
Application of a Validated Method for the Identification and Quantification of Mycotoxins in Wines Using UPLC-MS/MS

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Reagents
2.2. Samples
2.3. Instrument Conditions
2.4. Sample Preparation
2.5. Method Validation
2.5.1. Linearity
2.5.2. LOD and LOQ
2.5.3. Method Recovery
2.5.4. Uncertainty Measurement
3. Results and Discussion
3.1. UPLC-MS/MS Method Development
3.2. Sample Extraction Method
3.3. Method Validation
3.3.1. Linearity
3.3.2. Selectivity, LOD and LOQ
3.3.3. Accuracy, Precision, Matrix Effect
3.3.4. Uncertainty Measurement
3.4. Method Application to Wine Samples
3.5. Comparison of Present Method to Other Methods Reported in the Literature
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gruber-Dorninger, C.; Novak, B.; Nagl, V.; Berthiller, F. Emerging Mycotoxins: Beyond Traditionally Determined Food Contaminants. J. Agric. Food Chem. 2017, 65, 7052–7070. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Das, M.; Tripathi, A. Occurrence and toxicity of a fusarium mycotoxin, zearalenone. Crit. Rev. Food Sci. Nutr. 2020, 60, 2710–2729. [Google Scholar] [CrossRef] [PubMed]
- European Commission. Commission Regulation (EC) No 1881/2006 of 19 December 2006 Setting Maximum Levels for Certain Contaminants in Foodstuffs (Text with EEA Relevance); European Commission: Brussels, Belgium, 2006. [Google Scholar]
- EFSA Panel on Contaminants in the Food Chain. Statement on recent scientific information 616 on the toxicity of Ochratoxin A. EFSA J. 2010, 8, 1626. [Google Scholar]
- OIV. International Code of Oenological Practices. OIV Code Sheet Issue 2015/01, Chapter I.1.3–1. 2016. Available online: http://www.oiv.int/en/technical-standards-and-documents/oenologicalpractices/international-code-of-oenological-practices (accessed on 15 August 2017).
- Anesi, A.; Stocchero, M.; Dal Santo, S.; Commisso, M.; Zenoni, S.; Ceoldo, S.; Tornielli, G.B.; Siebert, T.E.; Herderich, M.; Pezzotti, M.; et al. Towards a scientific interpretation of the terroir concept: Plasticity of the grape berry metabolome. BMC Plant Biol. 2015, 15, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sohaib, H.; Bryce, A.; Adrian, B. Wine and Cardiovascular Health. Circulation 2017, 136, 1434–1448. [Google Scholar]
- Jara-Palacios, M.J. Wine Lees as a Source of Antioxidant Compounds. Antioxidants 2019, 8, 45. [Google Scholar] [CrossRef] [Green Version]
- Joseph, S.V.; Edirisinghe, I.; Burton-Freeman, B.M. Fruit Polyphenols: A Review of Anti-inflammatory Effects in Humans. Crit. Rev. Food Sci. Nutr. 2016, 56, 419–444. [Google Scholar] [CrossRef]
- Artero, A.; Tarín, J.J.; Cano, A. The impact of moderate wine consumption on health. Maturitas 2015, 80, 3–13. [Google Scholar] [CrossRef]
- Bonaccio, M.; Di Castelnuovo, A.; Costanzo, S.; Persichillo, M.; De Curtis, A.; Donati, M.B.; de Gaetano, G.; Iacoviello, L.; MOLI-SANI Study Investigators. Adherence to the traditional Mediterranean diet and mortality in subjects with diabetes. Prospective results from the MOLI-SANI study. Eur. J. Prev. Cardiol. 2016, 23, 400–407. [Google Scholar] [CrossRef]
- Di Lorenzo, A.; Bloise, N.; Meneghini, S.; Sureda, A.; Tenore, G.C.; Visai, L.; Arciola, C.R.; Daglia, M. Effect of Winemaking on the Composition of Red Wine as a Source of Polyphenols for Anti-Infective Biomaterials. Materials 2016, 9, 316. [Google Scholar] [CrossRef] [Green Version]
- Gepner, Y.; Golan, R.; Harman-Boehm, I.; Henkin, Y.; Schwarzfuchs, D.; Shelef, I.; Durst, R.; Kovsan, J.; Bolotin, A.; Leitersdorf, E.; et al. Effects of Initiating Moderate Alcohol Intake on Cardiometabolic Risk in Adults with Type 2 Diabetes: A 2-Year Randomized, Controlled 494 Trial. Ann. Intern. Med. 2015, 163, 569–579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giacosa, A.; Barale, R.; Bavaresco, L.; Faliva, M.A.; Gerbi, V.; La Vecchia, C.; Negri, E.; Opizzi, A.; Perna, S.; Pezzotti, M.; et al. Mediterranean Way of Drinking and Longevity. Crit. Rev. Food Sci. Nutr. 2016, 56, 635–640. [Google Scholar] [CrossRef] [PubMed]
- Giovinazzo, G.; Grieco, F. Functional Properties of Grape and Wine Polyphenols. Plant Foods Hum. Nutr. 2015, 70, 454–462. [Google Scholar] [CrossRef] [PubMed]
- De Salvo, K.B.; Olson, R.; Casavale, K.O. Dietary Guidelines for Americans. JAMA 2016, 315, 457–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lichtenstein, A.H.; Appel, L.J.; Brands, M.; Carnethon, M.; Daniels, S.; Franch, H.A.; Franklin, B.; Kris-Etherton, P.; Harris, W.S.; Howard, B.; et al. Diet and Lifestyle Recommendations Revision 2006. Circulation 2006, 114, 82–96. [Google Scholar] [CrossRef] [Green Version]
- Kumar Singh, A.; Cabral, C.; Kumar, R.; Ganguly, R.; Kumar Rana, H.; Gupta, A.; Rosaria Lauro, M.; Carbone, C.; Reis, F.; Pandey, A.K. Beneficial Effects of Dietary Polyphenols on Gut Microbiota and Strategies to Improve Delivery Efficiency. Nutrients 2019, 11, 2216. [Google Scholar] [CrossRef] [Green Version]
- Fraga, C.G.; Santos, J.A.; Moutinho-Pereira, J.; Carlos, C.; Silvestre, J.; Eiras-Dias, J.; Mota, T.; Malheiro, A.C. The effects of polyphenols and other bioactives on human health. Food Funct. 2019, 10, 514–528. [Google Scholar] [CrossRef] [Green Version]
- USDA. Nutrient Database. Available online: https://fdc.nal.usda.gov/ (accessed on 5 April 2022).
- OIV. World Vitiviniculture Situation. Wine Producers, 38th World Congress of Vine and Wine, Mainz. 2016. Available online: http://www.oiv.int/public/medias/5029/world-vitiviniculture-situation-2016.pdf (accessed on 15 August 2017).
- CYSTAT. Cyprus External Trade Statistics 2020 VOLUME IV Exports/Dispatches by Commodity and Country; CYSTAT: Lefkosia, Cyprus, 2020. [Google Scholar]
- CYSTAT. Cyprus External Trade Statistics 2019 Volume IV Exports/Dispatches by Commodity and Country; CYSTAT: Lefkosia, Cyprus, 2019. [Google Scholar]
- Khaneghah, A.M.; Fakhri, Y.; Abdi, L.; Coppa, C.F.S.C.; Franco, L.T.; de Oliveira, C.A.F. Mycotoxins in cereal-based products during 24 years (1983–2017): A global systematic review. Trends Food Sci. Technol. 2019, 91, 95–105. [Google Scholar] [CrossRef]
- Karakose, A.; Sanli, S.; Sanli, N.; Bulduk, I. Evaluation of patulin in commercial baby foods by solid phase extraction and liquid chromatography PDA detection. Czech J. Food Sci. 2015, 33, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Sadok, I.; Szmagara, A.; Staniszewska, M.M. The validated and sensitive HPLC-DAD method for determination of patulin in strawberries. Food Chem. 2018, 245, 364–370. [Google Scholar] [CrossRef]
- Zhu, W.; Ren, C.; Nie, Y.; Xu, Y. Quantification of ochratoxin A in Chinese liquors by a new solid-phase extraction clean-up combined with HPLC-FLD method. Food Control 2016, 64, 37–44. [Google Scholar] [CrossRef]
- Eke, Z.; Torkos, K. N,N-dimethyl-trimethylsilyl-carbamate as a derivatizing agent in gas chromatography of trichothecene mycotoxins. Microchem. J. 2004, 77, 43–46. [Google Scholar] [CrossRef]
- Mahmoud, A.F.; Escrivá, L.; Rodríguez-Carrasco, Y.; Moltó, J.C.; Berrada, H. Determination of trichothecenes in chicken liver using gas chromatography coupled with triple-quadrupole mass spectrometry. LWT 2018, 93, 237–242. [Google Scholar] [CrossRef]
- Kiszkiel-Taudul, I. Determination of antihistaminic pharmaceuticals in surface water samples by SPE-LC-MS/MS method. Microchem. J. 2021, 162, 105874. [Google Scholar] [CrossRef]
- Louppis, A.; Michalis, S.C.; Michael, G.K. An ultra-performance liquid chromatography-tandem mass spectrometric method for the identification and quantification of selected natural antioxidants in prickly pear samples. J. Food Compos. Anal. 2021, 104, 104155. [Google Scholar] [CrossRef]
- Naik, R.H.; Pallavi, M.S.; Bheemanna, M.; PavanKumar, K.; Reddy, V.C.S.; Udaykumar, N.R.; Paramasivam, M.; Yadav, S. Simultaneous determination of 79 pesticides in pigeon pea grains using GC–MS/MS and LC–MS/MS. Food Chem. 2021, 347, 128986. [Google Scholar]
- Nicolaou, A.G.; Stavrou, I.J.; Louppis, A.P.; Constantinou, M.S.; Kapnissi-Christodoulou, C. Application of an ultra-performance liquid chromatography-tandem mass spectrometric method for the detection and quantification of cannabis in cerumen samples. J. Chromatogr. A 2021, 1642, 462035. [Google Scholar] [CrossRef]
- Orso, D.; Floriano, L.; Ribeiro, L.C.; Bandeira, N.M.G.; Prestes, O.D.; Zanella, R. Simultaneous determination of multiclass pesticides and antibiotics in honey samples based on ultra-high performance liquid chromatography-tandem mass spectrometry. Food Anal. Methods 2015, 9, 1638–1653. [Google Scholar] [CrossRef]
- Zhou, H.; Liu, P.; Su, X.-C.; Liao, Y.-H.; Lei, N.-S.; Liang, Y.-H.; Zhou, S.-H.; Lin, W.-S.; Chen, J.; Feng, Y.-Q. Development and validation of the one-step purification method coupled to LC-MS/MS for simultaneous determination of four aflatoxins in fermented tea. Food Chem. 2021, 354, 129497. [Google Scholar] [CrossRef]
- Ellison, S.L.R.; Williams, E. EURACHEM/CITAC Guide. Quantifying Uncertainty in Analytical Measurement, 3rd ed.; EURACHEM/CITAC: London, UK, 2011; pp. 1–52. [Google Scholar]
- Galarini, R.; Moretti, S.; Saluti, G. Quality assurance and validation: General considerations and trends. In Chromatographic Analysis of the Environment: Mass Spectrometry Based Approaches, 4th ed.; Nollet, L.M.L., Lambropoulou, D.A., Eds.; CRC Press: Boca Raton, FL, USA, 2017; pp. 325–369. [Google Scholar]
- Silvestro, L.; Tarcomnicu, I.; Savu, S.R. Matrix Effects in Mass Spectrometry Combined with Separation Methods—Comparison HPLC, GC and Discussion on Methods to Control these Effects. In Tandem Mass Spectrometry-Molecular Characterization, 1st ed.; IntechOpen: London, UK, 2013. [Google Scholar]
- Van de Steeny, J.C.; Lambert, E.W. Comparison of Matrix Effects in HPLC-MS/MS and UPLC-MS/MS Analysis of Nine Basic Pharmaceuticals in Surface Waters. J. Am. Soc. Mass Spectrom. 2008, 19, 713–718. [Google Scholar] [CrossRef] [Green Version]
- Beltran, E.; Ibáñez, M.; Sancho, J.V.; Hernández, F. Determination of mycotoxins in different food commodities by ultra-high-pressure liquid chromatography coupled to triple quadrupole mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 1801–1809. [Google Scholar] [CrossRef] [PubMed]
- Giacinti, G.; Raynaud, C.; Capblancq, S.; Simon, V. Matrix-Matching as an Improvement Strategy for the Detection of Pesticide Residues. J. Food Sci. 2016, 5, 1342–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafontaine, C.; Shi, Y.; Espourteille, F.A. Multi-Class Antibiotic Screening of Honey Using Online Extraction with LC-MS/MS. Application Note 464; Thermo Fisher Scientific Inc.: Waltham, MA, USA, 2009. [Google Scholar]
- European Commission. Analytical Quality and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed. SANTE/2017/11813. Implemented by 1 January 2020; European Commission: Brussels, Belgium, 2017. [Google Scholar]
- Torovic, L.; Lakatoša, I.; Majkićc, T.; Bearac, I. Risk to public health related to the presence of ochratoxin A in wines from Fruška Gora. LWT 2020, 129, 109537. [Google Scholar] [CrossRef]
- Čepo, D.; Pelajić, M.; Vrček, I.V.; Krivohlavek, A.; Žuntar, I.; Karoglan, M. Differences in the levels of pesticides, metals, sulphites and ochratoxin A between organically and conventionally produced wines. Food Chem. 2018, 246, 394–403. [Google Scholar] [CrossRef]
- Dachery, B.; Veras, F.F.; Magro, L.D.; Manfroi, V.; Welke, J.E. Exposure risk assessment to ochratoxin A through consumption of juice and wine considering the effect of steam extraction time and vinification stages. Food Chem. Toxicol. 2017, 109, 237–244. [Google Scholar] [CrossRef]
- Remiro, R.; Irigoyen, A.; González-Peñas, E.; Lizarraga, E.; de Cerain, A.L. Levels of ochratoxins in Mediterranean red wines. Food Control 2013, 32, 63–68. [Google Scholar] [CrossRef]
- Bolton, S.; Mitchell, T.; Brannen, P.M.; Glenn, A.E. Assessment of Mycotoxins in Vitis vinifera Wines of the Southeastern United States. Am. J. Enol. Vitic. 2017, 68, 336–343. [Google Scholar] [CrossRef]
- Dias, J.V.; Maria da Graça, P.N.; Pizzutti, I.R.; Reichert, B.; Jung, A.A.; Cardoso, C.D. Simultaneous determination of pesticides and mycotoxins in wine by direct injection and liquid chromatography-tandem mass spectrometry analysis. Food Chem. 2019, 293, 83–91. [Google Scholar] [CrossRef]
- Freire, L.; Braga, P.A.; Furtado, M.M.; Delafiori, J.; Dias-Audibert, F.L.; Pereira, G.E.; Reyes, F.G.; Catharino, R.R.; Sant’Ana, A.S. From grape to wine: Fate of ochratoxin A during red, rose, and white winemaking process and the presence of ochratoxin derivatives in the final products. Food Control 2020, 113, 107167. [Google Scholar] [CrossRef]
- European Commission. Commission Regulation (EC) No 401/2006 of 23 February 2006 Laying Down the Methods of Sampling and Analysis for the Official Control of the Levels of Mycotoxins in Foodstuffs; European Commission: Brussels, Belgium, 2006. [Google Scholar]
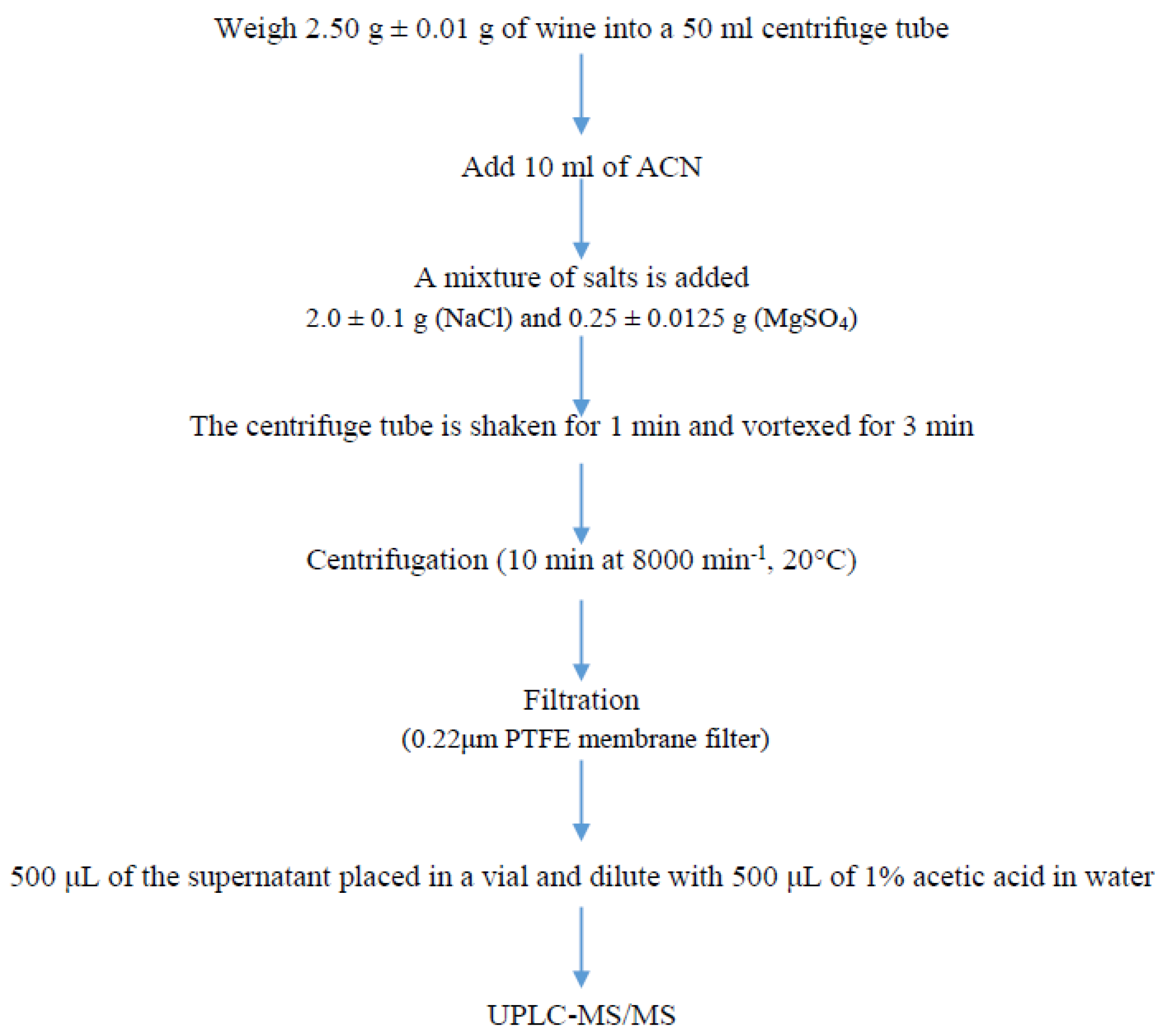

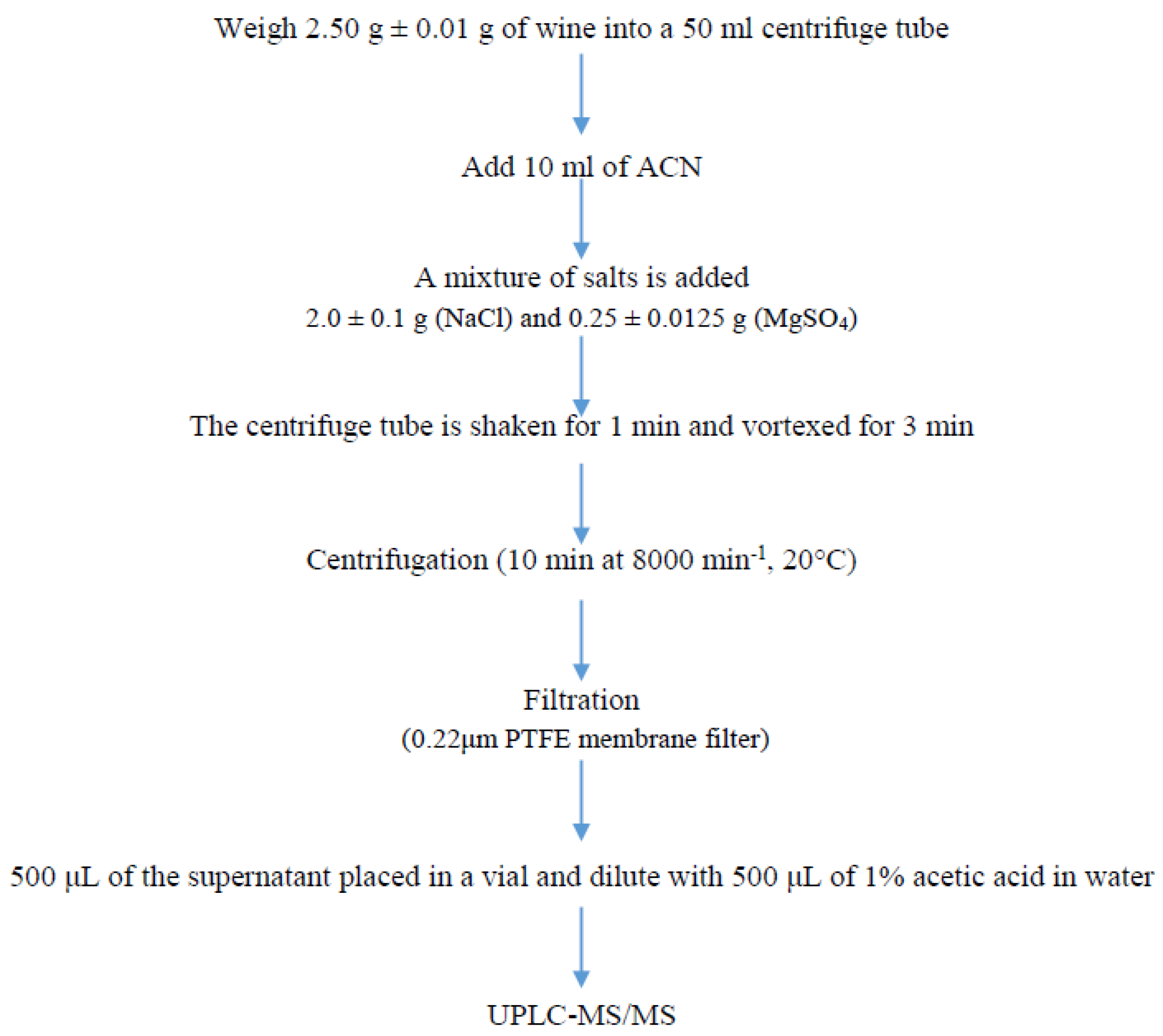{kind=link}
| Wine Type | Production Year | Container Type | Alcohol Strength |
|---|---|---|---|
| Rose Dry | 2020 | G | 12.5 |
| White Dry Xinisteri | 2020 | G | 13.5 |
| White Dry Xinisteri | 2020 | G | 12.5 |
| Lefkadha, Shivas (Dry Rose) | 2010 | G | 13.0 |
| Xinisteri | 2020 | G | 13.0 |
| Xinisteri | 2019 | G | 12.0 |
| Red Dry | 2018 | G | 13.5 |
| Maratheftiko, Lefkada | 2020 | G | 13.0 |
| Rose dry | 2019 | G | 13.5 |
| Red dry | 2019 | G | 13.0 |
| Cabernet Sauvignon | 2009 | G | 13.0 |
| White Dry | --- | G | 12.0 |
| Maratheftiko Red dry | 2017 | G | 13.0 |
| Maratheftiko Red dry | 2019 | G | 13.0 |
| Maratheftiko, Shiraz, dry | 2019 | G | 13.5 |
| Dry Rose | 2016 | P | 11.5 |
| Dry White | 2009 | P | 11.0 |
| Dry Red | 2006 | P | 11.5 |
| Medium sweet red | 2016 | P | 11.5 |
| White medium dry | 2012 | P | 11.5 |
| Medium red | --- | P | 12.0 |
| Dry red | --- | P | 12.0 |
| Red medium sweet | 2018 | P | 13.0 |
| Red dry | 2012 | P | 12.5 |
| Analyte | Precursor Ion | Quantifier Ion | Collision Energy (eV) | Qualifier Ion | Collision Energy (eV) |
|---|---|---|---|---|---|
| Ochratoxin A | 404.10 | 358.10 | 23 | 239.05 | 13 |
| Aflatoxin B1 | 313.25 | 241.05 | 35 | 285.15 | 21 |
| Aflatoxin B2 | 315.10 | 259.10 | 30 | 287.10 | 25 |
| Aflatoxin G1 | 329.10 | 283.10 | 25 | 243.10 | 25 |
| Aflatoxin G2 | 331.10 | 257.10 | 20 | 245.10 | 30 |
| Zearalenone | 319.20 | 185.10 | 23 | 187.15 | 20 |
| Analyte | Linear Regression (R2) | Retention Time (min) |
|---|---|---|
| Ochratoxin A | 0.9986 | 4.94 |
| Aflatoxin B1 | 0.9984 | 3.44 |
| Aflatoxin B2 | 0.9979 | 3.20 |
| Aflatoxin G1 | 0.9985 | 3.21 |
| Aflatoxin G2 | 0.9982 | 3.01 |
| Zearalenone | 0.9986 | 4.92 |
| Recovery (%) | |||||
|---|---|---|---|---|---|
| Analyte | LOD (μg kg−1) | LOQ (μg kg−1) | Conc. A (1 μg kg−1) | Conc. B (5 μg kg−1) | Conc. C (12 μg kg−1) |
| Ochratoxin A | 0.27 | 0.81 | 81 | 103 | 100 |
| Aflatoxin B1 | 0.03 | 0.08 | 86 | 106 | 107 |
| Aflatoxin B2 | 0.12 | 0.36 | 77 | 104 | 107 |
| Aflatoxin G1 | 0.03 | 0.10 | 82 | 108 | 103 |
| Aflatoxin G2 | 0.03 | 0.10 | 94 | 108 | 107 |
| Zearalenone | 0.06 | 0.19 | 91 | 107 | 103 |
| Conc. A | Conc. B | Conc. C | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Mean Concetration (μg kg−1) | SD (μg kg−1) | RSD (%) | MU (%) | Mean Concetration (μg kg−1) | SD (μg kg−1) | RSD (%) | MU (%) | Mean Concetration (μg kg−1) | SD (μg kg−1) | RSD (%) | MU (%) |
| Ochratoxin A | 0.81 | 0.04 | 4.62 | 6 | 5.15 | 0.07 | 1.37 | 6 | 11.98 | 0.23 | 1.92 | 6 |
| Aflatoxin B1 | 0.86 | 0.05 | 6.07 | 15 | 5.34 | 0.16 | 3.07 | 8 | 12.83 | 0.40 | 3.10 | 7 |
| Aflatoxin B2 | 0.77 | 0.04 | 4.90 | 21 | 5.37 | 0.16 | 2.93 | 15 | 12.85 | 0.16 | 1.26 | 8 |
| Aflatoxin G1 | 0.82 | 0.04 | 5.02 | 10 | 5.42 | 0.12 | 2.16 | 5 | 12.41 | 0.36 | 2.92 | 7 |
| Aflatoxin G2 | 0.94 | 0.07 | 7.35 | 7 | 5.38 | 0.13 | 2.47 | 8 | 12.84 | 0.50 | 3.88 | 7 |
| Zearalenone | 0.91 | 0.06 | 6.59 | 15 | 5.36 | 0.15 | 2.82 | 8 | 12.41 | 0.26 | 2.13 | 6 |
| Performance Characteristic | EU Regulation 401/2006 | Proposed Method |
|---|---|---|
| Recovery (B1, B2, G1, G2) 1 ppb | 70–110% | 77–94% |
| Recovery (B1, B2, G1, G2) 5 ppb | 70–110% | 104–108% |
| Recovery (B1, B2, G1, G2) 12 ppb | 80–110% | 103–107% |
| RSD | 0.66 × x RSDR (<15%) | 1.26–7.35% |
| Recovery ΖΕΝ (<50 ppb) | 60–120% | 91–107% |
| Recovery OΤA (≥1 ppb) | 70–110% | 81–103% |
| RSD (OTA) | ≤20% | 1.37–4.62% |
| RSD (ZEN) | ≤40% | 2.13–6.59% |
| Mycotoxins | Detection Method | Limit of Detection | Limit of Quantification | Extraction Method | Recoveries | Run Time | Reference |
|---|---|---|---|---|---|---|---|
| Aflatoxin B1, B2, G1, G2, Diacetoxyscirpenol, Deoxynivalenol, Fumonisin B1, B2, HT2-toxin, Ochratoxin A, T2-toxin, Zearalenone | UPLC-MS/MS | - | 1 and 50 μg kg−1 | acetonitrile and methanol mixture | 70–120% | 10 min | [49] |
| Ochatoxin A | HPLC-FLD | 0.01 μg L−1 | 0.02 μg L−1 | Immunoafinity column | 78% (white wine) and 70% (red wine) | - | [44] |
| Ochratoxin A | UPLC-MS/MS | 1 μg L−1 | 2 μg L−1 | dispersive solid-phase extraction (d-SPE) cleanup | 79–105% | 5 min | [50] |
| Ochratoxin A and fumonisins | LC-MS/MS | 0.1–10 μg L−1 | - | Immunoafinity column | - | 8 min | [48] |
| Ochatoxin A | HPLC-FLD | - | 0.5 μg L−1 | Immunoafinity column | - | 18 min | [45] |
| Ochatoxin A | HPLC-FLD | 0.05 μg L−1 | 0.08 μg L−1 | Immunoafinity column | 96% | - | [46] |
| Ochatoxin A | HPLC-FLD | 0.16 μg L−1 | 0.50 μg L−1 | Immunoafinity column | 74–93% | - | [47] |
| Ochratoxin A Aflatoxin B1, B2, G1, G2, Zearalenone | UPLC-MS/MS | 0.03–0.27 μg kg−1 | 0.08–2.43 μg kg−1 | Acetonitrile | 77–108% | 9 min | Proposed method |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Louppis, A.P.; Constantinou, M.S. Application of a Validated Method for the Identification and Quantification of Mycotoxins in Wines Using UPLC-MS/MS. Separations 2022, 9, 102. https://doi.org/10.3390/separations9040102
Louppis AP, Constantinou MS. Application of a Validated Method for the Identification and Quantification of Mycotoxins in Wines Using UPLC-MS/MS. Separations. 2022; 9(4):102. https://doi.org/10.3390/separations9040102
Chicago/Turabian StyleLouppis, Artemis P., and Michalis S. Constantinou. 2022. "Application of a Validated Method for the Identification and Quantification of Mycotoxins in Wines Using UPLC-MS/MS" Separations 9, no. 4: 102. https://doi.org/10.3390/separations9040102
APA StyleLouppis, A. P., & Constantinou, M. S. (2022). Application of a Validated Method for the Identification and Quantification of Mycotoxins in Wines Using UPLC-MS/MS. Separations, 9(4), 102. https://doi.org/10.3390/separations9040102