Pseudo-Stilbene- and Azobenzene-Type Systems for Optical Frequency Conversion: Estimating the First-Order Molecular Hyperpolarizability

 ,
,  ,
,  ,
,  and
and
Abstract
1. Introduction
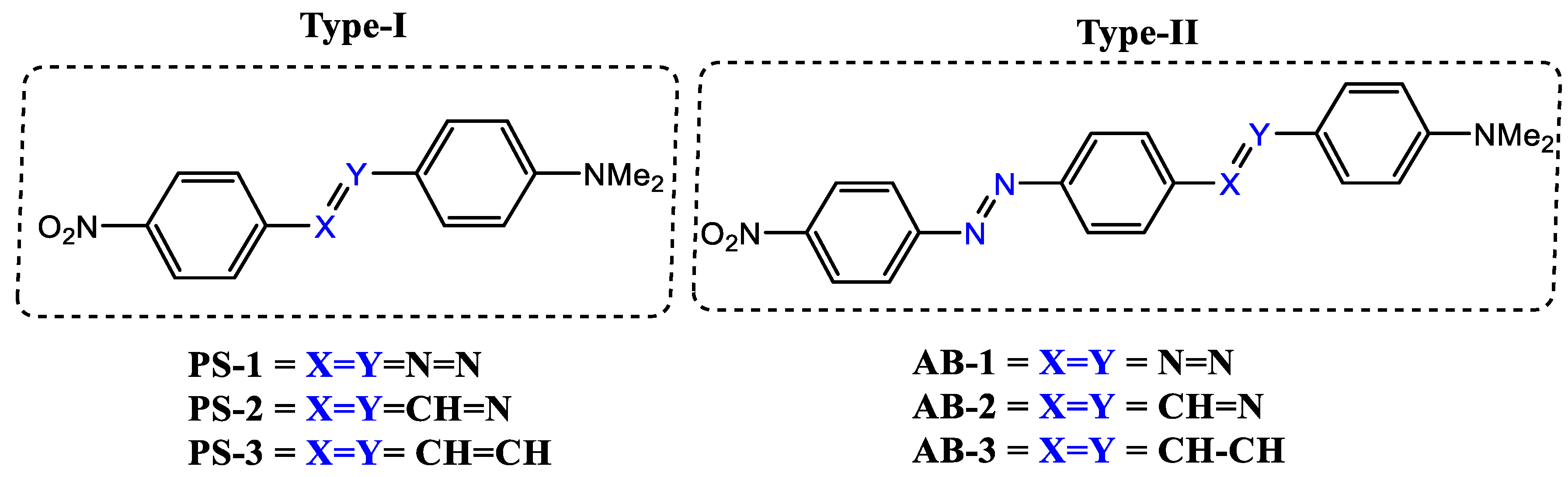
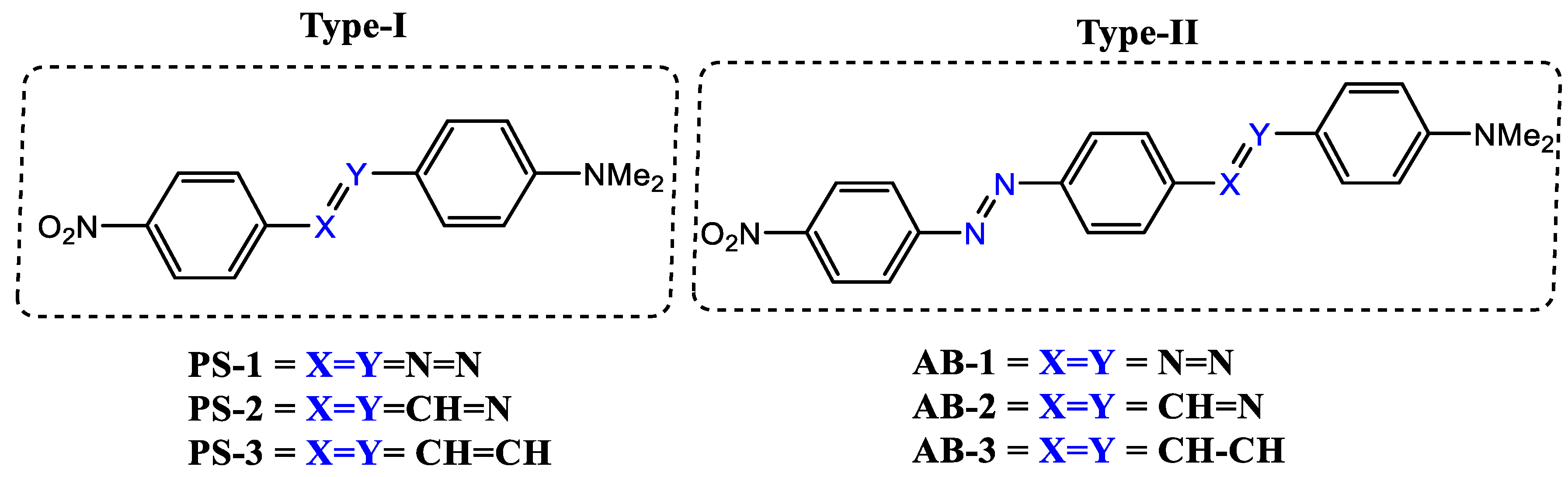
2. Investigated Compounds
3. Computational Methodology Details
4. Results and Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Agrell, E.; Karlsson, M.; Chraplyvy, A.R.; Richardson, D.J.; Krummrich, P.M.; Winzer, P.; Roberts, K.; Fischer, J.K.; Savory, S.J.; Eggleton, B.J. Roadmap of optical communications. J. Opt. 2016, 18, 63002. [Google Scholar] [CrossRef]
- Karothu, D.P.; Dushaq, G.; Ahmed, E.; Catalano, L.; Polavaram, S.; Ferreira, R.; Li, L.; Mohamed, S.; Rasras, M.; Naumov, P. Mechanically robust amino acid crystals as fiber-optic transducers and wide bandpass filters for optical communication in the near-infrared. Nat. Commun. 2021, 12, 1326. [Google Scholar] [CrossRef]
- Feldmann, A.; Gasser, O.; Lichtblau, F.; Pujol, E.; Poese, I.; Dietzel, C.; Wagner, D.; Wichtlhuber, M.; Tapiador, J.; Vallina-Rodriguez, N. A year in lockdown: How the waves of COVID-19 impact internet traffic. Commun. ACM 2021, 64, 101–108. [Google Scholar] [CrossRef]
- Castet, F.; Bogdan, E.; Plaquet, A.; Ducasse, L.; Champagne, B.; Rodriguez, V. Reference molecules for nonlinear optics: A joint experimental and theoretical investigation. J. Chem. Phys. 2012, 136, 24506. [Google Scholar] [CrossRef] [PubMed]
- Franken, P.A.; Hill, A.E.; Peters, C.W.; Weinreich, G. Generation of Optical Harmonics. Phys. Rev. Lett. 1961, 7, 118–119. [Google Scholar] [CrossRef]
- Chemla, D.S. Nonlinear Optical Properties of Organic Molecules and Crystals; Elsevier: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Radhakrishnan, T.P. Molecular structure, symmetry, and shape as design elements in the fabrication of molecular crystals for second harmonic generation and the role of molecules-in-materials. Acc. Chem. Res. 2008, 41, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Abegão, L.M.G.; Fonseca, R.D.; Santos, F.A.; Rodrigues, J.J.; Kamada, K.; Mendonça, C.R.; Piguel, S.; De Boni, L. First molecular electronic hyperpolarizability of series of π-conjugated oxazole dyes in solution: An experimental and theoretical study. RSC Adv. 2019, 9, 26476–26482. [Google Scholar] [CrossRef] [PubMed]
- Zhao, T.; Wang, C.; Hu, S.; Ji, S.; Hu, C.; Xu, K.; Teng, B. Structural design and characterization of a chalcone derivative crystal DAMO with strong SHG efficiency for NLO applications. Opt. Mater. 2021, 112, 110765. [Google Scholar] [CrossRef]
- De Araújo, R.S.; de Alcântara, A.M.; Abegão, L.M.G.; de Souza, Y.P.; Silva, A.C.B.; Machado, R.; Rodrigues, J.J.; Pliego, J.R.; d’Errico, F.; Valle, M.S.; et al. Second harmonic generation in pyrazoline derivatives of dibenzylideneacetones and chalcone: A combined experimental and theoretical approach. J. Photochem. Photobiol. A Chem. 2020, 388, 112147. [Google Scholar] [CrossRef]
- López, S.F.; Meza, M.P.; Hoyos, F.T. Study of the nonlinear optical properties of 4-nitroaniline type compounds by density functional theory calculations: Towards new NLO materials. Comput. Theor. Chem. 2018, 1133, 25–32. [Google Scholar] [CrossRef]
- Eggleton, B.J.; Vo, T.D.; Pant, R.; Schr, J.; Pelusi, M.D.; Choi, D.Y.; Madden, S.J.; Luther-Davies, B. Photonic chip based ultrafast optical processing based on high nonlinearity dispersion engineered chalcogenide waveguides. Laser Photon. Rev. 2012, 6, 97–114. [Google Scholar] [CrossRef]
- Castet, F.; Rodriguez, V.; Pozzo, J.-L.; Ducasse, L.; Plaquet, A.; Champagne, B. Design and characterization of molecular nonlinear optical switches. Acc. Chem. Res. 2013, 46, 2656–2665. [Google Scholar] [CrossRef]
- Wu, J.; Luo, J.; Jen, A.K.-Y. High-performance organic second-and third-order nonlinear optical materials for ultrafast information processing. J. Mater. Chem. C. 2020, 8, 15009–15026. [Google Scholar] [CrossRef]
- Santos, F.A.; Abegão, L.M.G.; Fonseca, R.D.; Alcântara, A.M.; Mendonça, C.R.; Alencar, M.A.R.C.; Valle, M.S.; Kamada, K.; De Boni, L.; Rodrigues, J.J. Nonlinear Optical Study in a Set of Dibenzylideneacetone Derivatives with Potential for Optical Frequency Conversion. Photonics 2020, 7, 8. [Google Scholar] [CrossRef]
- Pelosi, A.G.; Cocca, L.H.Z.; Abegão, L.M.G.; Sciuti, L.F.; Piguel, S.; De Boni, L.; Mendonça, C.R. Influence of electron-withdrawing groups in two-photon absorption of imidazopyridines derivatives. Dyes Pigments 2022, 198, 109972. [Google Scholar] [CrossRef]
- Ekbote, A.; Patil, P.S.; Maidur, S.R.; Chia, T.S.; Quah, C.K. Structure and nonlinear optical properties of (E)-1-(4-aminophenyl)-3-(3-chlorophenyl) prop-2-en-1-one: A promising new D-π-A-π-D type chalcone derivative crystal for nonlinear optical devices. J. Mol. Struct. 2017, 1129, 239–247. [Google Scholar] [CrossRef]
- Li, M.; Li, Y.; Zhang, H.; Wang, S.; Ao, Y.; Cui, Z. Molecular engineering of organic chromophores and polymers for enhanced bulk second-order optical nonlinearity. J. Mater. Chem. C. 2017, 5, 4111–4122. [Google Scholar] [CrossRef]
- Santos, F.A.; Cardoso, C.E.R.; Rodrigues, J.J., Jr.; De Boni, L.; Abegão, L.M.G. Nonlinear Optical Materials: Predicting the First-Order Molecular Hyperpolarizability of Organic Molecular Structures. Photonics 2023, 10, 545. [Google Scholar] [CrossRef]
- El Ouazzani, H.; Iliopoulos, K.; Pranaitis, M.; Krupka, O.; Smokal, V.; Kolendo, A.; Sahraoui, B. Second-and third-order nonlinearities of novel push− pull azobenzene polymers. J. Phys. Chem. B. 2011, 115, 1944–1949. [Google Scholar] [CrossRef]
- Szukalski, A.; Sahraoui, B.; Kulyk, B.; Lazar, C.A.; Manea, A.M.; Mysliwiec, J. Chemical structure versus second-order nonlinear optical response of the push–pull type pyrazoline-based chromophores. RSC Adv. 2017, 7, 9941–9947. [Google Scholar] [CrossRef]
- Wu, J.; Wilson, B.A.; Smith, D.W., Jr.; Nielsen, S.O. Towards an understanding of structure-nonlinearity relationships in triarylamine-based push-pull electro-optic chromophores: The influence of substituent and molecular conformation on molecular hyperpolarizabilities. J. Mater. Chem. C. 2014, 2, 2591–2599. [Google Scholar] [CrossRef]
- Misra, R.; Bhattacharyya, S.P. Intramolecular Charge Transfer: Theory and Applications; John Wiley & Sons: Hoboken, NJ, USA, 2018. [Google Scholar]
- Teo, K.Y.; Tiong, M.H.; Wee, H.Y.; Jasin, N.; Liu, Z.-Q.; Shiu, M.Y.; Tang, J.Y.; Tsai, J.-K.; Rahamathullah, R.; Khairul, W.M. The influence of the push-pull effect and a π-conjugated system in conversion efficiency of bis-chalcone compounds in a dye sensitized solar cell. J. Mol. Struct. 2017, 1143, 42–48. [Google Scholar] [CrossRef]
- Lee, S.; Jen, M.; Jang, T.; Lee, G.; Pang, Y. Twisted intramolecular charge transfer of nitroaromatic push–pull chromophores. Sci. Rep. 2022, 12, 6557. [Google Scholar] [CrossRef] [PubMed]
- Jaunet-lahary, T.; Chantzis, A.; Chen, K.J.; Laurent, A.D.; Jacquemin, D. Designing Efficient Azobenzene and Azothiophene Nonlinear Optical Photochromes. J. Phys. Chem. C 2014, 118, 28831–28841. [Google Scholar] [CrossRef]
- Jasmine, G.F.; Amalanathan, M.; Roy, S.D.D. Molecular structure and charge transfer contributions to nonlinear optical property of 2-Methyl-4-nitroaniline: A DFT study. J. Mol. Struct. 2016, 1112, 63–70. [Google Scholar] [CrossRef]
- Muniz-Miranda, F.; Pedone, A.; Muniz-Miranda, M. Spectroscopic and DFT investigation on the photo-chemical properties of a push-pull chromophore: 4-Dimethylamino-4′-nitrostilbene. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2018, 190, 33–39. [Google Scholar] [CrossRef]
- Sanyal, S.; Sissa, C.; Terenziani, F.; Pati, S.K.; Painelli, A. Superlinear amplification of the first hyperpolarizability of linear aggregates of DANS molecules. Phys. Chem. Chem. Phys. 2017, 19, 24979–24984. [Google Scholar] [CrossRef]
- Shen, T.; Wang, X.-N.; Lou, H.-X. Natural stilbenes: An overview. Nat. Prod. Rep. 2009, 26, 916–935. [Google Scholar] [CrossRef]
- Teka, T.; Zhang, L.; Ge, X.; Li, Y.; Han, L.; Yan, X. Stilbenes: Source plants, chemistry, biosynthesis, pharmacology, application and problems related to their clinical Application-A comprehensive review. Phytochemistry 2022, 197, 113128. [Google Scholar] [CrossRef]
- Likhtenshtein, G. Stilbenes: Applications in Chemistry, Life Sciences and Materials Science; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009. [Google Scholar]
- Barrett, C.J.; Mamiya, J.; Yager, K.G.; Ikeda, T. Photo-mechanical effects in azobenzene-containing soft materials. Soft Matter. 2007, 3, 1249–1261. [Google Scholar] [CrossRef]
- Day, P.N.; Nguyen, K.A.; Pachter, R. TDDFT Study of One-and Two-Photon Absorption Properties: Donor− π− Acceptor Chromophores. J. Phys. Chem. B. 2005, 109, 1803–1814. [Google Scholar] [CrossRef] [PubMed]
- Cariati, E.; Forni, A.; Biella, S.; Metrangolo, P.; Meyer, F.; Resnati, G.; Righetto, S.; Tordin, E.; Ugo, R. Tuning second-order NLO responses through halogen bonding. Chem. Commun. 2007, 2590–2592. [Google Scholar] [CrossRef] [PubMed]
- Iftime, M.-M.; Cozan, V.; Airinei, A.; Varganici, C.; Ailiesei, G.; Timpu, D.; Sava, I. Asymmetric azomethine amines with azobenzene moieties–liquid crystalline and optical properties. Liq. Cryst. 2019, 46, 1584–1594. [Google Scholar] [CrossRef]
- Stoilova, A.; Georgiev, A.; Nedelchev, L.; Nazarova, D.; Dimov, D. Structure-property relationship and photoinduced birefringence of the azo and azo-azomethine dyes thin films in PMMA matrix. Opt. Mater. 2019, 87, 16–23. [Google Scholar] [CrossRef]
- De Boni, L.; Rodrigues, J.J., Jr.; dos Santos, D.S., Jr.; Silva, C.; Balogh, D.T.; Oliveira, O.N., Jr.; Zilio, S.C.; Misoguti, L.; Mendonca, C.R. Two-photon absorption in azoaromatic compounds. Chem. Phys. Lett. 2002, 361, 209–213. [Google Scholar] [CrossRef]
- Dudek, M.; Tarnowicz-Staniak, N.; Deiana, M.; Pokładek, Z.; Samoć, M.; Matczyszyn, K. Two-photon absorption and two-photon-induced isomerization of azobenzene compounds. RSC Adv. 2020, 10, 40489–40507. [Google Scholar] [CrossRef]
- Shalin, N.I.; Fominykh, O.D.; Balakina, M.Y. E ff ect of acceptor moieties on static and dynamic fi rst hyperpolarizability of azobenzene chromophores. Chem. Phys. Lett. 2019, 717, 21–28. [Google Scholar] [CrossRef]
- De Boni, L.; Misoguti, L.; Zílio, S.C.; Mendonça, C.R. Degenerate two-photon absorption spectra in azoaromatic compounds. ChemPhysChem 2005, 6, 1121–1125. [Google Scholar] [CrossRef]
- Tonnelé, C.; Champagne, B.; Muccioli, L.; Castet, F. Nonlinear optical contrast in azobenzene-based self-assembled monolayers. Chem. Mater. 2019, 31, 6759–6769. [Google Scholar] [CrossRef]
- Qian, Y.; Wang, G.; Xiao, G.; Lin, B.; Cui, Y. The first-order molecular hyperpolarizability and thermal stability of charge-transfer azo diol and azo aldimine. Dyes Pigments 2007, 75, 460–465. [Google Scholar] [CrossRef]
- Guo, K.; Hao, J.; Zhang, T.; Zu, F.; Zhai, J.; Qiu, L.; Zhen, Z.; Liu, X.; Shen, Y. The synthesis and properties of novel diazo chromophores based on thiophene conjugating spacers and tricyanofuran acceptors. Dyes Pigments 2008, 77, 657–664. [Google Scholar] [CrossRef]
- Andrade, A.A.; Yamaki, S.B.; Misoguti, L.; Zilio, S.C.; Atvars, T.D.Z.; Oliveira, O.N., Jr.; Mendonça, C.R. Two-photon absorption in diazobenzene compounds. Opt. Mater. 2004, 27, 441–444. [Google Scholar] [CrossRef]
- Vivas, M.G.; Silva, D.L.; De Boni, L.; Bretonniere, Y.; Andraud, C.; Laibe-Darbour, F.; Mulatier, J.-C.; Zales, R.; Bartkowiak, W.; Canuto, S. Experimental and theoretical study on the one-and two-photon absorption properties of novel organic molecules based on phenylacetylene and azoaromatic moieties. J. Phys. Chem. B. 2012, 116, 14677–14688. [Google Scholar] [CrossRef]
- Day, P.N.; Pachter, R.; Nguyen, K.A. Analysis of nonlinear optical properties in donor–acceptor materials. J. Chem. Phys. 2014, 140, 184308. [Google Scholar] [CrossRef]
- Ohta, K.; Antonov, L.; Yamada, S.; Kamada, K. Theoretical study of the two-photon absorption properties of several asymmetrically substituted stilbenoid molecules. J. Chem. Phys. 2007, 127, 84504. [Google Scholar] [CrossRef] [PubMed]
- Krawczyk, P. DFT study of linear and nonlinear optical properties of donor-acceptor substituted stilbenes, azobenzenes and benzilideneanilines. J. Mol. Model. 2010, 16, 659–668. [Google Scholar] [CrossRef]
- Doraghi, F.; Yousefnejad, F.; Farzipour, S.; Aledavoud, S.P.; Larijani, B.; Mahdavi, M. Recent advances in synthesis of stilbene derivatives via cross-coupling reaction. Org. Biomol. Chem. 2023, 21, 1846–1861. [Google Scholar] [CrossRef]
- Zhang, Z.-H.; Peng, S.-Q.; Chi, S.; Chen, H.; Fan, L.; Liu, Y.; Ma, X.; Huang, M.-H. Isolated-alkene-linked porous organic polymers (BIT-POPs): Facile synthesis via ROMP and distinguishing overlapping signals in solid-state 13 C NMR. Polym. Chem. 2021, 12, 6745–6754. [Google Scholar] [CrossRef]
- Jorge, J.; Santos, K.F.D.P.; Timóteo, F.; Vasconcelos, R.R.P.; Cáceres, O.I.A.; Granja, I.J.A.; de Souza, J.; Monteiro, D.; Frizon, T.E.A.; Botteselle, G.D.V. Recent Advances on the Antimicrobial Activities of Schiff Bases and their Metal Complexes: An Updated Overview. Curr. Med. Chem. 2024. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16, Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988, 37, 785. [Google Scholar] [CrossRef]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [Google Scholar] [CrossRef]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cammi, R. Quantum mechanical continuum solvation models. Chem. Rev. 2005, 105, 2999–3094. [Google Scholar] [CrossRef]
- Pielak, K.; Bondu, F.; Sanguinet, L.; Rodriguez, V.; Champagne, B.; Castet, F. Second-order nonlinear optical properties of multiaddressable indolinooxazolidine derivatives: Joint computational and hyper-Rayleigh scattering investigations. J. Phys. Chem. C. 2017, 121, 1851–1860. [Google Scholar] [CrossRef]
- Brasselet, S.; Zyss, J. Multipolar molecules and multipolar fields: Probing and controlling the tensorial nature of nonlinear molecular media. JOSA B. 1998, 15, 257–288. [Google Scholar] [CrossRef]
- Khan, M.U.; Ibrahim, M.; Khalid, M.; Jamil, S.; Al-Saadi, A.A.; Janjua, M.R.S.A. Quantum chemical designing of indolo [3, 2, 1-jk] carbazole-based dyes for highly efficient nonlinear optical properties. Chem. Phys. Lett. 2019, 719, 59–66. [Google Scholar] [CrossRef]
- Mendis, B.A.S.; De Silva, K.M.N. A comprehensive study of linear and non-linear optical properties of novel charge transfer molecular systems. J. Mol. Struct. THEOCHEM 2004, 678, 31–38. [Google Scholar] [CrossRef]
- Vijayakumar, T.; Joe, I.H.; Nair, C.P.R.; Jayakumar, V.S. Efficient π electrons delocalization in prospective push–pull non-linear optical chromophore 4-[N, N-dimethylamino]-4′-nitro stilbene (DANS): A vibrational spectroscopic study. Chem. Phys. 2008, 343, 83–99. [Google Scholar] [CrossRef]
- Sun, M.; Chen, J.; Xu, H. Visualizations of transition dipoles, charge transfer, and electron-hole coherence on electronic state transitions between excited states for two-photon absorption. J. Chem. Phys. 2008, 128, 64106. [Google Scholar] [CrossRef] [PubMed]
- De La Torre, G.; Vázquez, P.; Agullo-Lopez, F.; Torres, T. Role of structural factors in the nonlinear optical properties of phthalocyanines and related compounds. Chem. Rev. 2004, 104, 3723–3750. [Google Scholar] [CrossRef] [PubMed]
- Choi, E.-Y.; Kim, P.-J.; Jazbinsek, M.; Kim, J.-T.; Lee, Y.S.; Gu, P.; Lee, S.W.; Kwon, O.-P. 4-Nitrophenylhydrazone Crystals with Large Quadratic Nonlinear Optical Response by Optimal Molecular Packing. Cryst. Growth Des. 2011, 11, 3049–3055. [Google Scholar] [CrossRef]
- Lou, A.J.-T.; Marks, T.J. A twist on nonlinear optics: Understanding the unique response of π-twisted chromophores. Acc. Chem. Res. 2019, 52, 1428–1438. [Google Scholar] [CrossRef]
- Oudar, J.L.; Zyss, J. Structural dependence of nonlinear-optical properties of methyl-(2, 4-dinitrophenyl)-aminopropanoate crystals. Phys. Rev. A. 1982, 26, 2016. [Google Scholar] [CrossRef]
- Sciuti, L.F.; Abegão, L.M.G.; Santos, C.H.D.D.; Cocca, L.H.Z.; da Costa, R.G.M.; Limberger, J.; Misoguti, L.; Mendonça, C.R.; De Boni, L. Modeling the First-Order Molecular Hyperpolarizability Dispersion from Experimentally Obtained One-and Two-Photon Absorption. J. Phys. Chem. A. 2022, 126, 2152–2159. [Google Scholar] [CrossRef]
- Rasool, F.; Hussain, A.; Yar, M.; Ayub, K.; Sajid, M.; Asif, H.M.; Imran, M.; Assiri, M.A. Nonlinear optical response of 9, 10-bis (phenylethynyl) anthracene mediated by electron donating and electron withdrawing substituents: A density functional theory approach. Mater. Sci. Semicond. Process. 2022, 148, 106751. [Google Scholar] [CrossRef]
- Orr, B.J.; Ward, J.F. Perturbation theory of the non-linear optical polarization of an isolated system. Mol. Phys. 1971, 20, 513–526. [Google Scholar] [CrossRef]
- Santos, F.A.; Abegão, L.M.G.; Fonseca, R.D.; Alcântara, A.M.; Mendonça, C.R.; Valle, M.S.; Alencar, M.; Kamada, K.; De Boni, L.; Rodrigues, J.J., Jr. Bromo-and chloro-derivatives of dibenzylideneacetone: Experimental and theoretical study of the first molecular hyperpolarizability and two-photon absorption. J. Photochem. Photobiol. A Chem. 2019, 369, 70–76. [Google Scholar] [CrossRef]
- Day, P.N.; Nguyen, K.A.; Pachter, R. Calculation of two-photon absorption spectra of donor-π-acceptor compounds in solution using quadratic response time-dependent density functional theory. J. Chem. Phys. 2006, 125, 94103. [Google Scholar] [CrossRef] [PubMed]
- Pal, A.K.; Duignan, T.J.; Autschbach, J. Calculation of linear and nonlinear optical properties of azobenzene derivatives with Kohn–Sham and coupled-cluster methods. Phys. Chem. Chem. Phys. 2018, 20, 7303–7316. [Google Scholar] [CrossRef] [PubMed]
- Patil, P.S.; Dharmaprakash, S.M.; Ramakrishna, K.; Fun, H.-K.; Kumar, R.S.S.; Rao, D.N. Second harmonic generation and crystal growth of new chalcone derivatives. J. Cryst. Growth 2007, 303, 520–524. [Google Scholar] [CrossRef]
- Azhar, S.M.; Anis, M.; Rabbani, G.; Shirsat, M.D.; Baig, M.I.; Hussaini, S.S.; AlFaify, S.; Khan, M.A. Growth of NH4H2PO4 crystal in urea environment to optimize linear-nonlinear optical traits for photonic device applications. Optik 2019, 185, 1247–1252. [Google Scholar] [CrossRef]
- Abegão, L.M.G.; Fonseca, R.D.; Santos, F.A.; Souza, G.B.; Barreiros, A.L.B.S.; Barreiros, M.L.; Alencar, M.; Mendonça, C.R.; Silva, D.L.; De Boni, L. Second-and third-order nonlinear optical properties of unsubstituted and mono-substituted chalcones. Chem. Phys. Lett. 2016, 648, 91–96. [Google Scholar] [CrossRef]
- Abegão, L.M.G.; Santos, F.A.; Fonseca, R.D.; Barreiros, A.L.B.S.; Barreiros, M.L.; Alves, P.B.; Costa, E.V.; Souza, G.B.; Alencar, M.A.R.C.; Mendonça, C.R. Chalcone-based molecules: Experimental and theoretical studies on the two-photon absorption and molecular first hyperpolarizability. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 227, 117772. [Google Scholar] [CrossRef]

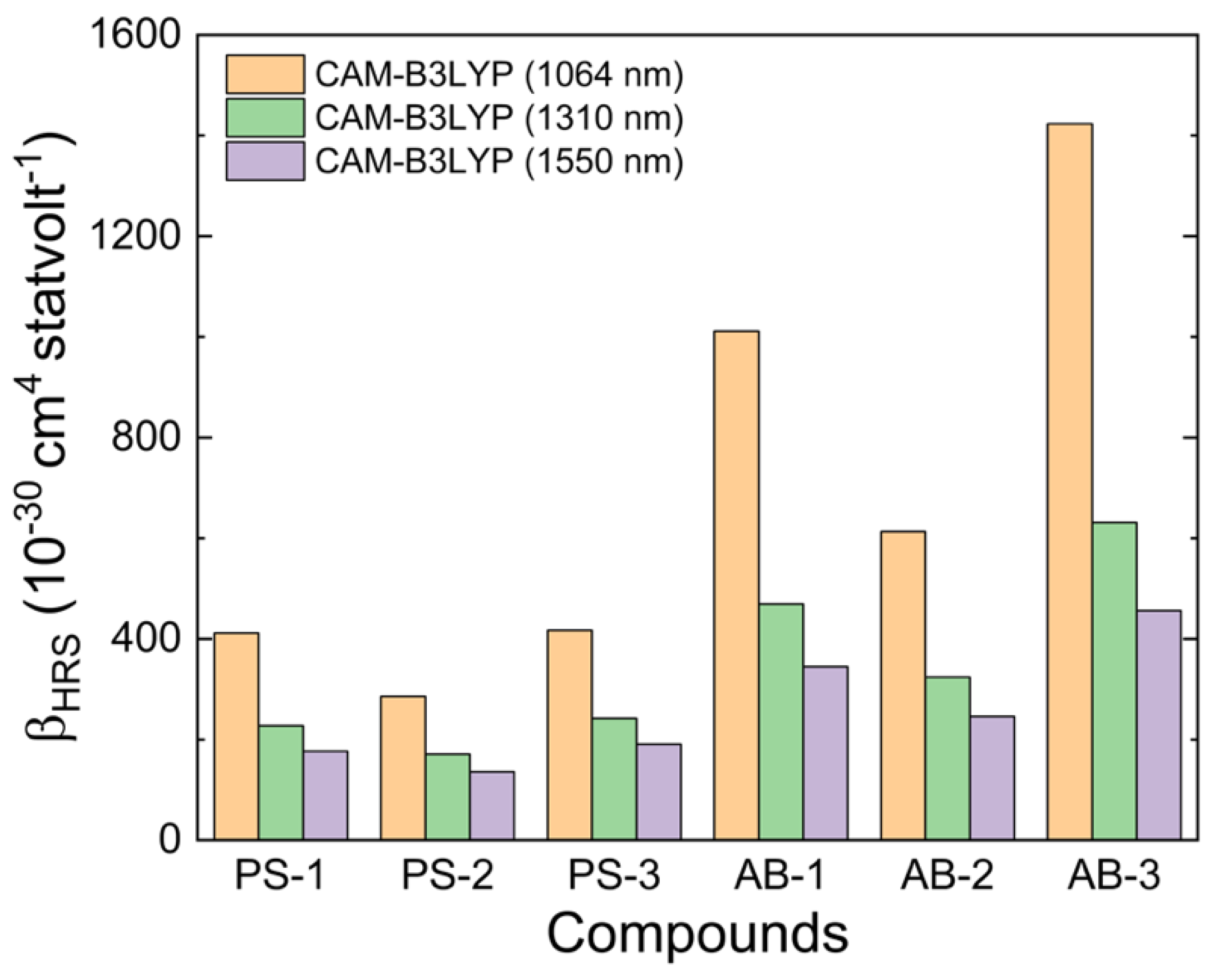

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Bond Length (Ǻ) | Bond Angle (Ǻ) | Torsion Angle (°) | ||||
|---|---|---|---|---|---|---|---|
| System Type I | PS-1 | C3-N1 | 1.418 | C3-N1-N2 | 114.59 | C3-N1-N2-C9 | 179.99 |
| N1-N2 | 1.250 | N1-N2-C9 | 116.24 | O17-N15-C6-C5 | −179.99 | ||
| N2-C9 | 1.392 | C6-N15-O17 | 117.57 | C20-N18-C12-C11 | −179.98 | ||
| C6-N15 | 1.461 | C6-N15-O16 | 117.66 | N2-C9-C10-C12 | −179.99 | ||
| C12-N18 | 1.372 | C12-N18-C19 | 124.19 | N1-N2-C10-C9 | −0.001 | ||
| N18-C19 | 1.459 | C12-N18-C20 | 123.93 | N1-N2-C3-C4 | −0.001 | ||
| N18-C20 | 1.457 | C5-C6-N15 | 118.95 | C12-C11-C10-N18 | −179.99 | ||
| N3-O1 | 1.217 | C11-C12-N18 | 121.63 | C6-C5-C4-N15 | 180.00 | ||
| N3-O2 | 1.217 | C12-C11-C10 | 120.56 | C5-C4-C6-N1 | 180.00 | ||
| PS-2 | C15-N3 | 1.268 | C3-C1-N2 | 122.04 | C9-N2-C1-C3 | 178.07 | |
| C14-C15 | 1.469 | C9-N2-C1 | 121.07 | C5-C6-N15-O17 | 179.97 | ||
| N3-C8 | 1.405 | C6-N15-O16 | 117.70 | C20-N18-C12-C11 | −169.59 | ||
| C5-N1 | 1.375 | C6-N15-O17 | 117.72 | N15-C6-C5-C4 | 179.95 | ||
| C11-N2 | 1.461 | C19-N18-C12 | 119.05 | O17-N15-C5-C4 | 178.79 | ||
| N1-C2 | 1.449 | C20-N18-C12 | 119.05 | C4-C3-C1-N2 | −11.62 | ||
| N1-C16 | 1.450 | N15-C6-C8 | 118.97 | O16-N15-C6-C5 | −0.02 | ||
| N2-O1 | 1.220 | N18-C12-C11 | 121.35 | N18-C12-C11-C10 | 178.80 | ||
| N2-O2 | 1.220 | C12-C11-C10 | 121.23 | C11-C10-C9-C2 | 179.72 | ||
| PS-3 | C1-C2 | 1.335 | C1-C2-C3 | 126.27 | C1-C2-C3-C9 | 179.96 | |
| C1-C3 | 1.460 | C1-C2-C9 | 127.36 | C5-C6-N15-O17 | 179.90 | ||
| C2-C9 | 1.457 | C6-N15-O16 | 117.80 | C20-N18-C12-C11 | −169.67 | ||
| C12-N18 | 1.376 | C6-N15-O17 | 117.82 | C6-C5-C4-C3 | −179.82 | ||
| N18-C19 | 1.446 | C12-N18-C19 | 119.43 | C1-C2-C3-C4 | −0.21 | ||
| N18-C16 | 1.446 | C12-N18-C20 | 119.25 | C19- N18-C12-C11 | 11.08 | ||
| N15-C6 | 1.461 | C11-C12-N18 | 121.37 | C5-C6-N15-O16 | −0.12 | ||
| N15-O17 | 1.218 | C4-C3-C1 | 123.46 | C6-C5-C4-N15 | −179.91 | ||
| N15-O18 | 1.218 | C2-C9-C10 | 123.95 | C10-C11-C12-N18 | 178.46 | ||
| System Type II | AB-1 | C3-N1 | 1.421 | C3-N1-N2 | 114.75 | C3-N1-N2-C9 | 179.99 |
| N1-N2 | 1.240 | N1-N2-C9 | 115.70 | C6-C5-N23-O25 | 179.99 | ||
| N2-C7 | 1.413 | C12-N15-N16 | 114.82 | C12-N15-N16-C17 | −180.00 | ||
| C9-N3 | 1.416 | N15-N16-C17 | 116.09 | N26-C20-C19-C18 | 179.08 | ||
| N15-N16 | 1.244 | C27-N26-C20 | 121.07 | C28-N26-C20-C19 | −173.93 | ||
| N15-C12 | 1.417 | C28-N26-C20 | 119.83 | N1-N2-C9-C10 | −0.04 | ||
| N16-C13 | 1.402 | C6-N23-O24 | 117.67 | N15-N16-C17-C18 | −0.37 | ||
| C20-N26 | 1.369 | C6-N23-O25 | 117.68 | C5-C4-C3-N1 | −179.99 | ||
| N26-C27 | 1.447 | C6-C5-N23 | 118.85 | C11-C10-C9-N2 | 179.99 | ||
| N26-C28 | 1.446 | C22-C21-C20 | 120.60 | N15-C12-C11-C10 | 179.99 | ||
| C6-N23 | 1.468 | C6-C5-C5 | 118.95 | C27-N26-C20-C19 | −6.73 | ||
| N23-O24 | 1.216 | C4-C5-C6-N23 | −179.99 | ||||
| N23-O25 | 1.216 | ||||||
| AB-2 | C3-N1 | 1.421 | C3-N1-N2 | 114.89 | C3-N1-N2-C9 | 179.60 | |
| N1-N2 | 1.239 | N1-N2-C9 | 115.72 | C12-C15-N16-C17 | −178.13 | ||
| N2-C9 | 1.416 | C12-C15-N16 | 122.52 | C25-N23-C19-C20 | 166.35 | ||
| C12-C15 | 1.467 | C17-N15-C1 | 120.67 | N23-C20-C19-C18 | 177.52 | ||
| C15-N16 | 1.268 | C20-N23-C25 | 119.03 | C19-C18-C17-N16 | 179.40 | ||
| N16-C17 | 1.406 | C20-N23-C24 | 118.95 | N1-N2-C9-C10 | −0.72 | ||
| C20-N23 | 1.378 | C6-N23-O28 | 118.12 | C18-C17-N16-C15 | −29.74 | ||
| N23-C25 | 1.449 | C5-C6-N23 | 118.78 | C25-N23-C20-C19 | 12.36 | ||
| N23-C24 | 1.450 | C19-C20-N23 | 121.51 | C5-C6-N26-O27 | 0.20 | ||
| C6-N26 | 1.463 | C5-C6-N26-O28 | −179.98 | ||||
| N26-O28 | 1.219 | C11-C12-C15-N16 | −178.88 | ||||
| N26-O27 | 1.219 | ||||||
| AB-3 | C3-N1 | 1.420 | C3-N1-N2 | 114.83 | C3-N1-N2-C9 | −179.96 | |
| N1-N2 | 1.242 | N1-N2-C9 | 116.06 | C12-C15-C16-C17 | 179.98 | ||
| N2-C9 | 1.409 | C12-C15-C16 | 126.23 | C24-N23-C20-C19 | −169.43 | ||
| C12-C15 | 1.460 | C15-C16-C17 | 127.24 | C25-N23-C20-C19 | −11.30 | ||
| C15-C16 | 1.338 | C24-N23-C24 | 119.33 | N23-C20-C19-C18 | −178.83 | ||
| C16-C17 | 1.457 | C24-N23-C25 | 119.50 | C5-C6-N26-O27 | 0.03 | ||
| C20-N23 | 1.373 | C6-N26-O27 | 118.15 | C5-C6-N26-O28 | −179.96 | ||
| N23-C25 | 1.449 | C6-N26-O28 | 118.16 | C4-C5-C6-N26 | 180.00 | ||
| N23-C24 | 1.450 | C19-C20-N23 | 121.41 | C16-C15-C12-C11 | −179.92 | ||
| N26-C6 | 1462 | C5-C6-N26 | 118.83 | C11-C10-C12-N2 | −179.98 | ||
| N26-O27 | 1.219 | C20-C19-C18 | 121.28 | C19-C18-C17-C16 | −179.96 | ||
| N4-O28 | 1.219 | C4-C5-C6 | 118.91 | ||||
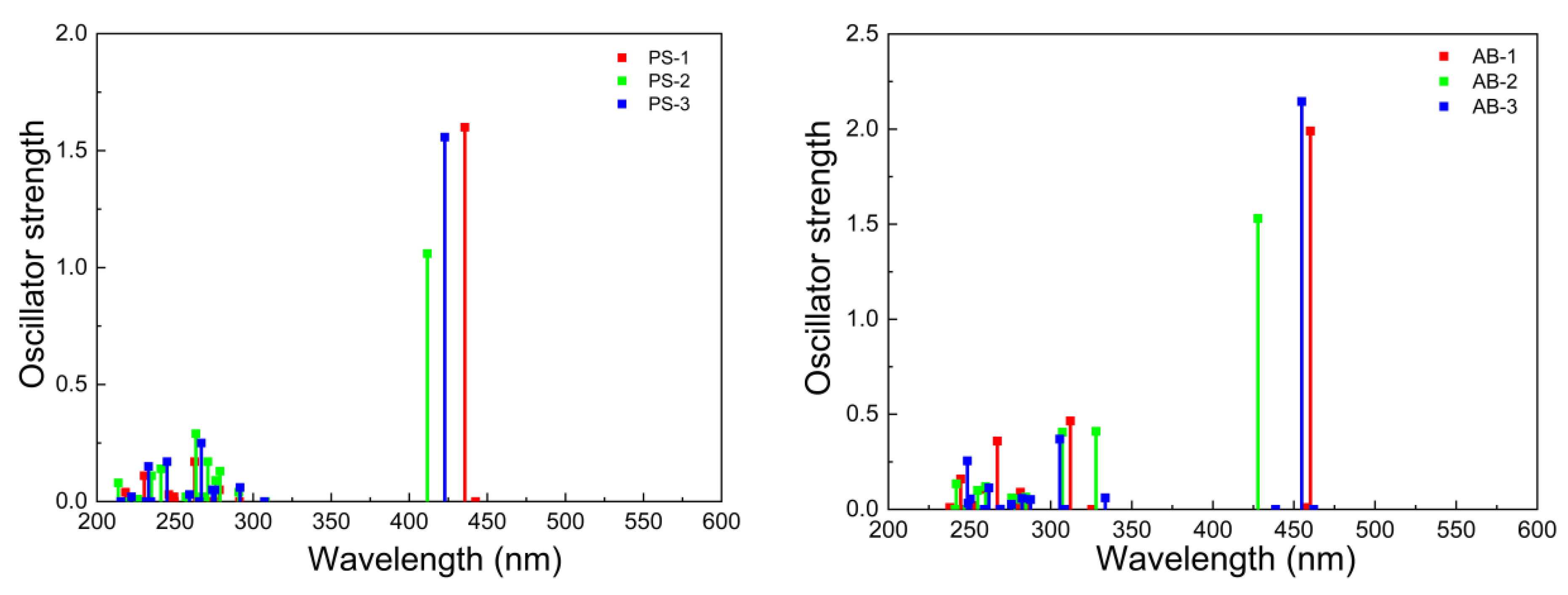
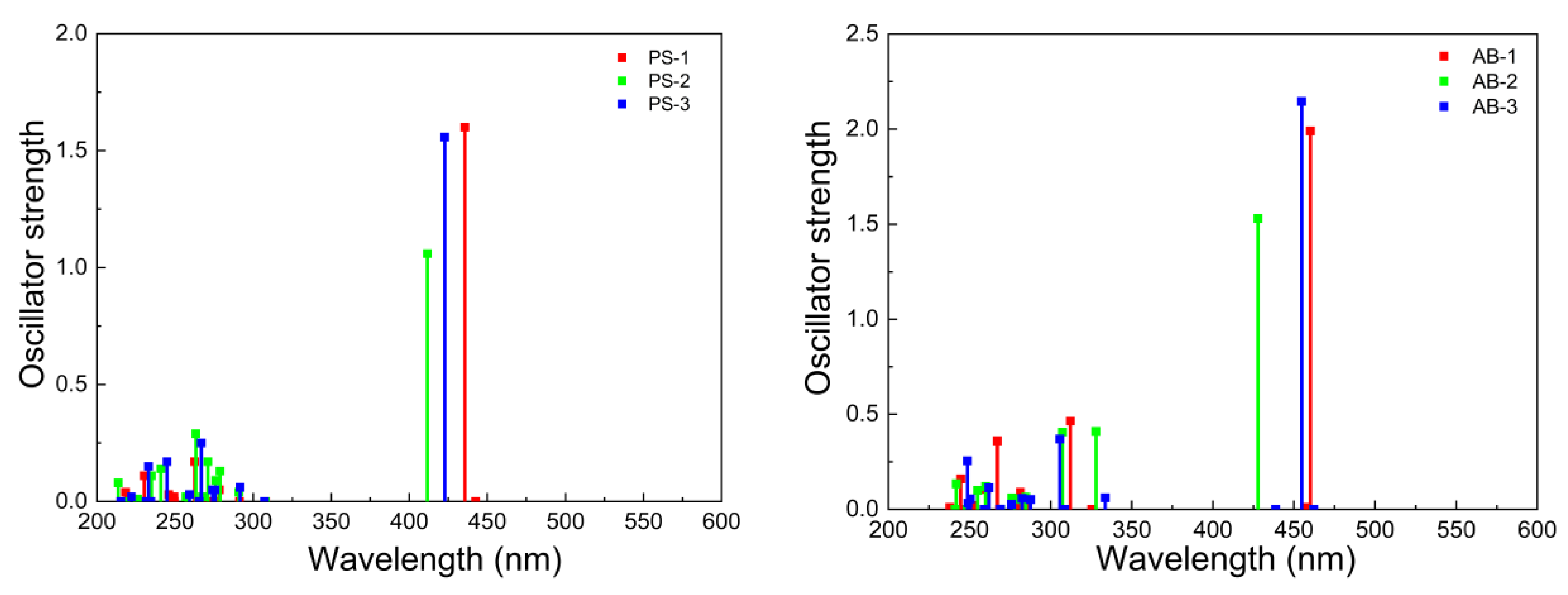
| Compounds | [eV] {Transition} | µ (D) | ||
|---|---|---|---|---|
| System Type I | PS-1 | 442 [2.80] {S0-S1} 436 [2.84] {S0-S2} | 0.00 1.61 | 13.86 |
| PS-2 | 412 [3.01] {S0-S1} | 1.06 | 10.52 | |
| PS-3 | 423 [2.93] {S0-S1} | 1.56 | 12.56 | |
| System Type II | AB-1 | 462 [2.68] {S0-S1} 455 [2.73] {S0-S2} | 0.00 2.15 | 14.03 |
| AB-2 | 462 [2.68] {S0-S1} 428 [2.90] {S0-S2} | 0.00 1.53 | 10.62 | |
| AB-3 | 460 [2.69] {S0-S1} | 1.99 | 13.23 | |
| Level of Theory | in a Vacuum | in DMSO | ||||
|---|---|---|---|---|---|---|
| PS-1 | PS-2 | PS-3 | PS-1 | PS-2 | PS-3 | |
| B3LYP | 73.2 a (144.30) b | 103.4 a (165.0) b | 95.9 a (152.1) b | 356.0 a (626.4) a | 434.6 a (938.5) b | 463.7 a (927.6) b |
| CAM-B3LYP | 50.0 a (74.6) b | 42.5 a (74.7) b | 50.5 (86.5) b | 181.5 a (113.2) b | 127.6 a (112.4) b | 187.4 a (123.4) b |
| Compounds | Center (-X=Y-) Torsion Angle (°) | NMe2 Torsion Angle (°) | µ (D) | ||
|---|---|---|---|---|---|
| System Type I | PS-1 | 179.99 | 179.98 | 13.86 | 411.6 |
| PS-2 | 178.07 | 169.59 | 10.52 | 285.5 | |
| PS-3 | 179.96 | 169.67 | 12.56 | 417.0 | |
| System Type II | AB-1 | 179.99 | 179.08 | 14.03 | 1011.3 |
| AB-2 | 179.60 | 166.35 | 10.62 | 613.1 | |
| AB-3 | 179.96 | 169.43 | 13.23 | 1423.5 | |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Araújo, R.S.; Rodrigues, J.J., Jr.; Alencar, M.A.R.C.; Rafique, J.; Saba, S.; Abegão, L.M.G. Pseudo-Stilbene- and Azobenzene-Type Systems for Optical Frequency Conversion: Estimating the First-Order Molecular Hyperpolarizability. Photonics 2024, 11, 283. https://doi.org/10.3390/photonics11030283
Araújo RS, Rodrigues JJ Jr., Alencar MARC, Rafique J, Saba S, Abegão LMG. Pseudo-Stilbene- and Azobenzene-Type Systems for Optical Frequency Conversion: Estimating the First-Order Molecular Hyperpolarizability. Photonics. 2024; 11(3):283. https://doi.org/10.3390/photonics11030283
Chicago/Turabian StyleAraújo, Raiane S., José J. Rodrigues, Jr., Márcio A. R. C. Alencar, Jamal Rafique, Sumbal Saba, and Luis M. G. Abegão. 2024. "Pseudo-Stilbene- and Azobenzene-Type Systems for Optical Frequency Conversion: Estimating the First-Order Molecular Hyperpolarizability" Photonics 11, no. 3: 283. https://doi.org/10.3390/photonics11030283
APA StyleAraújo, R. S., Rodrigues, J. J., Jr., Alencar, M. A. R. C., Rafique, J., Saba, S., & Abegão, L. M. G. (2024). Pseudo-Stilbene- and Azobenzene-Type Systems for Optical Frequency Conversion: Estimating the First-Order Molecular Hyperpolarizability. Photonics, 11(3), 283. https://doi.org/10.3390/photonics11030283