The images showcase the hippocampus and reticular thalamic nucleus regions of a GFP-labeled mouse brain slice. The comparison highlights that the contrast and quality of the bandpass filter images are comparable to those obtained with the traditional fluorescence microscope, demonstrating the efficacy of the hybrid bandpass filter in reducing the autofluorescence and enhancing the GFP signal detection. This improvement underscores the potential of the lensless device for detailed biological imaging applications, offering a portable and cost-effective alternative to conventional microscopy techniques.
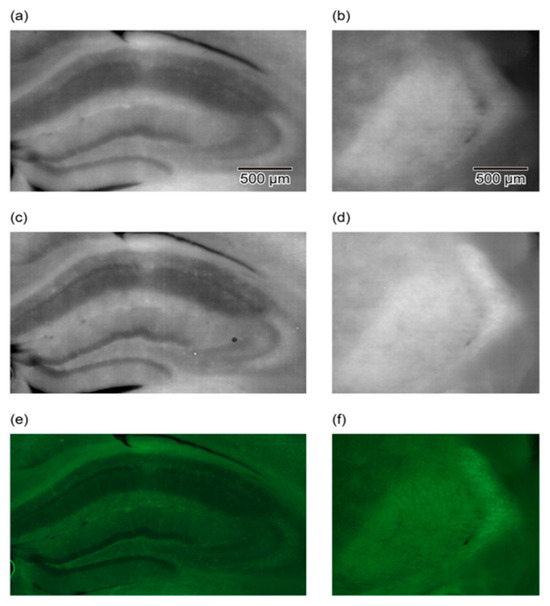
Figure 4.
Comparison of imaging results for hippocampus (a,c,e) and reticular thalamic nuclei (b,d,f) using LLFM and lens-based microscope. (a,b) Images obtained with a lensless imaging device featuring a hybrid longpass filter. This device includes a longpass interference filter to reflect the excitation light and an absorption filter to reject the scattered excitation light, enhancing the fluorescence detection. (c,d) Images of the same areas obtained with a lensless imaging device equipped with a hybrid bandpass filter. This improved filter setup incorporates both a shortpass interference filter and an absorption filter, effectively rejecting autofluorescence and scattered excitation light, resulting in clearer images with higher contrast and more detectable cell bodies. (e,f) Images captured with a conventional lens-based fluorescence microscope using a 5× objective lens with a numerical aperture (NA) of 0.1. These images serve as a benchmark to compare the performances of the lensless devices, showing similar levels of image clarity and contrast in detecting GFP-labeled cell bodies. The comparison demonstrates that the hybrid bandpass filter significantly enhances the performance of lensless fluorescence imaging, approaching the quality of conventional microscopy. Reproduced with permission from Ref. [49]. Copyright 2019, AIP Publishing.
Tissue imaging is a pivotal technique for visualizing the organization, structure, and function of tissues in health and disease, enabling early disease detection, accurate diagnosis, treatment planning, and the monitoring of disease progression [64,65]. LLFM emerges as a compelling approach for tissue imaging due to its enhanced accessibility, especially in resource-constrained environments or field settings where conventional microscopes may be impractical. Additionally, the wide field of view afforded by lensless imaging allows for the rapid screening and analysis of large tissue sections, facilitating the detection of localized abnormalities or rare events that may be missed with limited-field-of-view microscopes.
Beyond the applications described earlier, lensless fluorescence microscopy also shows promise in the detection and visualization of pathogenic organisms. The imaging of Giardia lamblia and Cryptosporidium parvum, two protozoan parasites known for causing enteric infections, highlighted the LLFM’s potential application in the screening of enteric parasites [51]. The parasites were first incubated with specific primary antibodies and then with fluorescently labeled secondary antibodies (Alexa Fluor® 488 for G. lamblia and Qdot® 625 for C. parvum). The samples were placed under the Talbot microscope, which used a 488 nm laser for the excitation, and fluorescence emissions were collected at a field of view of 4 mm × 4 mm with an 80 ms pixel dwell time. Separate fluorescence channels were used to distinguish the parasites, and the images were reconstructed using MATLAB. The imaging of pathogens is crucial as it enables the direct visualization of infectious agents, aiding in the accurate identification and understanding of disease mechanisms for the development of targeted treatments and prevention strategies [66,67,68]. Table 1 shows a list of all the techniques to make an LLFM with all possible applications.

Table 1.
Summarized list of all the techniques to make an LLFM with its specifications.
8. Conclusions
Lensless fluorescence microscopy (LLFM) has emerged as an alternative imaging technology, providing a unique blend of simplicity, cost-effectiveness, and high-throughput capabilities. Despite early limitations, subsequent advances in computational methods, hardware design, and signal processing have considerably improved the LLFM system performance, pushing the limits of the resolution, field-of-view, signal-to-noise ratio, and multicolor-imaging capabilities.
LLFM has attained resolutions comparable to those of conventional microscopes through the implementation of computational approaches such as deconvolution and compressive sensing, while advances in structured illumination, filter design, and light-guide plates have expanded the field of view and enhanced the signal detection. Notably, the best resolution achieved using a Fresnel zone plate for illumination in LLFM is 1 μm, and the best field of view has been achieved by converting the functionality of a flatbed scanner to lensless microscopy, resulting in an impressive 532 cm² area.
The wide range of applications highlighted in this study, including cellular imaging, tissue analysis, and pathogen detection, demonstrate LLFM’s adaptability and potential influence across several areas of biological research and clinical diagnostics. However, increasing the signal-to-noise ratio to image single cells remains a significant challenge and warrants more in-depth investigation. Furthermore, multicolor imaging with a high frame rate in LLFM is another open challenge that requires addressing. Secondly, in order to acquire comprehensive information from the sample, it is crucial to utilize multiple modalities. While, to some extent, LLFM has been successfully integrated with other modalities, such as holography [24,69], Raman microspectrometry, and scattering-based microscopy [70], there is still a need for more comprehensive multi-modal lensless imaging systems to acquire multiple pieces of information simultaneously.
While problems persist, the complementary integration of computational algorithms, optical engineering, neural networks, and sensor technologies opens up interesting possibilities for future LLFM developments. Ongoing research efforts to improve the resolution and image quality and to enable real-time multicolor imaging will undoubtedly propel this technology to new heights, providing scientists and clinicians with a powerful, accessible, and cost-effective imaging tool.
Author Contributions
Conceptualization, A.R.; methodology S.K., E.K.D. and A.R.; writing—original draft preparation, S.K., E.K.D. and A.R.; writing—review and editing, S.K. and A.R.; supervision, A.R. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by University of Toledo startup funds.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Roukos, V.; Pegoraro, G.; Voss, T.C.; Misteli, T. Cell Cycle Staging of Individual Cells by Fluorescence Microscopy. Nat. Protoc. 2015, 10, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Tian, Y.; Yu, J.; Yuan, Z. Fluorescent Probes for Biological Imaging. BioMed Res. Int. 2016, 2016, e3730486. [Google Scholar] [CrossRef] [PubMed]
- Peng, X.; Huang, X.; Du, K.; Liu, H.; Chen, L. High Spatiotemporal Resolution and Low Photo-Toxicity Fluorescence Imaging in Live Cells and in Vivo. Biochem. Soc. Trans. 2019, 47, 1635–1650. [Google Scholar] [CrossRef] [PubMed]
- Shim, S.-H.; Xia, C.; Zhong, G.; Babcock, H.P.; Vaughan, J.C.; Huang, B.; Wang, X.; Xu, C.; Bi, G.-Q.; Zhuang, X. Super-Resolution Fluorescence Imaging of Organelles in Live Cells with Photoswitchable Membrane Probes. Proc. Natl. Acad. Sci. USA 2012, 109, 13978–13983. [Google Scholar] [CrossRef] [PubMed]
- Nath, P.; Hamadna, S.S.; Karamchand, L.; Foster, J.; Kopelman, R.; Amar, J.G.; Ray, A. Intracellular Detection of Singlet Oxygen Using Fluorescent Nanosensors. Analyst 2021, 146, 3933–3941. [Google Scholar] [CrossRef] [PubMed]
- Karamchand, L.; Kim, G.; Wang, S.; Hah, H.J.; Ray, A.; Jiddou, R.; Lee, Y.-E.K.; Philbert, M.A.; Kopelman, R. Modulation of Hydrogel Nanoparticle Intracellular Trafficking by Multivalent Surface Engineering with Tumor Targeting Peptide. Nanoscale 2013, 5, 10327–10344. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Hoppe, A.D.; Seveau, S.; Swanson, J.A. Live Cell Fluorescence Microscopy to Study Microbial Pathogenesis. Cell. Microbiol. 2009, 11, 540–550. [Google Scholar] [CrossRef] [PubMed]
- Nath, P.; Mahtaba, K.R.; Ray, A. Fluorescence-Based Portable Assays for Detection of Biological and Chemical Analytes. Sensors 2023, 23, 5053. [Google Scholar] [CrossRef]
- Weidemann, T.; Mücksch, J.; Schwille, P. Fluorescence Fluctuation Microscopy: A Diversified Arsenal of Methods to Investigate Molecular Dynamics inside Cells. Curr. Opin. Struct. Biol. 2014, 28, 69–76. [Google Scholar] [CrossRef]
- Drummen, G.P.C. Fluorescent Probes and Fluorescence (Microscopy) Techniques—Illuminating Biological and Biomedical Research. Molecules 2012, 17, 14067–14090. [Google Scholar] [CrossRef]
- Petty, H.R. High Speed Microscopy in Biomedical Research. Opt. Photonics News OPN 2004, 15, 40–45. [Google Scholar] [CrossRef]
- Ozcan, A.; McLeod, E. Lensless Imaging and Sensing. Annu. Rev. Biomed. Eng. 2016, 18, 77–102. [Google Scholar] [CrossRef] [PubMed]
- McLeod, E.; Ozcan, A. Unconventional Methods of Imaging: Computational Microscopy and Compact Implementations. Rep. Prog. Phys. 2016, 79, 076001. [Google Scholar] [CrossRef] [PubMed]
- Daloglu, M.U.; Ray, A.; Collazo, M.J.; Brown, C.; Tseng, D.; Chocarro-Ruiz, B.; Lechuga, L.M.; Cascio, D.; Ozcan, A. Low-Cost and Portable UV Holographic Microscope for High-Contrast Protein Crystal Imaging. APL Photonics 2019, 4, 030804. [Google Scholar] [CrossRef]
- Daloglu, M.U.; Ray, A.; Gorocs, Z.; Xiong, M.; Malik, R.; Bitan, G.; McLeod, E.; Ozcan, A. Computational On-Chip Imaging of Nanoparticles and Biomolecules Using Ultraviolet Light. Sci. Rep. 2017, 7, 44157. [Google Scholar] [CrossRef] [PubMed]
- McLeod, E.; Dincer, T.U.; Veli, M.; Ertas, Y.N.; Nguyen, C.; Luo, W.; Greenbaum, A.; Feizi, A.; Ozcan, A. High-Throughput and Label-Free Single Nanoparticle Sizing Based on Time-Resolved On-Chip Microscopy. ACS Nano 2015, 9, 3265–3273. [Google Scholar] [CrossRef] [PubMed]
- Ray, A.; Khalid, M.A.; Demčenko, A.; Daloglu, M.; Tseng, D.; Reboud, J.; Cooper, J.M.; Ozcan, A. Holographic Detection of Nanoparticles Using Acoustically Actuated Nanolenses. Nat. Commun. 2020, 11, 171. [Google Scholar] [CrossRef] [PubMed]
- Kabir, M.A.; Kharel, A.; Malla, S.; Kreis, Z.J.; Nath, P.; Wolfe, J.N.; Hassan, M.; Kaur, D.; Sari-Sarraf, H.; Tiwari, A.K.; et al. Automated Detection of Apoptotic versus Nonapoptotic Cell Death Using Label-Free Computational Microscopy. J. Biophotonics 2021, 15, e202100310. [Google Scholar] [CrossRef]
- Zhang, Y.; Ouyang, M.; Ray, A.; Liu, T.; Kong, J.; Bai, B.; Kim, D.; Guziak, A.; Luo, Y.; Feizi, A.; et al. Computational Cytometer Based on Magnetically Modulated Coherent Imaging and Deep Learning. Light Sci. Appl. 2019, 8, 91. [Google Scholar] [CrossRef]
- Verdaasdonk, J.S.; Stephens, A.D.; Haase, J.; Bloom, K. Bending the Rules: Widefield Microscopy and the Abbe Limit of Resolution. J. Cell. Physiol. 2014, 229, 132–138. [Google Scholar] [CrossRef]
- Arpali, S.A.; Arpali, C.; Coskun, A.F.; Chiang, H.-H.; Ozcan, A. High-Throughput Screening of Large Volumes of Whole Blood Using Structured Illumination and Fluorescent on-Chip Imaging. Lab Chip 2012, 12, 4968–4971. [Google Scholar] [CrossRef]
- Han, C.; Pang, S.; Bower, D.V.; Yiu, P.; Yang, C. Wide Field-of-View On-Chip Talbot Fluorescence Microscopy for Longitudinal Cell Culture Monitoring from within the Incubator. Anal. Chem. 2013, 85, 2356–2360. [Google Scholar] [CrossRef] [PubMed]
- Mudraboyina, A.K.; Blockstein, L.; Luk, C.C.; Syed, N.I.; Yadid-Pecht, O. A Novel Lensless Miniature Contact Imaging System for Monitoring Calcium Changes in Live Neurons. IEEE Photonics J. 2014, 6, 3900115. [Google Scholar] [CrossRef]
- Coskun, A.F.; Su, T.-W.; Ozcan, A. Wide Field-of-View Lens-Free Fluorescent Imaging on a Chip. Lab Chip 2010, 10, 824–827. [Google Scholar] [CrossRef] [PubMed]
- Candes, E.J.; Tao, T. Near-Optimal Signal Recovery From Random Projections: Universal Encoding Strategies? IEEE Trans. Inf. Theory 2006, 52, 5406–5425. [Google Scholar] [CrossRef]
- Candès, E.J.; Romberg, J.K.; Tao, T. Stable Signal Recovery from Incomplete and Inaccurate Measurements. Commun. Pure Appl. Math. 2006, 59, 1207–1223. [Google Scholar] [CrossRef]
- Coskun, A.F.; Sencan, I.; Su, T.-W.; Ozcan, A. Lensless Wide-Field Fluorescent Imaging on a Chip Using Compressive Decoding of Sparse Objects. Opt. Express 2010, 18, 10510–10523. [Google Scholar] [CrossRef] [PubMed]
- Coskun, A.F.; Sencan, I.; Su, T.-W.; Ozcan, A. Lensfree Fluorescent On-Chip Imaging of Transgenic Caenorhabditis Elegans over an Ultra-Wide Field-of-View. PLoS ONE 2011, 6, e15955. [Google Scholar] [CrossRef] [PubMed]
- Coskun, A.F.; Su, T.-W.; Sencan, I.; Ozcan, A. Lensless Fluorescent Microscopy on a Chip. JoVE (J. Vis. Exp.) 2011, 54, e3181. [Google Scholar] [CrossRef]
- Bishara, W.; Su, T.-W.; Coskun, A.F.; Ozcan, A. Lensfree On-Chip Microscopy over a Wide Field-of-View Using Pixel Super-Resolution. Opt. Express 2010, 18, 11181. [Google Scholar] [CrossRef]
- Adams, J.K.; Yan, D.; Wu, J.; Boominathan, V.; Gao, S.; Rodriguez, A.V.; Kim, S.; Carns, J.; Richards-Kortum, R.; Kemere, C.; et al. In Vivo Lensless Microscopy via a Phase Mask Generating Diffraction Patterns with High-Contrast Contours. Nat. Biomed. Eng. 2022, 6, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Adams, J.K.; Boominathan, V.; Avants, B.W.; Vercosa, D.G.; Ye, F.; Baraniuk, R.G.; Robinson, J.T.; Veeraraghavan, A. Single-Frame 3D Fluorescence Microscopy with Ultraminiature Lensless FlatScope. Sci. Adv. 2017, 3, e1701548. [Google Scholar] [CrossRef] [PubMed]
- Pang, S.; Han, C.; Lee, L.M.; Yang, C. Fluorescence Microscopy Imaging with a Fresnel Zone Plate Array Based Optofluidic Microscope. Lab Chip 2011, 11, 3698–3702. [Google Scholar] [CrossRef] [PubMed]
- Semple, J.I.; Garcia-Verdugo, R.; Lehner, B. Rapid Selection of Transgenic C. elegans Using Antibiotic Resistance. Nat. Methods 2010, 7, 725–727. [Google Scholar] [CrossRef]
- Rohde, C.B.; Zeng, F.; Gonzalez-Rubio, R.; Angel, M.; Yanik, M.F. Microfluidic System for On-Chip High-Throughput Whole-Animal Sorting and Screening at Subcellular Resolution. Proc. Natl. Acad. Sci. USA 2007, 104, 13891–13895. [Google Scholar] [CrossRef]
- Orth, A.; Crozier, K.B. High Throughput Multichannel Fluorescence Microscopy with Microlens Arrays. Opt. Express 2014, 22, 18101. [Google Scholar] [CrossRef]
- Chokshi, T.V.; Bazopoulou, D.; Chronis, N. An Automated Microfluidic Platform for Calcium Imaging of Chemosensory Neurons in Caenorhabditis Elegans. Lab Chip 2010, 10, 2758–2763. [Google Scholar] [CrossRef]
- Coucheron, D.A.; Helle, Ø.I.; Øie, C.I.; Tinguely, J.-C.; Ahluwalia, B.S. High-Throughput Total Internal Reflection Fluorescence and Direct Stochastic Optical Reconstruction Microscopy Using a Photonic Chip. J. Vis. Exp. 2019, 153, e60378. [Google Scholar] [CrossRef]
- Göröcs, Z.; Ling, Y.; Yu, M.D.; Karahalios, D.; Mogharabi, K.; Lu, K.; Wei, Q.; Ozcan, A. Giga-Pixel Fluorescent Imaging over an Ultra-Large Field-of-View Using a Flatbed Scanner. Lab Chip 2013, 13, 4460–4466. [Google Scholar] [CrossRef]
- Haider, S.A.; Cameron, A.; Siva, P.; Lui, D.; Shafiee, M.J.; Boroomand, A.; Haider, N.; Wong, A. Fluorescence Microscopy Image Noise Reduction Using a Stochastically-Connected Random Field Model. Sci. Rep. 2016, 6, 20640. [Google Scholar] [CrossRef]
- Zhang, Y.; Khan, A.A.; Vigil, G.D.; Howard, S.S. Investigation of Signal-to-Noise Ratio in Frequency-Domain Multiphoton Fluorescence Lifetime Imaging Microscopy. J. Opt. Soc. Am. A 2016, 33, B1–B11. [Google Scholar] [CrossRef]
- Shanmugam, A.; Salthouse, C.D. Lensless Fluorescence Imaging with Height Calculation. J. Biomed. Opt. 2014, 19, 016002. [Google Scholar] [CrossRef] [PubMed]
- Dao, L.; Lucotte, B.; Glancy, B.; Chang, L.-C.; Hsu, L.-Y.; Balaban, R.S. Use of independent component analysis to improve signal-to-noise ratio in multi-probe fluorescence microscopy. J. Microsc. 2014, 256, 133–144. [Google Scholar] [CrossRef]
- Martinelli, L.; Choumane, H.; Ha, K.-N.; Sagarzazu, G.; Goutel, C.; Weisbuch, C.; Gacoin, T.; Benisty, H. Sensor-Integrated Fluorescent Microarray for Ultrahigh Sensitivity Direct-Imaging Bioassays: Role of a High Rejection of Excitation Light. Appl. Phys. Lett. 2007, 91, 083901. [Google Scholar] [CrossRef]
- Lee, S.A.; Ou, X.; Lee, J.E.; Yang, C. Chip-Scale Fluorescence Microscope Based on a Silo-Filter Complementary Metal-Oxide Semiconductor Image Sensor. Opt. Lett. 2013, 38, 1817–1819. [Google Scholar] [CrossRef]
- Sasagawa, K.; Kimura, A.; Haruta, M.; Noda, T.; Tokuda, T.; Ohta, J. Highly Sensitive Lens-Free Fluorescence Imaging Device Enabled by a Complementary Combination of Interference and Absorption Filters. Biomed. Opt. Express 2018, 9, 4329–4344. [Google Scholar] [CrossRef] [PubMed]
- Sasagawa, K.; Ohta, Y.; Kawahara, M.; Haruta, M.; Tokuda, T.; Ohta, J. Wide Field-of-View Lensless Fluorescence Imaging Device with Hybrid Bandpass Emission Filter. AIP Adv. 2019, 9, 035108. [Google Scholar] [CrossRef]
- Rustami, E.; Sasagawa, K.; Sugie, K.; Ohta, Y.; Takehara, H.; Haruta, M.; Tashiro, H.; Ohta, J. Thin and Scalable Hybrid Emission Filter via Plasma Etching for Low-Invasive Fluorescence Detection. Sensors 2023, 23, 3695. [Google Scholar] [CrossRef]
- Gregor, I.; Butkevich, E.; Enderlein, J.; Mojiri, S. Instant Three-Color Multiplane Fluorescence Microscopy. Biophys. Rep. 2021, 1, 100001. [Google Scholar] [CrossRef]
- Kaya, M.; Stein, F.; Rouwkema, J.; Khalil, I.S.M.; Misra, S. Serial Imaging of Micro-Agents and Cancer Cell Spheroids in a Microfluidic Channel Using Multicolor Fluorescence Microscopy. PLoS ONE 2021, 16, e0253222. [Google Scholar] [CrossRef]
- Pang, S.; Han, C.; Erath, J.; Rodriguez, A.; Yang, C. Wide Field-of-View Talbot Grid-Based Microscopy for Multicolor Fluorescence Imaging. Opt. Express 2013, 21, 14555–14565. [Google Scholar] [CrossRef] [PubMed]
- Shin, H.; Yoon, G.-W.; Choi, W.; Lee, D.; Choi, H.; Jo, D.S.; Choi, N.; Yoon, J.-B.; Cho, I.-J. Miniaturized Multicolor Fluorescence Imaging System Integrated with a PDMS Light-Guide Plate for Biomedical Investigation. Npj Flex. Electron. 2023, 7, 7. [Google Scholar] [CrossRef]
- Gao, L.; Ma, Y.; Huang, L.; Sen, C.; Burri, S.; Bruschini, C.; Yang, X.; Cameron, R.; Fishbein, G.; Gomperts, B.; et al. Light-Field Tomographic Fluorescence Lifetime Imaging Microscopy; Research Square: Durham, NC, USA, 2023. [Google Scholar]
- Suhling, K.; Hirvonen, L.M.; Levitt, J.A.; Chung, P.-H.; Tregidgo, C.; Le Marois, A.; Rusakov, D.A.; Zheng, K.; Ameer-Beg, S.; Poland, S.; et al. Fluorescence Lifetime Imaging (FLIM): Basic Concepts and Some Recent Developments. Med. Photonics 2015, 27, 3–40. [Google Scholar] [CrossRef]
- Datta, R.; Heaster, T.M.; Sharick, J.T.; Gillette, A.A.; Skala, M.C. Fluorescence Lifetime Imaging Microscopy: Fundamentals and Advances in Instrumentation, Analysis, and Applications. J. Biomed. Opt. 2020, 25, 071203. [Google Scholar] [CrossRef] [PubMed]
- Datta, R.; Gillette, A.; Stefely, M.; Skala, M.C. Recent Innovations in Fluorescence Lifetime Imaging Microscopy for Biology and Medicine. J. Biomed. Opt. 2021, 26, 070603. [Google Scholar] [CrossRef] [PubMed]
- Sensors|Free Full-Text|A 72 × 60 Angle-Sensitive SPAD Imaging Array for Lens-Less FLIM. Available online: https://www.mdpi.com/1424-8220/16/9/1422 (accessed on 6 June 2024).
- Khoubafarin, S.; Nath, P.; Popofski, H.; Ray, A. Two-Dimensional Microlens Array for Low-Cost High-Resolution Bio-Imaging. In Proceedings of the Optics and Biophotonics in Low-Resource Settings IX, San Francisco, CA, USA, 6 March 2023; SPIE: Bellingham, WA, USA, 2023; Volume 12369, pp. 29–35. [Google Scholar]
- Khoubafarin, S.; Kharel, A.; Malla, S.; Nath, P.; Kaur, D.; Tiwari, A.K.; Ray, A. Monitoring the Efficacy of Chemotherapeutic Drugs Using Dark Field Imaging. In Proceedings of the Label-Free Biomedical Imaging and Sensing (LBIS) 2022, San Francisco, CA, USA, 2 March 2022; SPIE: Bellingham, WA, USA, 2022; Volume 11972, pp. 31–38. [Google Scholar]
- Khoubafarin, S.; Kharel, A.; Malla, S.; Nath, P.; Irving, R.E.; Kaur, D.; Tiwari, A.K.; Ray, A. Label-Free Identification of Cell Death Mechanism Using Scattering-Based Microscopy and Deep Learning. J. Phys. D Appl. Phys. 2023, 56, 485401. [Google Scholar] [CrossRef]
- Mellem, J.E.; Brockie, P.J.; Madsen, D.M.; Maricq, A.V. Action Potentials Contribute to Neuronal Signaling in C. Elegans. Nat. Neurosci. 2008, 11, 865–867. [Google Scholar] [CrossRef] [PubMed]
- Pinkston-Gosse, J.; Kenyon, C. DAF-16/FOXO Targets Genes That Regulate Tumor Growth in Caenorhabditis elegans. Nat. Genet. 2007, 39, 1403–1409. [Google Scholar] [CrossRef]
- Lehner, B.; Crombie, C.; Tischler, J.; Fortunato, A.; Fraser, A.G. Systematic Mapping of Genetic Interactions in Caenorhabditis Elegans Identifies Common Modifiers of Diverse Signaling Pathways. Nat. Genet. 2006, 38, 896–903. [Google Scholar] [CrossRef]
- Rahman, A.; Jahangir, C.; Lynch, S.M.; Alattar, N.; Aura, C.; Russell, N.; Lanigan, F.; Gallagher, W.M. Advances in Tissue-Based Imaging: Impact on Oncology Research and Clinical Practice. Expert Rev. Mol. Diagn. 2020, 20, 1027–1037. [Google Scholar] [CrossRef]
- Wang, S.; Larina, I.V. 8—High-Resolution Imaging Techniques in Tissue Engineering. In Monitoring and Evaluation of Biomaterials and their Performance In Vivo; Narayan, R.J., Ed.; Woodhead Publishing: Cambridge, UK, 2017; pp. 151–180. ISBN 978-0-08-100603-0. [Google Scholar]
- Gordon, O.; Ruiz-Bedoya, C.A.; Ordonez, A.A.; Tucker, E.W.; Jain, S.K. Molecular Imaging: A Novel Tool to Visualize Pathogenesis of Infections In Situ. mBio 2019, 10, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Fischer, E.R.; Hansen, B.T.; Schwartz, C.L.; Nair, V. Microscopy Tools and Techniques Used in the Study of Infectious Disease Agents. Microsc. Microanal. 2018, 24, 1340–1341. [Google Scholar] [CrossRef][Green Version]
- Ady, J.; Fong, Y. Imaging for Infection: From Visualization of Inflammation to Visualization of Microbes. Surg. Infect. 2014, 15, 700–707. [Google Scholar] [CrossRef] [PubMed]
- Coskun, A.F.; Sencan, I.; Su, T.W.; Ozcan, A. Wide-Field Lensless Fluorescent Microscopy Using a Tapered Fiber-Optic Faceplate on a Chip. Analyst 2011, 136, 3512–3518. [Google Scholar] [CrossRef] [PubMed]
- Strola, S.; Schultz, E.; Allier, C.; DesRoches, B.; Lemmonier, J.; Dinten, J.-M. Raman Microspectrometer Combined with Scattering Microscopy and Lensless Imaging for Bacteria Identification. In Proceedings of the Advanced Biomedical and Clinical Diagnostic Systems XI, San Francisco, CA, USA, 3–5 February 2013; SPIE: Bellingham, WA, USA, 2013; Volume 8572. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).