Abstract
Experimental nonlinear optics (NLO) is usually expensive due to the high-end photonics and electronic devices needed to perform experiments such as incoherent second harmonic generation in liquid phase, multi-photon absorption, and excitation. Nevertheless, exploring NLO responses of organic and inorganic compounds has already opened a world of new possibilities. For example, NLO switches, NLO frequency converters, and a new way to obtain biological images through the incoherent second harmonic generation (SHG) originate from first-order molecular hyperpolarizability (β). The microscopic effect of the coherent or incoherent SHG is, in fact, the β. Therefore, estimating β without using expensive photonic facilities will optimize time- and cost-efficiency to predict if a specific molecular structure can generate light with double its incident frequency. In this work, we have simulated the β values of 27 organic compounds applying density functional theory (PBE0, TPSSh, wB97XD, B3LYP, CAM-B3LYP, and M06-2X) and Hartree–Fock methods using the Gaussian software package. The predicted β was compared with the experimental analogs obtained by the well-known Hyper–Rayleigh Scattering (HRS) technique. The most reliable functionals were CAM-B3LYP and M06-2X, with an unsigned average error of around 25%. Moreover, we have developed post-processing software—Hyper-QCC, providing an effortless, fast, and reliable way to analyze the Gaussian output files.
1. Introduction
Nonlinear optics (NLO) requires comprehensive and expensive pieces of equipment, such as high-sensitivity—optoelectronics, and of course, coherent, monochromatic, and high-intensity light sources—LASERS. For example, a fully operational and complete infrastructure to determine the second- and third-order NLO phenomena, such as second harmonic generation (SHG) and two-photon absorption (TPA) with ultra-short pulses, should apport an investment of around a half-million dollars. Despite this, the first reported experiment that confirmed the SHG of light [1] of a quartz crystal used a ruby laser, a few optomechanical parts, and a photographic plate, which cost only a fraction of the investment mentioned above. However, a bulk compound is not always available to determine the SHG. The compounds are usually available in their raw state, e.g., powder-synthesized materials to be dissolved in a solvent medium. Therefore, if one is interested in finding the potential of raw materials to be used for optical frequency conversion, the first-order molecular hyperpolarizability in solution must be quantified. For example, suppose a solute–solvent medium is able to generate SHG. In that case, it should be called an incoherent second harmonic generation (ISHG) phenomenon because of the non-directional emission of light. The ISHG can be quantified by evaluating the dynamic first-order molecular hyperpolarizability (βHRS), wherein the HRS in the subscript is due to the Hyper–Rayleigh Scattering (HRS) [2] technique. This technique requires a sophisticated photonics facility with the above investment value, mainly if ultra-short pulses are employed.
The massive number of yearly publications on organic and organometallic molecular structures [3,4,5,6,7,8,9,10,11,12] indicates the existence of thousands of new materials available to be characterized with NLO experiments. As mentioned before, NLO experiments are expensive, unfortunately, making them undoable for many research groups. Still, there is an inexpensive solution to estimate the βHRS both in gas and liquid phase. Such a solution is based on quantum chemical calculations (QCC), which require only computational resources, comprising a good equilibrium between hardware and software. Until now, Moore’s law has correctly predicted that a central processing unit’s (CPU) processing power will double every two years [13]. The consequence of Moore’s law was the commercialization of powerful CPUs with accessible prices for the public in general, allowing easy access to a particular type of simulation in physics. Therefore, combining the Gaussian software package [14] with powerful CPUs to perform QCC is a valid option to predict the first-order molecular hyperpolarizability [15,16,17] without the need for an expensive photonics facility. One of the significant advantages of using computational simulations instead of conducting experiments to estimate NLO properties is that besides using the already-existent synthesized compound, its novel derivatives, not yet synthesized, can also be investigated. Therefore, such a procedure will benefit chemists in optimizing the cost associated with the production of new materials, particularly those used in photonic devices and techniques.
The investment level needed to run QCC to estimate the first-order molecular hyperpolarizability of organic molecules depends mainly on the molecule’s size and how fast the QCC need to be acquired. For example, in previous works, we used a desktop (nonserver) system with 8 cores and 16 GB of RAM to calculate molecules with less than 50 atoms [7,18,19]. However, to perform QCC in large molecules (~100 atoms), a minimum setup of 32 cores and 128 GB RAM will be recommended. In addition, the Gaussian software package is also an investment that must be considered. Nevertheless, the investment to predict first-order molecular hyperpolarizability is a fraction of the necessary investment to acquire the experimental values. Still, the reliability of theoretical results will depend on the method used in the calculations, as one will verify in this work.
Due to the evolution of optical communication networks, the search and development of novel compounds with NLO performance have significant importance in improving this field [8,20,21]. For example, frequency optical converters are one of the main components of optical multiplexing [22,23,24]. Another area taking advantage of the optical frequency conversion is the biological field, in which incoherent SHG and third harmonic generation (THG) are being used to improve imaging diagnostics [25,26,27,28]. Therefore, the βHRS estimation is essential to discover and improve raw materials with the potential to be used in optical communications or biological fields. As mentioned before, the Gaussian software package and the hardware type will improve the readiness of the QCC. Still, to compute the final value of the first-order molecular hyperpolarizability in a solvent medium, one must consider the components of the first-order tensor retrieved by the Gaussian software. Therefore, post-processing calculations will improve the time efficiency and the error-free association with the βHRS final result. Moreover, if a βHRS spectrum is necessary to understand better the βHRS magnitude behavior as a function of the wavelength, post-processing software will drastically improve the final analysis readiness.
In this work, we have used QCC via the Gaussian software package to predict βHRS values of 27 compounds in solvent media from different organic families. The equivalent βHRS tensor components, third-order rank tensors, were obtained by density functional theory [29] and Hartree–Fock [30] methods. The theoretical results were compared with their experimental analogs, retrieved from the literature, using the HRS technique. Such a comparison allows us to determine the most reliable level of theory to predict the βHRS values for these types of molecular structures. Moreover, we have developed and introduced a homemade post-processing software developed in Python—Hyper-QCC, that analyzes the output files from the QCC employed with the Gaussian software in a reliable and fast way. Finally, we wish to stress that Hyper-QCC alone does not perform QCC, but it can help in achieving the final βHRS values in an effortless, fast, and reliable way.
2. Materials and Methods
2.1. Investigated Compounds
All the samples investigated in this work have experimental βHRS values reported in the literature. For a better reader experience, the 27 compounds were designated as Ai, Bi, and Ci, with “i” varying from 1 to 9. The A compounds, which have the lowest experimental βHRS (<30 × 10−30 cm4 statVolt−1) reported are A1 (dibenzoylmethane) [31]; A2 (nitrobenzyl pyridine derivative) [32]; A3 and A4 (s-aniline derivatives) [33]; A5 and A6 (diethylamino derivatives) [34]; and A7, A8, and A9 (indolinooxazolidine derivatives) [35]. The B compounds have the second-highest experimental βHRS (>30 and <80 × 10−30 cm4 statVolt−1) reported: B1 and B2 (imidazole derivatives); B3 (triazine derivative) [33]; B4, B5, B6, and B7 (dibenzylideneacetone derivatives) [36,37]; B8 (oxazoles derivative) [38]; and B9 (chalcones derivative) [39]. Finally, the C compounds are the ones with the highest experimental βHRS (>80 × 10−30 cm4 statVolt−1) values: C1, C3, and C5 (dimethylamino derivatives); C2 (sulfone phthalein); C4 (diethylamino derivative) [34]; C6 (azulenic-barbituric) [40]; C7 (nitrobenzyl pyridine derivative) [32]; and C8 and C9 (thiophene-incorporated polyene) [41]. All molecular structures mentioned above are depicted in Figure 1.
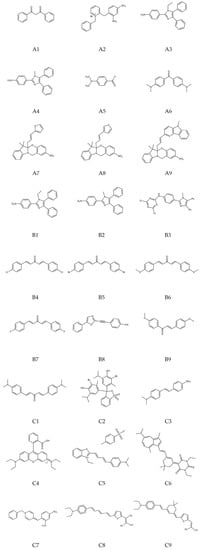
Figure 1.
The first-order molecular hyperpolarizability values have been calculated for all depicted molecular structures.
2.2. First-Order Hyperpolarizability and Quantum Chemical Calculations
As previously mentioned, the HRS is one of the experimental techniques used to measure the intensity of the incoherently scattered frequency-doubled light generated after the laser beam interaction with a chromophore in an isotropic solution. The relation between scattered optical intensity at double frequency (I2ω) and the optical intensity of the incident beam (Iω) is given by Equation (1), in which G is a parameter that includes experimental factors such as the scattering geometry, local field factors at ω and 2ω, and photon collection efficiency [42]. N represents the molar concentration of the used samples, while is the orientational average first molecular electronic hyperpolarizability squared. The subscripts s and c refer to the solvent and chromophore, respectively.
Although, in this work, the βHRS values were determined theoretically, the reader needs to understand, in general terms, how to experimentally calculate βHRS once the concept of the orientational average is essential to achieve the full result of the estimated βHRS via QCC. Thus, let us consider a plane-polarized incident light beam at ω frequency, while the observation of the 2ω frequency is made perpendicular to the propagation plane. The full HRS intensity could be described by Equation (2), in which and correspond to the orientationally averaged tensor components.
In order to theoretically estimate βHRS, one should consider the relationship between the average molecular positions and the molecular first-order hyperpolarizability tensor components without assuming Kleinman’s conditions of symmetry [38,43,44]. Such a relationship is given by Equations (3) and (4), in which the “ZZZ” and “ZXX” are the so-called laboratory coordinates, and the “x”,”y”, and “z” are the molecular coordinates. The indices ζ, η, and ξ depict the molecular frame Cartesian axes x, y, and z.
The βHRS tensor (third-order rank tensor) can be calculated using two formalisms: Cartesian [43] and mixed spherical–Cartesian [42]. The most significant difference between the formalisms usually appears when calculating the βHRS of compounds with low-symmetry molecular structures, as reported in the literature [45]. For example, for low-symmetry molecular structures, it is more accurate to calculate βHRS using mixed spherical–Cartesian [15]. In addition, one of the drawbacks of Cartesian formalism is related to the arbitrary orientation of the Cartesian axes invoked for theoretical calculations, which could be a source of misfits with experimental data, leading to less accurate predicted results [19,42].
The Gaussian 16 software package [14] provides all tensor components, which must be carefully introduced in the above equations to correctly obtain the full HRS intensity, described by Equation (2). In this work, we have used the rotational invariance concept combined with the mixed spherical–Cartesian formalism [15] to estimate the βHRS once we have a mix of high and low symmetry of the investigated molecular structures. In this type of formalism, the orientationally averaged first-order hyperpolarizability is expressed by Equation (5), in which the molecular βHRS tensor is decomposed as the sum of a dipolar (J = 1) and an octupolar (J = 3) tensorial form. The relationships between the dipolar and octupolar components and the Cartesian components of βHRS are described in Equations (6) and (7). Therefore, the post-processing software (Hyper-QCC) we developed, which will be freely distributed, will use the mixed spherical–Cartesian formalism to estimate the final theoretical values for the static and dynamic first-order hyperpolarizabilities.
The tensor components were obtained through QCC performed using the density functional theory (DFT) [29] method and Hartree–Fock (HF) [30] method, totalizing seven functionals: B3LYP, CAM-B3LYP, M06-2X, PBE0, TPSSh, wB97XD, and HF. All functionals were combined with a triple-zeta split valence Pople basis set [46] with two additional polarization functions for non-hydrogen atoms, two additional polarization functions on hydrogen atoms, and diffuse functions on hydrogen and non-hydrogen atoms: 6-311++G(2d,2p).
The well-known exchange–correlation functional Becke, three-parameter, Lee–Yang–Parr (B3LYP) [47] is, as far as we know, one of the most-used hybrid density functional in the literature. Vosko, Wilk, and Nusair originally implemented it, and it was later refined by Becke in 1993. The main reason for its wide use is associated with its accuracy in predicting the electronic structure of a wide range of molecular systems. However, its long-range corrected version, which includes the Coulomb-attenuating method (CAM-B3LYP) [48], released in 2004 by Yanai, Tew, and Handy, revealed a better accuracy, particularly in predicting optical spectroscopic parameters, such as first-order molecular hyperpolarizabilities [16,17,19,49]. Later, in 2006 a variation of a hybrid functional by Truhlar and Zhao (M06-2X) [50] was released. M06-2X has not been widely used compared with the two previously mentioned functionals, but it has been revealed to be one of the most suitable functionals for studying non-covalent interactions. CAM-B3LYP and M06-2X have been considered the two most suitable functionals to calculate spectroscopic parameters related to linear and nonlinear optical spectroscopy, such as one- and two-photon absorption [51,52,53] and first- and second-hyperpolarizabilities [54,55,56,57].
The combination of the Perdew–Burke–Ernzerhof (PBE) exchange–correlation functional [58] with a fraction of the exact Hartree–Fock (HF) exchange allowed Adamo and Barone to develop the extension of PBE, designated as PBE0 [59]. While released nearly simultaneously with B3LYP in 1996, PBE0 is not as commonly applied as B3LYP but is still a mainstay of DFT functionals. As such, PBE0 has certainly been and can be employed for the computation of static polarizabilities and hyperpolarizabilities [60,61,62,63]. Henceforth, the PBE0 functional will be designated as PBE1PBE since the Gaussian keyword to run the PBE0 functional is PBE1PBE, as already mentioned elsewhere [64].
The extension of the Tao–Perdew–Staroverov–Scuseria (TPSS) functional is designated as TPSSh [65] and is also an exchange–correlation functional. TPSSh is one of the most recent functionals used in this work, released in 2008, and is known to improve the accuracy of the method for describing electronic properties such as ionization potentials and charge transfer excitations. TPSSh uses a fraction of the exact exchange from the PBE functional, and few theoretical works have used this method to calculate hyperpolarizabilities [66]. Still, to the best of our knowledge, we did not find any comparison with the experimental dynamic first-order molecular hyperpolarizability. Therefore, in this work, we will have the opportunity to compare the theoretical values obtained with TPSSh with the experimental ones.
wB97XD is a dispersion-corrected density functional theory [67,68], an extension of B97, and a hybrid density functional developed by Adrian Becke [69], released in 2008. The wB97XD functional includes a dispersion correction to account for the long-range van der Waals interactions between molecules, with an empirical scaling factor to improve the method’s accuracy. Therefore, wB97XD is considered a functional suitable for predicting properties related to non-covalent interactions, such as molecular binding energies and intermolecular distances [70,71,72]. In addition, some works in the literature use this functional to predict linear and nonlinear optical properties [73,74,75,76]. However, to the best of our knowledge, there is no direct comparison with experimental results with dynamic first-order molecular hyperpolarizabilities.
Because all the previously described functionals use the DFT method with an exact exchange of the Hartree–Fock (HF) method, we have also decided to include HF in our calculations. Since the primordial years of QCC, HF has been widely used in several fields. Even though it has several limitations, e.g., not taking into account the effects of electron correlation, HF is still used to predict optical spectroscopic parameters [77,78,79,80]. Neglecting the effects of electron correlation could be a drawback regarding reliability, but it is an advantage in terms of computational cost when compared with hybrid functionals.
The computational methodology consists of two steps: the molecular structure geometry optimization and computing the tensor components of the first-order molecular hyperpolarizability. The first computational step is the geometry optimization calculation in solvent medium. All Cartesian coordinates obtained with the geometry optimization can be found in the Supplementary Materials (SM) in Table S1. The solvation effect was obtained by employing a polarizable continuum model (PCM) using the integral equation formalism variant (IEF-PCM) [81]. The level of theory CAM-B3LYP was the only method used to calculate the energy minima of all compounds. The second step is to calculate the static (ω = 0 a.u.) and dynamic (ω ≠ 0 a.u.) tensor components, i.e., βζηξ. The Gaussian output files provided in the second step are post-processed by Hyper-QCC to achieve β0 and βHRS. All static and dynamic first-order molecular hyperpolarizability values in solvent medium are available in the SM, Tables S2–S5.
Regarding the solvent media, we would like to emphasize that the objective of the presented work is to compare the experimental first-order molecular hyperpolarizabilities with the theoretical ones. Thus, the solvents used in IEF-PCM were the ones used in the Hyper–Rayleigh scattering technique: ethanol, dioxane, dichloromethane, toluene, chloroform, and acetonitrile. In addition, we would like to point out that compound C5 comprises a cationic-conjugated molecule and anionic counterion. In this particular case, we have calculated C5 in both forms, i.e., the cationic-conjugated molecule with its counterion and the cationic-conjugated molecule separated from its counterion. Moreover, there are countless possibilities for the geometry starting point. In Section 4 of the SM, one can find detailed information regarding the C5 results in both forms and the strategy used to define the geometry starting point.
To understand how accurate the functionals used in this work are to predict the βHRS, firstly, we calculated the relative error (RE) for each compound, expressed by Equation (8), in which and are the theoretical and experimental first-order molecular hyperpolarizabilities, respectively. Secondly, we calculated the mean error for each functional considering the RE for each compound, expressed by Equation (9) and henceforth designated as unsigned average error (UAE), in which n is the total number of compounds. The smaller the UAE, the higher the accuracy performance of a specific functional for the investigated compounds in this work.
2.3. Hyper-QCC: Post-Processing Software
Once again, we would like to emphasize that the post-processing software—Hyper-QCC, developed and used in this work, requires the Gaussian software package to calculate the components’ tensor. Therefore, only after the methodology described in the previous section, Hyper-QCC can be used to achieve the final values of the static and dynamic first-order molecular hyperpolarizability. The main objective of using Hyper-QCC is to process the components of the second-order tensor from the first-order molecular hyperpolarizability obtained with the Gaussian program package in an effortless, fast, and reliable way.
Hyper-QCC was fully implemented using Python due to its ease when converting to multiple operating systems. The framework also retains several native libraries for graphical user interface (GUI) building and software compilation support. The GUI library chosen was PyQt6, and Pyinstaller was used as the compiler. Hyper-QCC will be available for MacOS and Windows users in its first version. Hyper-QCC was built to calculate the final values of β0 and βHRS without complexity, requiring only a few technical parameters from the user. The tensor components are calculated in the background, and the results are displayed in seconds. A simplified diagram of interactions required when using Hyper-QCC is shown in Figure 2, while an exemplification on how to use the Hyper-QCC can be found in the SM—Figures S1–S4. Hyper-QCC can be downloaded after a form submission available at https://photonicsresearchgroup.org/hyper-qcc (accessed on 7 March 2023).
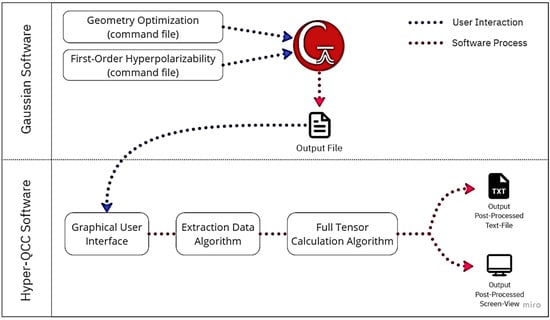
Figure 2.
Schematic diagram of the working principle of the post-processing software used in this work—Hyper-QCC.
3. Results and Discussion
3.1. First-Order Molecular Hyperpolarizability
The advances in theoretical models combined with the power evolution of CPUs allow researchers to predict physical spectroscopy quantities through QCC without using expensive experimental facilities. For example, estimating NLO properties, such as the first-molecular hyperpolarizability and two-photon absorption of organic and organometallic compounds, has become common in recent years [62,82,83,84,85,86,87,88]. However, to have a certain level of reliability on the simulated results, one should compare if the level of theory and the post-processing methods used can follow their experimental analogs. We have used the UAE to evaluate the performance of each functional, i.e., we have calculated the relative error between theoretical and experimental dynamic first-order molecular hyperpolarizability for each investigated compound and then computed the average for all compounds for each functional. The functionals with higher reliability, i.e., with higher performance in terms of UAE, are the ones with UAE values within or close to the experimental uncertainty. Moreover, the computational cost (CC) benchmark is also available.
A golden rule for non-theoretical researchers considers that the most accurate result is the experimental one; therefore, a simulated physical quantity should be at least in the same order of magnitude as its experimental analog. Still, one should be careful when applying such a rule, especially when analyzing NLO effects, since some observed variables, such as the pulse width and the intensity of the interaction light (ω), could influence the results. Besides the intrinsic properties of the incident light, particularly regarding the HRS experimental technique, one of the most common mistakes is to evaluate the first-molecular hyperpolarizability of compounds with fluorescence emission around the 2ω spectral region, which could be triggered by two-photon excitation and lead to an overestimated βHRS value. Conversely, a βHRS underestimated experimental value can be achieved if the compound has non-zero absorbance in the spectral region of 2ω.
The results and discussion are focused on βHRS, i.e., the predicted values for the dynamic first-order molecular hyperpolarizability using QCC, once they can be directly compared with their experimental analogs. The experimental βHRS values used in this work were obtained exclusively by the HRS technique. The static first-order molecular hyperpolarizability (β0) can be derived through experimental methods, such as one- and two-photon absorption. In a previous work [83], we calculated β0 by gathering experimental spectroscopic parameters from one- and two-photon absorption spectra. For example, in a two-level energy system, those spectroscopic parameters are transition dipole moments from the ground state to excited states (μ0m, μ0n), the energy needed to achieve them (ωom, ωon), and the difference of the permanent dipole moment between these two excited states (ηnm). All these spectroscopic parameters can be combined in the following equation, [89,90], to obtain the experimental static first-order molecular hyperpolarizability. However, for the investigated compounds, we did not find the experimental results on β0 for the majority of the compounds.
The theoretical βHRS values were estimated with seven types of functionals, such as HF, B3LYP, TPSSh, PEB1PEB, wB97xD, CAM-B3LYP, and M06-2X, by using the same basis set (6-311++G(2d,2p)). However, only two functionals (CAM-B3LYP and M06-2X) achieved an acceptable performance in terms of UAE, i.e., these two functionals predicted the βHRS values within or close to the experimental relative mean error of all compounds. All calculated results are presented in Tables S2–S5 in the SM. Figure 3a,b,c depicts the theoretical dynamic first-order molecular hyperpolarizability values obtained with CAM-B3LYP/6-311++G(2d,2p) (red circles) and M06-2X/6-311++G(2d,2p) (blue diamonds) using a frequency of 1064 nm. The experimental data (black squares) with their associated uncertainty are also depicted in the mentioned figures. The UAE can be verified for all calculated functionals in Figure 3d.
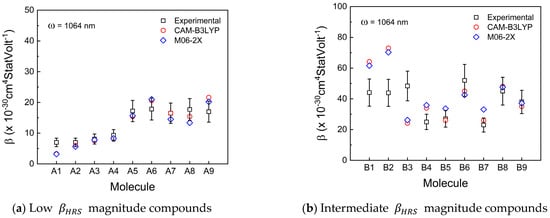
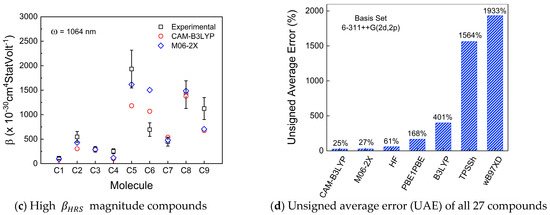
Figure 3.
Dynamic first-order molecular hyperpolarizability (βHRS) comparison between experimental (black squares) and theoretical CAM-B3LYP/6311++G(2d,2p) (red circles) and M06-2X/6-311++G(2d,2p) (blue diamonds) for (a) low, (b) intermediate, and (c) high βHRS magnitude compounds investigated in this work. The unsigned average error (UAE) values of all used functionals are depicted in (d).
Both wB97XD and TPSSh are the functionals with higher UAE values (Figure 3d) than all used functionals. The UAE values for both functionals represent a severe drawback regarding performance reliability once the theoretical βHRS averaged values from 27 compounds are 2 orders of magnitude separated from their experimental analogs. Regarding the CC, wB97XD is one of the functionals with better performance, as shown in Figure 4 (red bar). Contrary to this, TPSSh (yellow bar in Figure 4) has the worst CC general performance. Therefore, for these types of molecule families, TPSSh presented the lowest βHRS prediction accuracy, which could justify the lack of works in the literature using these functionals to compare experimental and theoretical βHRS values.
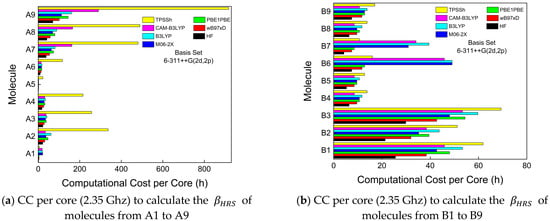
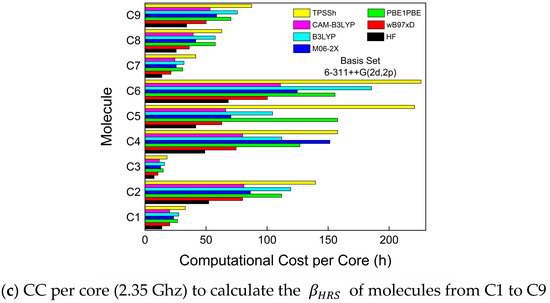
Figure 4.
Computational cost (CC), per core, of all functionals used to investigate the first-order molecular hyperpolarizabilities of all compounds in this work. The hardware to run Gaussian 16 was under a high-performance computing center/Linux. A total of 64 cores (2.35 GHz) with 128 GB of RAM were used to perform the QCC of this work.
The B3LYP functional is known to overestimate some of the nonlinear optical properties, particularly βHRS [91,92,93], and in this study, such behavior was also confirmed for most of the simulated compounds. B3LYP recorded the third-highest UAE value and its CC (cyan bar in Figure 4) was surprisingly higher than excepted. Usually, B3LYP has a CC lower than CAM-B3LYP, which, in some compounds, was not observed, such as compounds from groups B and C. However, B3LYP has been widely reported in the literature to simulate linear and nonlinear spectroscopic parameters [16,17,19,49,79,94,95,96,97,98], as mentioned in Section 2.2. The fourth-highest UAE value belongs to PBE1PBE, which is a functional with the second-best CC general performance along with wB97XD. The βHRS prediction accuracy when using PBE1PBE is still above one order of magnitude compared with the experimental values.
The HF method presented the best CC performance (black bars in Figure 4), as expected, due to the absence of electron correlation effects, which implies fewer calculations and less computational cost. Still, even with these significant limitations, the HF method was able to present a better prediction accuracy performance compared to the PBE1PBE functional, which is surprisingly positive for HF. In fact, HF UAE values are below 100%, meaning that the βHRS prediction accuracy is in the same order of magnitude as the experimental values. Therefore, we can conclude that it is more advantageous to choose HF instead of PBE1PBE, B3LYP, TPSSh, or wB97XD to perform this calculation once we significantly reduce CC and have a higher prediction accuracy.
On the other hand, CAM-B3LYP and M06-2X achieved the best UAE performance as expected once these two functionals have been pointed out in previous works that are more suitable to accurately predict dynamic hyperpolarizabilities on organic molecular structures [54,55,56,57,93,99,100,101]. Regarding the CC, in most cases, CAM-B3LYP (magenta bars in Figure 4) and M06-2X (blue bars in Figure 4) needed approximately the same time to complete the βHRS calculations.
Having low values of UAE is crucial for QCC accuracy; however, a balance between accuracy and CC is highly advantageous for obvious reasons. For example, changes in the basis set could reduce the CC and keep approximately the same UAE. Therefore, we performed QCC with a double-zeta split valence (6-31++G(2d,2p)) instead of the triple-zeta split valence, combined with the CAM-B3LYP functional for all 27 investigated compounds. As a result, an average reduction of 2.5 h per core was observed, with similar UAE values, i.e., for most compounds, the relative error between the βHRS was less than 10%, indicating that double-zeta split valence can be used in future works since its CC is significantly lower and the UAE is approximately the same. All results are presented in Table S6 in the SM. We also tested different polarization functions, such as (2df, 2pd) with all used functionals, to understand how CC and UAE will behave. No UAE significant improvements were observed, and worse, the CC increased. All results can be found in Tables S7–S9 in the SM. In summary, we can conclude that for the investigated compounds, decreasing from a triple-zeta to double-zeta split valence will keep approximately the same βHRS accuracy with a lower CC, while increasing the polarization functions from (2d, 2p) to (2df, 2pd) does not improve the βHRS accuracy and increases the CC.
So far, the presented and discussed results concern a single wavelength, i.e., the βHRS was calculated using a constant incident light frequency, e.g., ω = 1064 nm. However, a spectral behavior of the first-order molecular hyperpolarizability could be interesting to evaluate in which spectral region the compound will have the higher potential to be used as a frequency optical converter. Therefore, the Hyper-QCC software was also prepared to post-process multiple incident frequencies obtained from a single Gaussian .log file. Figure 5 presents the comparison spectra of theoretical and experimental first-order molecular hyperpolarizability of the azulenic-barbituric compound. Unfortunately, there are not so many experimental works published exploring the βHRS spectrum, probably due to the more complex experimental apparatus needed to obtain the faint light of the incoherent second harmonic generation (ISGH). For this purpose, e.g., an optical parametric oscillator will be needed to tune the incident light into different frequencies (ωi). Moreover, a set of laser line filters or a monochromator device will also be needed to collect the correct emission of 2ωi. As one could understand, it is much more inexpensive and faster to predict βHRS by performing QCC combined with a post-processing software such as Hyper-QCC, particularly when one wishes to understand the spectral behavior of βHRS.
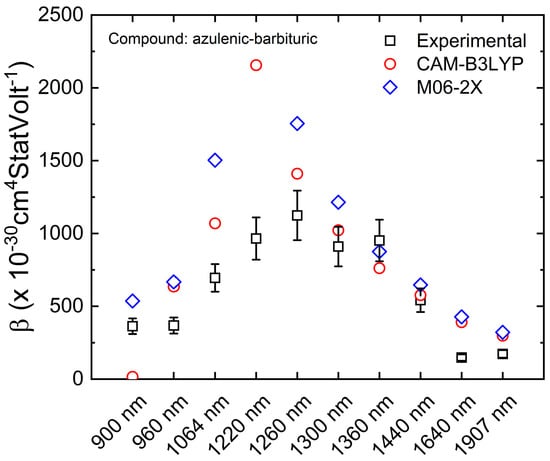
Figure 5.
Azulenic-barbituric (C6) dynamic first-order molecular hyperpolarizability (βHRS) spectra comparison between experimental (black squares) and theoretical using the level of theory CAM-B3LYP/6-311++G(2d,2p) (red circles) and M06-2X/6-311++G(2d,2p) (blue diamonds).
3.2. Applications
The ISHG is becoming an important tool in the field of bioimaging, without using any injection dye, as reported by Hui Mingalone et. al. [25], in which they have used an incident laser wavelength (1028 nm) on collagen cells to obtain an image formed by the second-harmonic generated light emitted by collagen. Although, to obtain such images, expensive equipment, such as a two-photon femtosecond microscope, is always needed. Still, before trying the bioimaging acquisition of biological cells, one can, for example, simulate the βHRS response, using the methods described in this paper, in an easy and straightforward way to evaluate or predict the ISHG, particularly by using the Hyper-QCC software.
The search for optical frequency converters [36,37,38,39,102,103,104] or optical switches [98,105,106] is a hot topic for the lasers and optical communications field. Two of the most commercial crystals able to convert the frequency of light into its double are Beta Barium Borate (BBO) and Lithium Triborate (LBO) [107,108,109]. Still, it is crucial to continue the search for new organic/organometallic raw materials that will have better SHG efficiency compared with the mentioned ones. Therefore, QCC combined with Hyper-QCC will give the ability for chemists, physicists, and materials scientists to evaluate the βHRS of several raw compounds before starting the expensive and time-costly processes of synthesis and optical characterization, at least before the growing crystal process.
4. Conclusions
In summary, in this work, we searched the literature for experimental dynamic first-order molecular hyperpolarizability values of organic compounds. We selected only the experimental data acquired with sub-nanosecond laser pulses to have the most reliable experimental nonlinear optical data, i.e., with less probability of having thermal effects masking the electronic ones. Therefore, we were restricted to the organic compounds available from the experimental data published in the literature. Consequently, we categorized the compounds as groups designated by A, B, and C. The molecules with the lowest experimental dynamic first-order hyperpolarizabilities were assigned to Group A, the intermediate experimental values to Group B, and the highest to Group C.
We used the Gaussian software package to perform quantum chemical calculations using different levels of theory to simulate both static and dynamic first-order molecular hyperpolarizabilities. We have chosen seven functionals, PBE0, TPSSh, wB97XD, B3LYP, CAM-B3LYP, M06-2X, and HF, combined with one basis set, 6-311++G(2d,2p). We have developed a post-processing software—Hyper-QCC, which will be freely distributed, to process the components of the third-order tensor from the first-order molecular hyperpolarizability obtained with the Gaussian program package in an effortless, fast, and reliable way.
Finally, CAM-B3LYP and M06-2X functionals are suitable to accurately predict dynamic hyperpolarizabilities on organic molecular structures, particularly those belonging to the family of molecules investigated in this work. In addition, this work shows that quantum chemical calculations with the proper post-processing software could optimize time efficiency and cost and can be used, e.g., to establish the initial step of developing nonlinear optical frequency conversion devices [110,111,112]. Moreover, collagen’s incoherent second harmonic generation phenomenon has already been tested and used to achieve biological images [25]. Therefore, quantum chemical calculations using the Gaussian software package combined with Hyper-QCC can also be used to predict the potential of biological compounds to generate an incoherent second harmonic.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/photonics10050545/s1. Table S1: Optimized geometry in solvent for compounds used to perform the calculation of the static (β0) and dynamic (βHRS) first hyperpolarizability β (in 10−30 cm4 statvolt−1). The values below correspond to the functional CAM-B3LYP with the basis set 6-311++G(2d,2p). Table S2: Theoretical values for static (β0) and dynamic at 1064 nm (βHRS) first-order molecular hyperpolarizability (in 10−30 cm4 statvolt−1) in a solvent medium of the investigated compounds. The computational cost (CC) is represented in terms of one single core, and the units are in hours. However, 64 cores (2.35 GHz) with 128 GB of RAM were used to perform the calculations. Table S3: Theoretical values for static (β0) and dynamic at 1064 nm (βHRS) first-order molecular hyperpolarizability (in 10−30 cm4 statvolt−1) in a solvent medium of the investigated compounds. The computational cost (CC) is represented in terms of one single core, and the units are in hours. However, 64 cores (2.35 GHz) with 128 GB of RAM were used to perform the calculations. Table S4: Theoretical values for static (β0) and dynamic at 1064 nm (βHRS) first-order molecular hyperpolarizability (in 10−30 cm4 statvolt−1) in a solvent medium of the investigated compounds. The computational cost (CC) is represented in terms of one single core, and the units are in hours. However, 64 cores (2.35 GHz) with 128 GB of RAM were used to perform the calculations. Table S5: Theoretical values for static (β0) and dynamic at 1064 nm (βHRS) first-order molecular hyperpolarizability (in 10−30 cm4 statvolt−1) in a solvent medium of the investigated compounds. The computational cost (CC) is represented in terms of one single core, and the units are in hours. However, 64 cores (2.35 GHz) with 128 GB of RAM were used to perform the calculations. Table S6: All βHRS values are in 10−30 cm4 statvolt−1 units, and computational cost per core (CC) is in hours. A total of 64 cores were used in the calculations. The relative error was calculated considering the following equation: RE = |βDZ-βTZ |/βTZ. Table S7: The following data were obtained for compound A7. All β0 and βHRS values are in 10−30 cm4 statvolt−1 units, and computational cost per core (CC) is in hours. A total of 64 cores were used in the calculations. The relative error was calculated considering the following equation: RE = |βHRS-βHRS-EXP. |/βHRS-EXP. Table S8: The following data were obtained for compound B6. All β0 and βHRS values are in 10−30 cm4 statvolt−1 units, and computational cost per core (CC) is in hours. A total of 64 cores were used in the calculations. The relative error was calculated considering the following equation: RE = |βHRS-βHRS-EXP. |/βHRS-EXP. Table S9: The following data were obtained for compound C8. All β0 and βHRS values are in 10−30 cm4 statvolt−1 units, and computational cost per core (CC) is in hours. A total of 64 cores were used in the calculations. The relative error was calculated considering the following equation: RE = |βHRS-βHRS-EXP. |/βHRS-EXP. Table S10: All β0 and βHRS values are in 10−30 cm4 statvolt−1 units. Figure S1. Hyper-QCC first section example. The solvent box is marked since the calculations were carried out using dimethylformamide (DMF) as a medium. Hyper-QCC also offers support to multiple wavelength calculations, but in this case, the single wavelength is coherent with our logfile. Figure S2. Hyper-QCC second section example. In this section, it is possible to retrieve the parameters chosen prior to the calculations. It is also possible to visualize the CPUs utilized and their performance. Figure S3. Hyper-QCC results table. The column “Static” for β0 and the wavelength column displaying βHRS. Figure S4. Hyper-QCC results table for multiple tensor logfile. The column “Static” for β0 and the multiple wavelengths are distributed in the columns displaying βHRS. Figure S5. Optimized geometry used in the calculations of compound C5.
Author Contributions
Conceptualization, L.M.G.A.; methodology, L.M.G.A., F.A.S. and C.E.R.C.; validation, L.M.G.A. and F.A.S.; formal analysis, L.D.B. and J.J.R.J.; investigation, L.M.G.A. and F.A.S.; resources, L.M.G.A.; writing—original draft preparation, F.A.S. and C.E.R.C.; writing—review and editing, L.M.G.A., L.D.B. and J.J.R.J.; visualization, F.A.S. and L.M.G.A.; supervision, L.M.G.A.; project administration, L.M.G.A.; funding acquisition, L.M.G.A. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Brazilian Council for Scientific and Technological Development (CNPq) with the following grant numbers: 311439/2021-7, 406190/2021-6, and 440225/2021-3.
Data Availability Statement
Not applicable to this article.
Acknowledgments
We thank the computer support from LaMCAD/UFG (Laboratório Multiusuário de Computação de Alto Desempenho/Universidade Federal de Goiás).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Franken, P.A.; Hill, A.E.; Peters, C.; Weinreich, G. Generation of optical harmonics. Phys. Rev. Lett. 1961, 7, 118. [Google Scholar] [CrossRef]
- Clays, K.; Persoons, A. Hyper-Rayleigh scattering in solution. Phys. Rev. Lett. 1991, 66, 2980. [Google Scholar] [CrossRef] [PubMed]
- Maser, A.; Gmeiner, B.; Utikal, T.; Götzinger, S.; Sandoghdar, V. Few-photon coherent nonlinear optics with a single molecule. Nat. Photonics 2016, 10, 450–453. [Google Scholar] [CrossRef]
- Lacroix, P.G.; Malfant, I.; Lepetit, C. Second-order nonlinear optics in coordination chemistry: An open door towards multi-functional materials and molecular switches. Coord. Chem. Rev. 2016, 308, 381–394. [Google Scholar] [CrossRef]
- Lou, A.J.-T.; Marks, T.J. A twist on nonlinear optics: Understanding the unique response of π-twisted chromophores. Acc. Chem. Res. 2019, 52, 1428–1438. [Google Scholar] [CrossRef]
- Parthasarathy, V.; Pandey, R.; Das, P.K.; Castet, F.; Blanchard-Desce, M. Linear and Nonlinear Optical Properties of Tricyanopropylidene-Based Merocyanine Dyes: Synergistic Experimental and Theoretical Investigations. ChemPhysChem 2018, 19, 187–197. [Google Scholar] [CrossRef]
- de Araújo, R.S.; de Alcântara, A.M.; Abegao, L.M.; de Souza, Y.P.; Silva, A.C.B.; Machado, R.; Rodrigues, J.J., Jr.; Júnior, J.R.P.; d’Errico, F.; Valle, M.S. Second harmonic generation in pyrazoline derivatives of dibenzylideneacetones and chalcone: A combined experimental and theoretical approach. J. Photochem. Photobiol. A Chem. 2020, 388, 112147. [Google Scholar] [CrossRef]
- Gu, B.; Zhao, C.; Baev, A.; Yong, K.-T.; Wen, S.; Prasad, P.N. Molecular nonlinear optics: Recent advances and applications. Adv. Opt. Photonics 2016, 8, 328–369. [Google Scholar] [CrossRef]
- Zhu, W.; Zhu, L.; Sun, L.; Zhen, Y.; Dong, H.; Wei, Z.; Hu, W. Uncovering the intramolecular emission and tuning the nonlinear optical properties of organic materials by cocrystallization. Angew. Chem. Int. Ed. 2016, 55, 14023–14027. [Google Scholar] [CrossRef]
- Semin, S.; Li, X.; Duan, Y.; Rasing, T. Nonlinear optical properties and applications of fluorenone molecular materials. Adv. Opt. Mater. 2021, 9, 2100327. [Google Scholar] [CrossRef]
- Colombo, A.; Dragonetti, C.; Guerchais, V.; Roberto, D. An excursion in the second-order nonlinear optical properties of platinum complexes. Coord. Chem. Rev. 2021, 446, 214113. [Google Scholar] [CrossRef]
- Ullah, F.; Ayub, K.; Mahmood, T. Remarkable second and third order nonlinear optical properties of organometallic C6Li6–M3O electrides. New J. Chem. 2020, 44, 9822–9829. [Google Scholar] [CrossRef]
- Schaller, R.R. Moore’s law: Past, present and future. IEEE Spectr. 1997, 34, 52–59. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Petersson, G.A.; Nakatsuji, H.; et al. Gaussian 16 Rev. C.01; Gaussian, Inc.: Wallingford, CT, USA, 2016. [Google Scholar]
- Castet, F.; Bogdan, E.; Plaquet, A.; Ducasse, L.; Champagne, B.; Rodriguez, V. Reference molecules for nonlinear optics: A joint experimental and theoretical investigation. J. Chem. Phys. 2012, 136, 024506. [Google Scholar] [CrossRef]
- Li, X.; Zhang, Y.; Lu, J. Remarkably enhanced first hyperpolarizability and nonlinear refractive index of novel graphdiyne-based materials for promising optoelectronic applications: A first-principles study. Appl. Surf. Sci. 2020, 512, 145544. [Google Scholar] [CrossRef]
- Pielak, K.; Bondu, F.; Sanguinet, L.; Rodriguez, V.; Champagne, B.; Castet, F. Second-order nonlinear optical properties of multiaddressable indolinooxazolidine derivatives: Joint computational and hyper-Rayleigh scattering investigations. J. Phys. Chem. C 2017, 121, 1851–1860. [Google Scholar] [CrossRef]
- Abegão, L.M.; Fonseca, R.D.; Santos, F.A.; Souza, G.B.; Barreiros, A.L.B.; Barreiros, M.L.; Alencar, M.; Mendonca, C.R.; Silva, D.L.; De Boni, L. Second-and third-order nonlinear optical properties of unsubstituted and mono-substituted chalcones. Chem. Phys. Lett. 2016, 648, 91–96. [Google Scholar] [CrossRef]
- Silva, D.L.; Fonseca, R.D.; Vivas, M.G.; Ishow, E.; Canuto, S.; Mendonca, C.R.; De Boni, L. Experimental and theoretical investigation of the first-order hyperpolarizability of a class of triarylamine derivatives. J. Chem. Phys. 2015, 142, 064312. [Google Scholar] [CrossRef]
- Yang, X.; Lin, X.; Zhao, Y.S.; Yan, D. Recent advances in micro-/nanostructured metal–organic frameworks towards photonic and electronic applications. Chem.–A Eur. J. 2018, 24, 6484–6493. [Google Scholar] [CrossRef]
- Reshef, O.; De Leon, I.; Alam, M.Z.; Boyd, R.W. Nonlinear optical effects in epsilon-near-zero media. Nat. Rev. Mater. 2019, 4, 535–551. [Google Scholar] [CrossRef]
- Kariduraganavar, M.Y.; Doddamani, R.V.; Waddar, B.; Parne, S.R. Nonlinear Optical Responsive Molecular Switches. In Nonlinear Optics: From Solitons to Similaritons; Books on Demand: Paris, France, 2021; Volume 187. [Google Scholar]
- Wang, Y.; Yang, H.; Dong, W.; Lei, L.; Xu, J.; Zhang, X.; Xu, P. Highly Nonlinear Organic-Silicon Slot Waveguide for Ultrafast Multimode All-Optical Logic Operations. IEEE Photonics J. 2020, 12, 1–12. [Google Scholar] [CrossRef]
- Jiang, X.; Zhang, L.; Liu, S.; Zhang, Y.; He, Z.; Li, W.; Zhang, F.; Shi, Y.; Lü, W.; Li, Y. Ultrathin metal–organic framework: An emerging broadband nonlinear optical material for ultrafast photonics. Adv. Opt. Mater. 2018, 6, 1800561. [Google Scholar] [CrossRef]
- Mingalone, C.K.H.; Liu, Z.; Hollander, J.M.; Garvey, K.D.; Gibson, A.L.; Banks, R.E.; Zhang, M.; McAlindon, T.E.; Nielsen, H.C.; Georgakoudi, I. Bioluminescence and second harmonic generation imaging reveal dynamic changes in the inflammatory and collagen landscape in early osteoarthritis. Lab. Investig. 2018, 98, 656–669. [Google Scholar] [CrossRef] [PubMed]
- Cheng, H.-B.; Li, Y.; Tang, B.Z.; Yoon, J. Assembly strategies of organic-based imaging agents for fluorescence and photoacoustic bioimaging applications. Chem. Soc. Rev. 2020, 49, 21–31. [Google Scholar] [CrossRef] [PubMed]
- Parodi, V.; Jacchetti, E.; Osellame, R.; Cerullo, G.; Polli, D.; Raimondi, M.T. Nonlinear optical microscopy: From fundamentals to applications in live bioimaging. Front. Bioeng. Biotechnol. 2020, 8, 1174. [Google Scholar] [CrossRef]
- Khadria, A.; de Coene, Y.; Gawel, P.; Roche, C.; Clays, K.; Anderson, H.L. Push–pull pyropheophorbides for nonlinear optical imaging. Org. Biomol. Chem. 2017, 15, 947–956. [Google Scholar] [CrossRef]
- Kohn, W.; Sham, L.J. Self-consistent equations including exchange and correlation effects. Phys. Rev. 1965, 140, A1133. [Google Scholar] [CrossRef]
- Hartree, D.R.; Hartree, W. Self-consistent field, with exchange, for beryllium. Proc. R. Soc. London Ser. A—Math. Phys. Sci. 1935, 150, 9–33. [Google Scholar]
- Dwivedi, Y.; Tamashiro, G.; De Boni, L.; Zilio, S.C. Nonlinear optical characterizations of dibenzoylmethane in solution. Opt. Commun. 2013, 293, 119–124. [Google Scholar] [CrossRef]
- Houbrechts, S.; Clays, K.; Persoons, A.; Pikramenou, Z.; Lehn, J.-M. Hyper-Rayleigh scattering investigation of nitrobenzyl pyridine model compounds for optical modulation of the hyperpolarisability. Chem. Phys. Lett. 1996, 258, 485–489. [Google Scholar] [CrossRef]
- Srinivas, K.; Prabhakar, C.; Sitha, S.; Bhanuprakash, K.; Rao, V.J. Enhanced molecular first hyperpolarizability in s-triazine derivatives: Combined experimental and computational studies. J. Mol. Struct. 2014, 1075, 118–123. [Google Scholar] [CrossRef]
- Kaatz, P.; Shelton, D.P. Polarized hyper-Rayleigh light scattering measurements of nonlinear optical chromophores. J. Chem. Phys. 1996, 105, 3918–3929. [Google Scholar] [CrossRef]
- Beaujean, P.; Bondu, F.; Plaquet, A.; Garcia-Amoros, J.; Cusido, J.; Raymo, F.M.; Castet, F.; Rodriguez, V.; Champagne, B. Oxazines: A new class of second-order nonlinear optical switches. J. Am. Chem. Soc. 2016, 138, 5052–5062. [Google Scholar] [CrossRef]
- Santos, F.A.; Abegão, L.M.; Fonseca, R.D.; Alcântara, A.M.; Mendonça, C.R.; Alencar, M.A.; Valle, M.S.; Kamada, K.; De Boni, L.; Rodrigues, J.J., Jr. Nonlinear optical study in a set of dibenzylideneacetone derivatives with potential for optical frequency conversion. Photonics 2020, 7, 8. [Google Scholar] [CrossRef]
- Santos, F.A.; Abegao, L.M.; Fonseca, R.D.; Alcantara, A.M.; Mendonca, C.R.; Valle, M.S.; Alencar, M.; Kamada, K.; De Boni, L.; Rodrigues, J., Jr. Bromo-and chloro-derivatives of dibenzylideneacetone: Experimental and theoretical study of the first molecular hyperpolarizability and two-photon absorption. J. Photochem. Photobiol. A Chem. 2019, 369, 70–76. [Google Scholar] [CrossRef]
- Abegao, L.M.; Fonseca, R.D.; Santos, F.A.; Rodrigues, J.J.; Kamada, K.; Mendonça, C.R.; Piguel, S.; De Boni, L. First molecular electronic hyperpolarizability of series of π-conjugated oxazole dyes in solution: An experimental and theoretical study. RSC Adv. 2019, 9, 26476–26482. [Google Scholar] [CrossRef]
- Abegao, L.M.; Santos, F.A.; Fonseca, R.D.; Barreiros, A.L.; Barreiros, M.L.; Alves, P.B.; Costa, E.V.; Souza, G.B.; Alencar, M.A.; Mendonca, C.R. Chalcone-based molecules: Experimental and theoretical studies on the two-photon absorption and molecular first hyperpolarizability. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2020, 227, 117772. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Liu, S.; Wang, C.C.; Wang, C. Dispersion of the first hyperpolarizability of a strongly charge-transfer chromophore investigated by tunable wavelength hyper-Rayleigh scattering. J. Chem. Phys. 2001, 114, 7103–7108. [Google Scholar] [CrossRef]
- Hsu, C.-C.; Shu, C.-F.; Huang, T.-H.; Wang, C.; Lin, J.-L.; Wang, Y.-K.; Zang, Y.-L. Hyper-Rayleigh scattering of thiophene-incorporated polyene chromophores with π-configuration locking. Chem. Phys. Lett. 1997, 274, 466–472. [Google Scholar] [CrossRef]
- Brasselet, S.; Zyss, J. Multipolar molecules and multipolar fields: Probing and controlling the tensorial nature of nonlinear molecular media. JOSA B 1998, 15, 257–288. [Google Scholar] [CrossRef]
- Bogdan, E.; Plaquet, A.; Antonov, L.; Rodriguez, V.; Ducasse, L.; Champagne, B.; Castet, F. Solvent effects on the second-order nonlinear optical responses in the keto− enol equilibrium of a 2-Hydroxy-1-naphthaldehyde derivative. J. Phys. Chem. C 2010, 114, 12760–12768. [Google Scholar] [CrossRef]
- de Wergifosse, M.; Grimme, S. Nonlinear-response properties in a simplified time-dependent density functional theory (sTD-DFT) framework: Evaluation of the first hyperpolarizability. J. Chem. Phys. 2018, 149, 024108. [Google Scholar] [CrossRef]
- Verbiest, T.; Clays, K.; Rodriguez, V. Second-Order Nonlinear Optical Characterization Techniques: An Introduction; CRC Press: Boca Raton, FL, USA, 2009. [Google Scholar]
- Krishnan, R.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785. [Google Scholar] [CrossRef] [PubMed]
- Yanai, T.; Tew, D.P.; Handy, N.C. A new hybrid exchange–correlation functional using the Coulomb-attenuating method (CAM-B3LYP). Chem. Phys. Lett. 2004, 393, 51–57. [Google Scholar] [CrossRef]
- Wang, Y.-F.; Li, J.; Huang, J.; Qin, T.; Liu, Y.-M.; Zhong, F.; Zhang, W.; Li, Z.-R. Long-Range Charge Transfer Driven by External Electric Field in Alkalides M–LCaL–M (M= Li or Na, L= All-cis 1, 2, 3, 4, 5, 6-Hexafluorocyclohexane): Facially Polarized Janus-Type Second Order Nonlinear Molecular Optical Switches. J. Phys. Chem. C 2019, 123, 23610–23619. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar]
- Kita, H.; Yamakado, R.; Fukuuchi, R.; Konishi, T.; Kamada, K.; Haketa, Y.; Maeda, H. Switching of Two-Photon Optical Properties by Anion Binding of Pyrrole-Based Boron Diketonates through Conformation Change. Chem.–A Eur. J. 2020, 26, 3404–3410. [Google Scholar] [CrossRef]
- Klausen, M.; Dubois, V.; Clermont, G.; Tonnelé, C.; Castet, F.; Blanchard-Desce, M. Dual-wavelength efficient two-photon photorelease of glycine by π-extended dipolar coumarins. Chem. Sci. 2019, 10, 4209–4219. [Google Scholar] [CrossRef]
- Samanta, P.K.; Alam, M.M.; Misra, R.; Pati, S.K. Tuning of hyperpolarizability, and one-and two-photon absorption of donor–acceptor and donor–acceptor–acceptor-type intramolecular charge transfer-based sensors. Phys. Chem. Chem. Phys. 2019, 21, 17343–17355. [Google Scholar] [CrossRef]
- Shalin, N.I.; Fominykh, O.D.; Balakina, M.Y. Effect of acceptor moieties on static and dynamic first hyperpolarizability of azobenzene chromophores. Chem. Phys. Lett. 2019, 717, 21–28. [Google Scholar] [CrossRef]
- Besalú-Sala, P.; Sitkiewicz, S.P.; Salvador, P.; Matito, E.; Luis, J.M. A new tuned range-separated density functional for the accurate calculation of second hyperpolarizabilities. Phys. Chem. Chem. Phys. 2020, 22, 11871–11880. [Google Scholar] [CrossRef]
- Champagne, B.; Beaujean, P.; De Wergifosse, M.; Cardenuto, M.H.; Liégeois, V.; Castet, F. Quantum Chemical Methods for Predicting and Interpreting Second-Order Nonlinear Optical Properties: From Small to Extended π-Conjugated Molecules. In Frontiers of Quantum Chemistry; Springer: Singapore, 2018; pp. 117–138. [Google Scholar]
- Pielak, K.; Bondu, F.; Sanguinet, L.; Rodriguez, V.; Castet, F.; Champagne, B. Acido-triggered switching of the second-order nonlinear optical properties of a ferrocenyl-containing indolino-oxazolidine derivative. Dye. Pigment. 2019, 160, 641–646. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Adamo, C.; Barone, V. Toward chemical accuracy in the computation of NMR shieldings: The PBE0 model. Chem. Phys. Lett. 1998, 298, 113–119. [Google Scholar] [CrossRef]
- Suponitsky, K.Y.; Liao, Y.; Masunov, A.E. Electronic hyperpolarizabilities for donor−acceptor molecules with long conjugated bridges: Calculations versus experiment. J. Phys. Chem. A 2009, 113, 10994–11001. [Google Scholar] [CrossRef] [PubMed]
- Suponitsky, K.Y.; Tafur, S.; Masunov, A.E. Applicability of hybrid density functional theory methods to calculation of molecular hyperpolarizability. J. Chem. Phys. 2008, 129, 044109. [Google Scholar] [CrossRef]
- Liu, Y.; Yuan, Y.; Tian, X.; Yuan, J.; Sun, J. High first-hyperpolarizabilities of thiobarbituric acid derivative-based donor-π-acceptor nonlinear optical-phores: Multiple theoretical investigations of substituents and conjugated bridges effect. Int. J. Quantum Chem. 2020, 120, e26176. [Google Scholar] [CrossRef]
- Johnson, L.E.; Dalton, L.R.; Robinson, B.H. Optimizing calculations of electronic excitations and relative hyperpolarizabilities of electrooptic chromophores. Acc. Chem. Res. 2014, 47, 3258–3265. [Google Scholar] [CrossRef]
- Adamo, C.; Cossi, M.; Scalmani, G.; Barone, V. Accurate static polarizabilities by density functional theory: Assessment of the PBE0 model. Chem. Phys. Lett. 1999, 307, 265–271. [Google Scholar] [CrossRef]
- Tao, J.; Perdew, J.P.; Staroverov, V.N.; Scuseria, G.E. Climbing the density functional ladder: Nonempirical meta–generalized gradient approximation designed for molecules and solids. Phys. Rev. Lett. 2003, 91, 146401. [Google Scholar] [CrossRef] [PubMed]
- Şirikci, G.; Ancın, N.A.; Öztaş, S.G. Theoretical studies of organotin (IV) complexes derived from ONO-donor type schiff base ligands. J. Mol. Model. 2015, 21, 221. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Systematic optimization of long-range corrected hybrid density functionals. J. Chem. Phys. 2008, 128, 084106. [Google Scholar] [CrossRef] [PubMed]
- Chai, J.-D.; Head-Gordon, M. Long-range corrected hybrid density functionals with damped atom–atom dispersion corrections. Phys. Chem. Chem. Phys. 2008, 10, 6615–6620. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. V. Systematic optimization of exchange-correlation functionals. J. Chem. Phys. 1997, 107, 8554–8560. [Google Scholar] [CrossRef]
- Sutradhar, D.; Chandra, A.K.; Zeegers-Huyskens, T. Theoretical study of the interaction of fluorinated dimethyl ethers and the ClF and HF molecules. Comparison between halogen and hydrogen bonds. Int. J. Quantum Chem. 2016, 116, 670–680. [Google Scholar] [CrossRef]
- Safdari, F.; Raissi, H.; Shahabi, M.; Zaboli, M. DFT calculations and molecular dynamics simulation study on the adsorption of 5-fluorouracil anticancer drug on graphene oxide nanosheet as a drug delivery vehicle. J. Inorg. Organomet. Polym. Mater. 2017, 27, 805–817. [Google Scholar] [CrossRef]
- Lohith, T.; Hema, M.; Karthik, C.; Sandeep, S.; Mallesha, L.; Alsaiari, N.S.; Sridhar, M.; Katubi, K.M.; Abualnaja, K.M.; Lokanath, N. Persistent prevalence of non-covalent interaction in pyrimidine containing sulfonamide derivative: A quantum computational analysis. J. Mol. Struct. 2022, 1266, 133378. [Google Scholar] [CrossRef]
- Hadji, D.; Rahmouni, A. Molecular structure, linear and nonlinear optical properties of some cyclic phosphazenes: A theoretical investigation. J. Mol. Struct. 2016, 1106, 343–351. [Google Scholar] [CrossRef]
- Ejuh, G.; Tchangnwa Nya, F.; Ottou Abe, M.; Jean-Baptiste, F.; Ndjaka, J. Electronic structure, physico-chemical, linear and non linear optical properties analysis of coronene, 6B-, 6N-, 3B3N-substituted C24H12 using RHF, B3LYP and wB97XD methods. Opt. Quantum Electron. 2017, 49, 382. [Google Scholar] [CrossRef]
- Al-Hamdani, U.J.; Hassan, Q.M.; Zaidan, A.M.; Sultan, H.; Hussain, K.A.; Emshary, C.; Alabdullah, Z.T. Optical nonlinear properties and all optical switching in a synthesized liquid crystal. J. Mol. Liq. 2022, 361, 119676. [Google Scholar] [CrossRef]
- Prakasam, M.; Anbarasan, P. Second order hyperpolarizability of triphenylamine based organic sensitizers: A first principle theoretical study. RSC Adv. 2016, 6, 75242–75250. [Google Scholar] [CrossRef]
- Karakas, A.; Karakaya, M.; Ceylan, Y.; El Kouari, Y.; Taboukhat, S.; Boughaleb, Y.; Sofiani, Z. Ab-initio and DFT methodologies for computing hyperpolarizabilities and susceptibilities of highly conjugated organic compounds for nonlinear optical applications. Opt. Mater. 2016, 56, 8–17. [Google Scholar] [CrossRef]
- Maidur, S.R.; Patil, P.S.; Rao, S.V.; Shkir, M.; Dharmaprakash, S. Experimental and computational studies on second-and third-order nonlinear optical properties of a novel D-π-A type chalcone derivative: 3-(4-methoxyphenyl)-1-(4-nitrophenyl) prop-2-en-1-one. Opt. Laser Technol. 2017, 97, 219–228. [Google Scholar] [CrossRef]
- Castet, F.; Rodriguez, V.; Pozzo, J.-L.; Ducasse, L.; Plaquet, A.; Champagne, B. Design and characterization of molecular nonlinear optical switches. Acc. Chem. Res. 2013, 46, 2656–2665. [Google Scholar] [CrossRef] [PubMed]
- Tolbin, A.Y.; Dzuban, A.V.; Shestov, V.I.; Gudkova, Y.I.; Brel, V.K.; Tomilova, L.G.; Zefirov, N.S. Peripheral functionalisation of a stable phthalocyanine J-type dimer to control the aggregation behaviour and NLO properties: UV-Vis, fluorescence, DFT, TDHF and thermal study. RSC Adv. 2015, 5, 8239–8247. [Google Scholar] [CrossRef]
- Tomasi, J.; Mennucci, B.; Cancès, E. The IEF version of the PCM solvation method: An overview of a new method addressed to study molecular solutes at the QM ab initio level. J. Mol. Struct. THEOCHEM 1999, 464, 211–226. [Google Scholar] [CrossRef]
- Tamer, Ö.; Şimşek, M.; Avcı, D.; Atalay, Y. First and second order hyperpolarizabilities of flavonol derivatives: A density functional theory study. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2022, 283, 121728. [Google Scholar] [CrossRef]
- Sciuti, L.F.; Abegão, L.M.; Dos Santos, C.H.; Zucolotto Cocca, L.H.; da Costa, R.G.; Limberger, J.; Misoguti, L.; Mendonça, C.R.; De Boni, L. Modeling the First-Order Molecular Hyperpolarizability Dispersion from Experimentally Obtained One-and Two-Photon Absorption. J. Phys. Chem. A 2022, 126, 2152–2159. [Google Scholar] [CrossRef]
- Lescos, L.; Sitkiewicz, S.P.; Beaujean, P.; Blanchard-Desce, M.; Champagne, B.; Matito, E.; Castet, F. Performance of DFT functionals for calculating the second-order nonlinear optical properties of dipolar merocyanines. Phys. Chem. Chem. Phys. 2020, 22, 16579–16594. [Google Scholar] [CrossRef]
- Devi, C.L.; Yesudas, K.; Makarov, N.S.; Rao, V.J.; Bhanuprakash, K.; Perry, J.W. Combined experimental and theoretical study of one-and two-photon absorption properties of D–π–A–π–D type bis (carbazolylfluorenylethynyl) arene derivatives: Influence of aromatic acceptor bridge. Dye. Pigment. 2015, 113, 682–691. [Google Scholar] [CrossRef]
- Matczyszyn, K.; Olesiak-Banska, J.; Nakatani, K.; Yu, P.; Murugan, N.A.; Zalesny, R.; Roztoczynska, A.; Bednarska, J.; Bartkowiak, W.; Kongsted, J. One-and Two-Photon Absorption of a Spiropyran–Merocyanine System: Experimental and Theoretical Studies. J. Phys. Chem. B 2015, 119, 1515–1522. [Google Scholar] [CrossRef]
- Kang, G.; Nasiri Avanaki, K.; Mosquera, M.A.; Burdick, R.K.; Villabona-Monsalve, J.P.; Goodson, T., III; Schatz, G.C. Efficient modeling of organic chromophores for entangled two-photon absorption. J. Am. Chem. Soc. 2020, 142, 10446–10458. [Google Scholar] [CrossRef] [PubMed]
- Abegão, L.M.; Fonseca, R.D.; Ramos, T.N.; Mahuteau-Betzer, F.; Piguel, S.; Joatan, R.J., Jr.; Mendonca, C.R.; Canuto, S.; Silva, D.L.; De Boni, L. Oxazole dyes with potential for photoluminescence bioprobes: A two-photon absorption study. J. Phys. Chem. C 2018, 122, 10526–10534. [Google Scholar] [CrossRef]
- Marder, S.R.; Beratan, D.N.; Cheng, L.-T. Approaches for optimizing the first electronic hyperpolarizability of conjugated organic molecules. Science 1991, 252, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Orr, B.; Ward, J. Perturbation theory of the non-linear optical polarization of an isolated system. Mol. Phys. 1971, 20, 513–526. [Google Scholar] [CrossRef]
- Nénon, S.; Champagne, B. SCC-DFTB calculation of the static first hyperpolarizability: From gas phase molecules to functionalized surfaces. J. Chem. Phys. 2013, 138, 204107. [Google Scholar] [CrossRef]
- Hu, Y.-Y.; Sun, S.-L.; Muhammad, S.; Xu, H.-L.; Su, Z.-M. How the number and location of lithium atoms affect the first hyperpolarizability of graphene. J. Phys. Chem. C 2010, 114, 19792–19798. [Google Scholar] [CrossRef]
- Srivastava, R.; Al-Omary, F.A.; El-Emam, A.A.; Pathak, S.K.; Karabacak, M.; Narayan, V.; Chand, S.; Prasad, O.; Sinha, L. A combined experimental and theoretical DFT (B3LYP, CAM-B3LYP and M06-2X) study on electronic structure, hydrogen bonding, solvent effects and spectral features of methyl 1H-indol-5-carboxylate. J. Mol. Struct. 2017, 1137, 725–741. [Google Scholar] [CrossRef]
- Rasool, F.; Hussain, A.; Yar, M.; Ayub, K.; Sajid, M.; Asif, H.M.; Imran, M.; Assiri, M.A. Nonlinear optical response of 9, 10-bis (phenylethynyl) anthracene mediated by electron donating and electron withdrawing substituents: A density functional theory approach. Mater. Sci. Semicond. Process. 2022, 148, 106751. [Google Scholar] [CrossRef]
- Kodikara, M.S.; Stranger, R.; Humphrey, M.G. Long-Range Corrected DFT Calculations of First Hyperpolarizabilities and Excitation Energies of Metal Alkynyl Complexes. ChemPhysChem 2018, 19, 1537–1546. [Google Scholar] [CrossRef]
- Capobianco, A.; Centore, R.; Noce, C.; Peluso, A. Molecular hyperpolarizabilities of push–pull chromophores: A comparison between theoretical and experimental results. Chem. Phys. 2013, 411, 11–16. [Google Scholar] [CrossRef]
- de Wergifosse, M.; Champagne, B. Electron correlation effects on the first hyperpolarizability of push–pull π-conjugated systems. J. Chem. Phys. 2011, 134, 074113. [Google Scholar] [CrossRef]
- Pielak, K.; Tonnelé, C.; Sanguinet, L.; Cariati, E.; Righetto, S.; Muccioli, L.; Castet, F.; Champagne, B. Dynamical behavior and second harmonic generation responses in acido-triggered molecular switches. J. Phys. Chem. C 2018, 122, 26160–26168. [Google Scholar] [CrossRef]
- Tonnelé, C.; Champagne, B.; Muccioli, L.; Castet, F. Second-order nonlinear optical properties of Stenhouse photoswitches: Insights from density functional theory. Phys. Chem. Chem. Phys. 2018, 20, 27658–27667. [Google Scholar] [CrossRef] [PubMed]
- Bondu, F.; Quertinmont, J.; Rodriguez, V.; Pozzo, J.L.; Plaquet, A.; Champagne, B.; Castet, F. Second-Order Nonlinear Optical Properties of a Dithienylethene–Indolinooxazolidine Hybrid: A Joint Experimental and Theoretical Investigation. Chem.–A Eur. J. 2015, 21, 18749–18757. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.; El-Emam, A.A.; Pathak, S.K.; Srivastava, R.; Shukla, V.K.; Prasad, O.; Sinha, L. Experimental and theoretical DFT (B3LYP, X3LYP, CAM-B3LYP and M06-2X) study on electronic structure, spectral features, hydrogen bonding and solvent effects of 4-methylthiadiazole-5-carboxylic acid. Mol. Simul. 2019, 45, 1029–1043. [Google Scholar] [CrossRef]
- Custodio, J.M.; Gotardo, F.; Vaz, W.F.; D’Oliveira, G.D.; de Almeida, L.R.; Fonseca, R.D.; Cocca, L.H.; Perez, C.N.; Oliver, A.G.; De Boni, L. Benzenesulfonyl incorporated chalcones: Synthesis, structural and optical properties. J. Mol. Struct. 2020, 1208, 127845. [Google Scholar] [CrossRef]
- Fonseca, R.D.; Vivas, M.G.; Silva, D.L.; Eucat, G.; Bretonnière, Y.; Andraud, C.; De Boni, L.; Mendonca, C.R. First-order hyperpolarizability of triphenylamine derivatives containing cyanopyridine: Molecular branching effect. J. Phys. Chem. C 2018, 122, 1770–1778. [Google Scholar] [CrossRef]
- Vivas, M.G.; Barboza, C.A.; Germino, J.C.; Fonseca, R.D.; Silva, D.L.; Vazquez, P.A.; Atvars, T.D.; Mendonca, C.R.; De Boni, L. Molecular Structure–Optical Property Relationship of Salicylidene Derivatives: A Study on the First-Order Hyperpolarizability. J. Phys. Chem. A 2020, 125, 99–105. [Google Scholar] [CrossRef]
- Tonnelé, C.; Pielak, K.; Deviers, J.; Muccioli, L.; Champagne, B.; Castet, F. Nonlinear optical responses of self-assembled monolayers functionalized with indolino–oxazolidine photoswitches. Phys. Chem. Chem. Phys. 2018, 20, 21590–21597. [Google Scholar] [CrossRef] [PubMed]
- Chai, Z.; Hu, X.; Wang, F.; Niu, X.; Xie, J.; Gong, Q. Ultrafast all-optical switching. Adv. Opt. Mater. 2017, 5, 1600665. [Google Scholar] [CrossRef]
- Röcker, C.; Weinert, P.; Villeval, P.; Lupinski, D.; Delaigue, M.; Hönninger, C.; Weber, R.; Graf, T.; Ahmed, M.A. Nonlinear absorption in lithium triborate frequency converters for high-power ultrafast lasers. Opt. Express 2022, 30, 5423–5438. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.-y.; Huang, J.Y.; Wang, H.; Wong, K.; Wong, G. Second-harmonic generation from regeneratively amplified femtosecond laser pulses in BBO and LBO crystals. JOSA B 1998, 15, 200–209. [Google Scholar] [CrossRef]
- Akbari, R.; Major, A. Optical, spectral and phase-matching properties of BIBO, BBO and LBO crystals for optical parametric oscillation in the visible and near-infrared wavelength ranges. Laser Phys. 2013, 23, 035401. [Google Scholar] [CrossRef]
- Karuppasamy, P.; Kamalesh, T.; Anitha, K.; Kalam, S.A.; Pandian, M.S.; Ramasamy, P.; Verma, S.; Rao, S.V. Synthesis, crystal growth, structure and characterization of a novel third order nonlinear optical organic single crystal: 2-amino 4, 6-dimethyl pyrimidine 4-nitrophenol. Opt. Mater. 2018, 84, 475–489. [Google Scholar] [CrossRef]
- Liu, J.; Ouyang, C.; Huo, F.; He, W.; Cao, A. Progress in the enhancement of electro-optic coefficients and orientation stability for organic second-order nonlinear optical materials. Dye. Pigment. 2020, 181, 108509. [Google Scholar] [CrossRef]
- Liu, X.; Yang, Z.; Wang, D.; Cao, H. Molecular structures and second-order nonlinear optical properties of ionic organic crystal materials. Crystals 2016, 6, 158. [Google Scholar] [CrossRef]







Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).