1. Introduction
“I joined Prof. Cavallini’s research group in 1984, as medical student at Sapienza University of Rome. I had followed the one-year course in biochemistry held by Prof. Carlo De Marco, one of his collaborators, and decided to carry out the experimental thesis at the Department of Biochemical Sciences. I was thus introduced to Doriano Cavallini, who invited me to follow him to his laboratories, located in an L-shaped corridor, whose entrance was at the back of the department building. An entire wall of the long corridor was occupied by a wooden cupboard with glass doors; I was struck by the hundreds of chemical compounds and reagents and the most disparate glassware they contained. He introduced me to his young collaborators, who assigned me the task of developing, under their guidance, a method for the determination of mercaptolactate in human urine [
1]. Since then, I have been working with Cavallini and his group for more than 20 years [
2]. The purification of the mammalian liver enzyme involved in the formation of cystathionine ketimine was my first article published with the Cavallini research school [
3]. Years later, I received the Cavallini black notebook, as a gift, where he had noted over the years the several chemical syntheses developed or carried out in his laboratory”
Sulfur constitutes one of the fundamental elements in living organisms. The amount of sulfur is around 140 g in a 70 Kg healthy human adult man; thus, it is the seventh most abundant mineral in body. Sulfur can occur in different and often unusual oxidation states. Indeed, the versatility of sulfur biochemistry and biology lies in the ability of sulfur to cycle through a variety of biologically relevant oxidation states ranging from −2, as in hydrogen sulfide (H
2S), to +6, as in sulfate. Sulfur biological compounds exert important functions in all living organisms, and their transformations are involved in free radical scavenging, tissue protection, the modulation of enzyme activity, and the regulation of gene expression both in plants and animals. The metabolism of sulfur-containing amino acids consists of a variety of reactions and pathways, with several intermediates and products whose biochemical significance still needs to be fully elucidated [
4].
Cavallini’s work over 60 years (
Figure 1) was certainly one of the propellants in this field; he made very important contributions to the knowledge of the metabolic interplay between sulfur-containing compounds and the involved enzymes, and of sulfur metabolism diseases. His outstanding ability in chemical synthesis, in a time where very few compounds were commercially available, allowed for the identification of many intermediate metabolites. He was the principal author of more than 250 scientific papers and articles, in which he described for the first time some new derivatives of amino acids such as thiotaurine, alanine thiosulfonate, thiocysteine, cystamine disulfoxide, cystaldimine, thialysine, selenalysine, and selenaproline, and a whole series of cyclic sulfur-containing compounds that he named ketimines [
5]. A paper from 1981 reports the enzymatic in vitro synthesis of cystine ketimine (a seven-membered sulfur-nitrogen-containing cyclic compound), produced by oxidation of L-cystine by L-amino acid oxidase [
6]. It is the first paper concerned with ketimines, and the first one out of more than 70 papers and 18 years research of his group [
7]. Nowadays, this class of natural compounds has been well characterized and studied, although many aspects remain to be disclosed [
8,
9,
10,
11,
12]. The discovery of persulfide groups, and of sulfoselenide groups, in proteins [
13,
14,
15,
16], is also noteworthy, and this has been a very active research field in recent years [
17,
18,
19,
20]. More significant than his written contributions to the development of metabolic biochemistry, however, are the ideas that he passed on to many of his coworkers and still active “fellows”, which produced an avalanche of further investigations. We, his latest coworkers, received “Cavallini black notebook” as a gift, where he had noted over the years all the techniques he had learned and used, and the several chemical syntheses developed or carried out in his laboratory.
In this paper, we report the synthesis of sulfur biomolecules mostly developed by Cavallini school. All the reported chemical syntheses were taken from the “Cavallini black notebook”. Some syntheses were originally developed by Cavallini and his early pupils, others were transcribed by Cavallini from original papers. These last have been retranslated into English, taking into account the original works and the observations that Cavallini himself reported in notebook. Many of the chemical compounds of which we report the synthesis are not yet commercially available. Moreover, the chemical preparations of these sulfur-containing compounds are currently difficult to find and often not available in digital form. We hope that our aim to share these preparations will be useful to researchers interested in sulfur chemistry, biochemistry and metabolism.
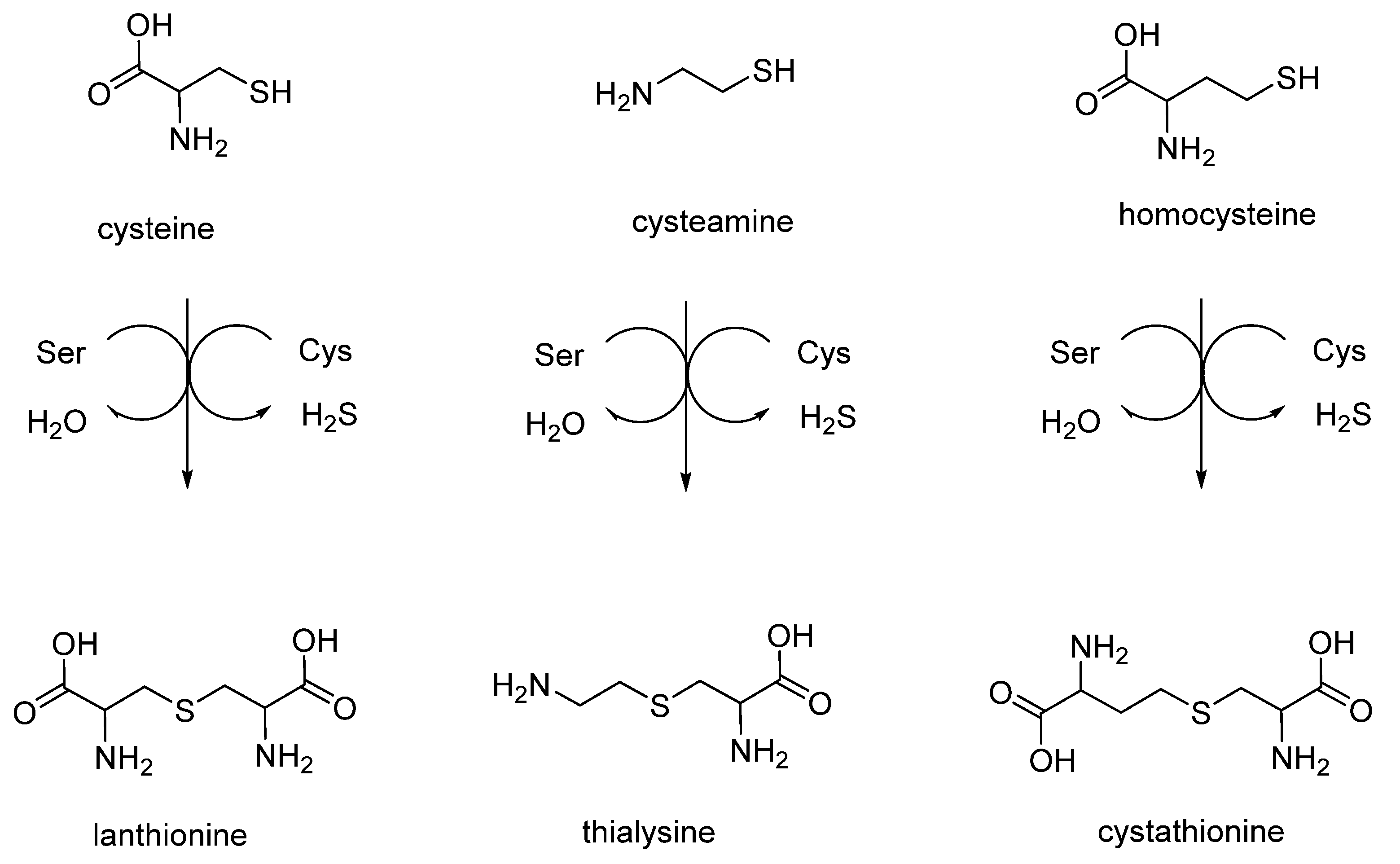
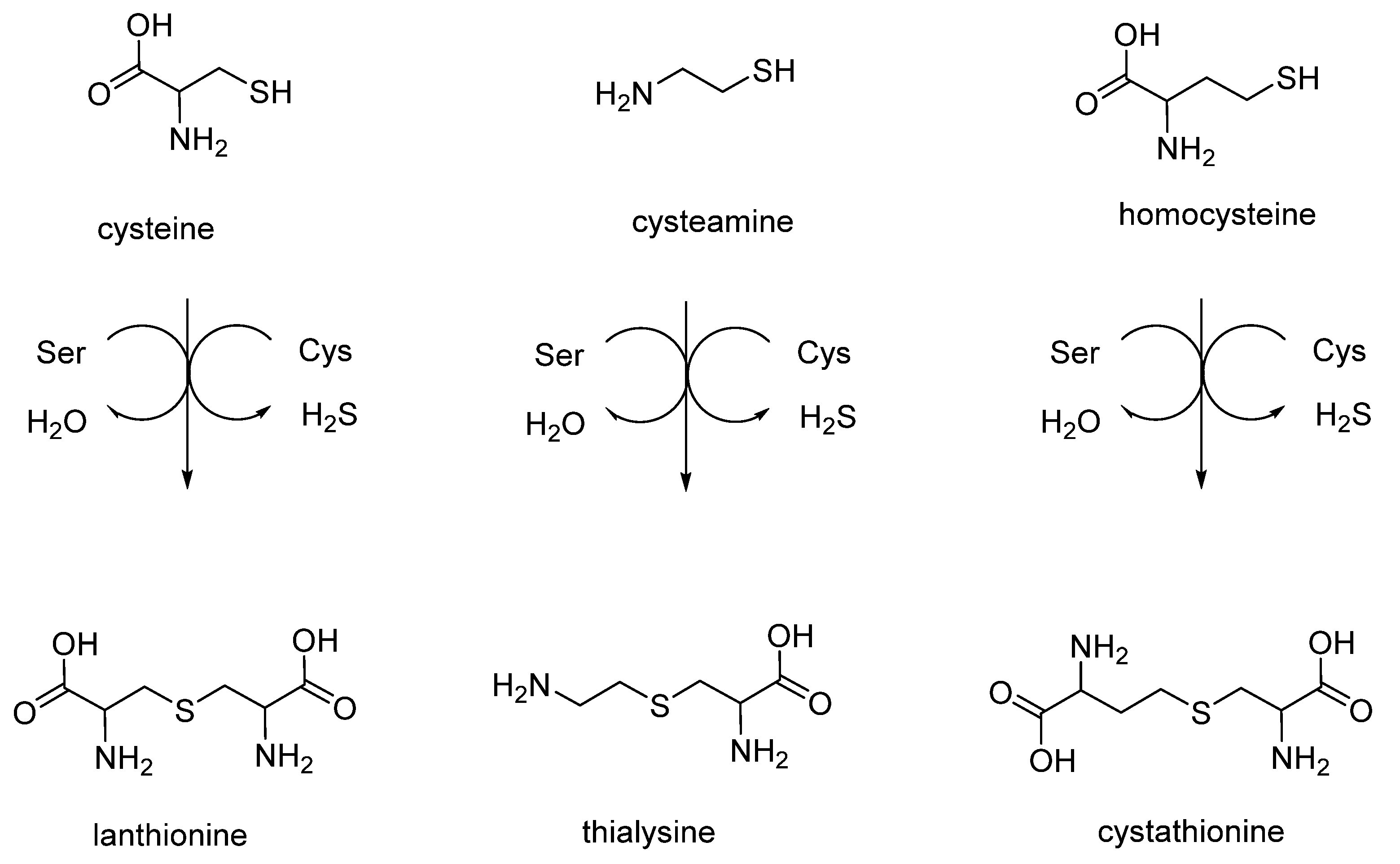
2. Linear Thioethers (Lanthionine, Cystathionine and Thialysine)
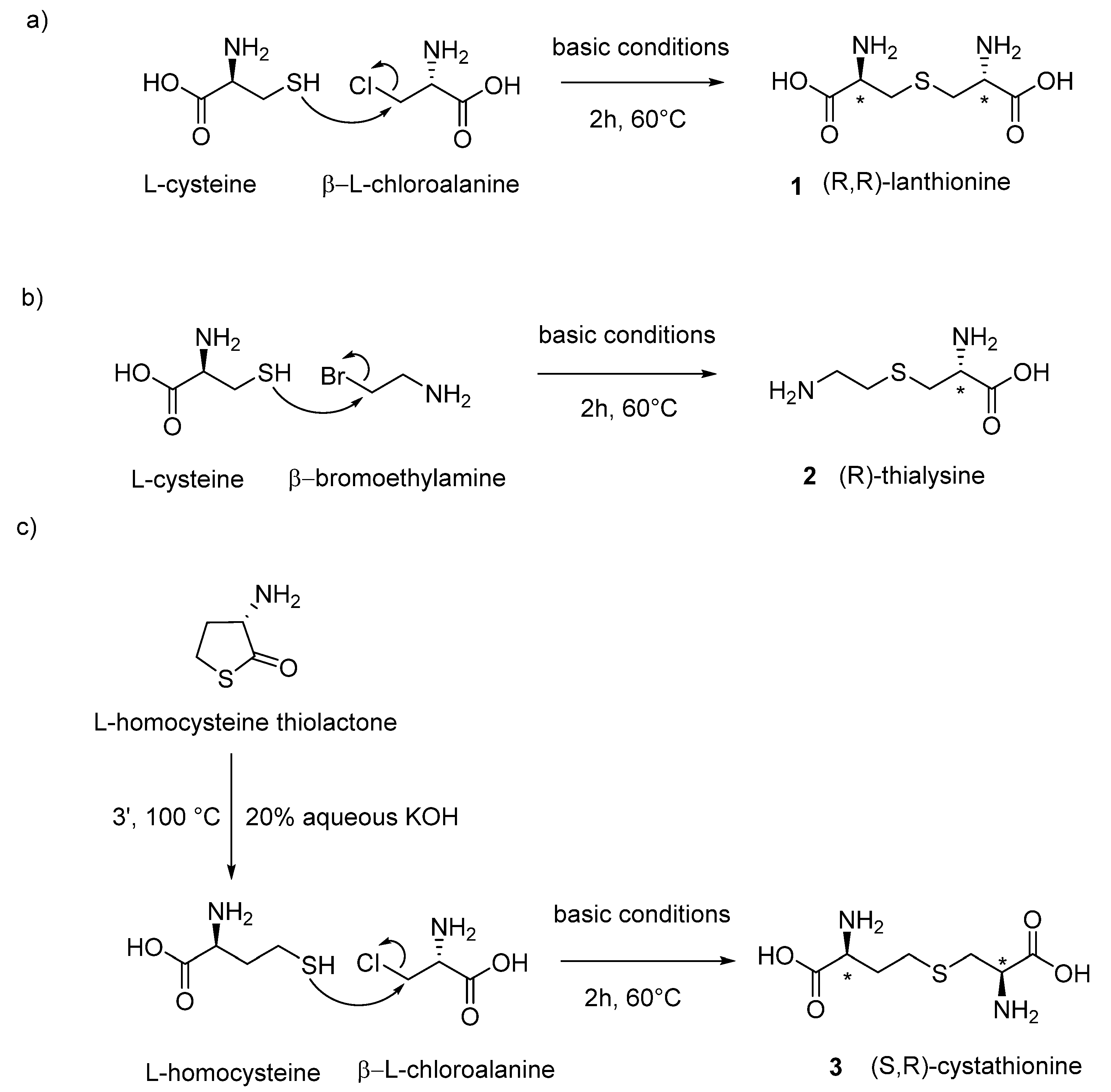
Linear lanthionines are produced by different organisms, including mammals and humans, and are of significant importance and interest in the still unexplored field of sulfur metabolism. Different synthetic strategies were set up to produce biologically important thioethers lanthionines derived in vivo from sulfur amines and amino acid metabolism (
Figure 2).
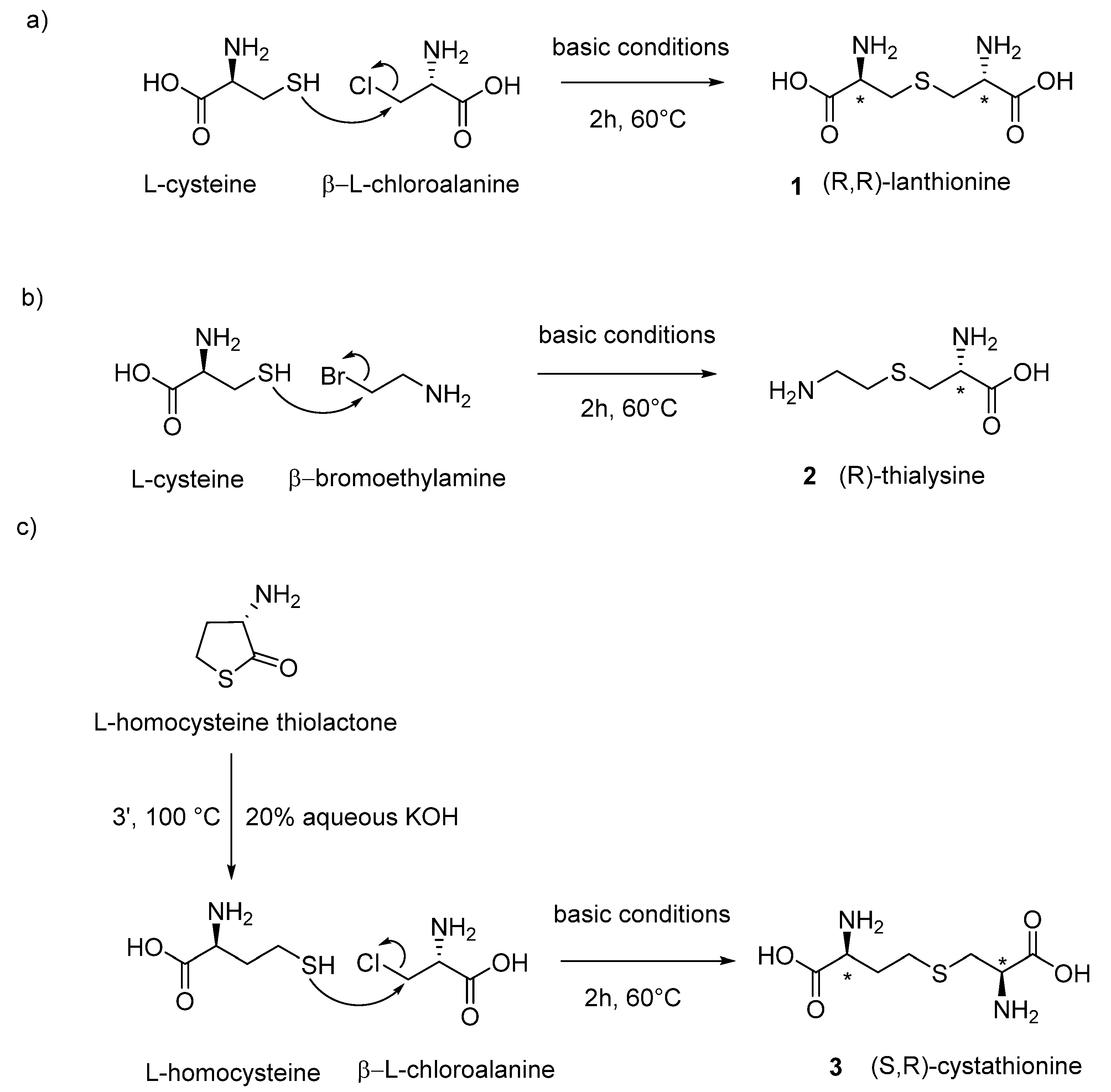
The chemical synthesis of linear lanthionines utilized in Cavallini’s laboratory is similar to the strategy adopted by Brown and Du Vigneaud [
21] and involves the use of two reagents in aqueous basic conditions. In two cases, β-chloroalanine is utilized as one portion of the product that, depending on the use of different thiols as second reagent, can give rise to lanthionine or cystathionine. In the case of thialysine, the synthetic strategy involves as starting materials bromoethylamine and L-cysteine, which condensate to form aminoethylcysteine (also called thialysine). In
Figure 3, the general synthetic conditions for the synthesis of biological stereoisomeric products (L-series reagent as precursors) are reported.
(
1)
L-Lanthionine: L-cysteine free base (210 mg, 2.0 mmol) was slowly added (4 h) under nitrogen atmosphere to a solution of 210 mg of β-L-chloroalanine in 2 mL of deoxygenated water. The pH of the solution was maintained between 8 and 9 by adding small amounts of concentrated NaOH (20µL). The reaction was then left under stirring at 50 °C. After 2 days, the reaction was negative to the Folin–Marenzi test for the presence of free thiols. After cooling, the white precipitate was washed three times with absolute ethanol and diethyl ether. After the last wash, 130 mg of a white powder was obtained [
21,
22].
(
2)
L-Thialysine (aminoethylcysteine): L-cysteine hydrochloride (5 g in 10 mL of deoxygenated water) was kept under a flow of nitrogen for a period of 15 min. Maintaining the nitrogen atmosphere, 10 mL of water containing 6 g of KOH was added. At this solution, 7 g of β-bromoethylamine hydrobromide was maintained for a period of 10 min in water bath at 60–70 °C. Next, the solution was left at room temperature under nitrogen for 3 h and neutralized with hydrobromidric acid and 80 mL of ethanol added. After 4–5 h at 0 °C, the solution was filtrated and concentrated to 20 mL under reduced pressure; then, 60 mL of alcohol was added and the suspension dissolved by warming and adding a small amount of water. After a night at 0 °C, the solution was filtered from a new precipitate of KBr. The supernatant, which contains thialysine as a mixed hydrobromide and hydrochloride salt, was chromatographed through a Dowex 50 column (2.5 cm × 40 cm, acid form H
+, 80–100 mesh) to eliminate HBr and HCl. After washing of the column with 500 mL of water, the product was eluted with 200 mL of 1 N NH
3. After drying under reduced pressure, the oily residue was dissolved in 20 mL of water and neutralized with concentrated HCl. Then, 80 mL of ethanol was added, and the product precipitated by slowly adding, with vigorous shaking, 100 mL acetone. The oily precipitate, by shaking and rubbing, solidified into a semi-crystalline mass. After few days at 0 °C, the supernatant was removed, and the solid mass was mechanically detached from the walls of the container, broken in the presence of acetone, filtered and dried (4.8 g, 75% yield). The products were recrystallized by using the following procedure: 1 g of product was suspended in 95% ethanol, and then boiling water was added in a small portion until complete solubilization. Solution was filtered, and acetone was added by shaking until a slight permanent turbidity was reached. The solution was clarified by boiling and left at room temperature for a few hours. Acetone was then added in drops until a slight turbidity was obtained again: after rubbing the walls and after 24 h at 0 °C, the compound crystallized spontaneously. Recrystallization with acetone was repeated 4–5 times to obtain 5.0 g of the pure product [
23].
(
3)
L-Cystathionine: 11.6 mmol of L-homocysteine thiolactone (1.8 g) was treated with 23.2 mmol of KOH (930 mg) in a volume of 5 mL of degassed water for 3 min at 100 °C to allow the opening of the thiolactone ring. The solution was heated in a water bath to 60 °C under nitrogen atmosphere. Slowly, during a period of 2 h, 10 mmol of β-L-chloroalanine was added (1.6 g). After 2 h, solid pellets of KOH were added until they become alkali, and they were left to stay for 9–10 h. A considerable amount of KCl precipitated, and 20 mL of oxygen-free water was added for complete solubilization of the precipitated KCl. The nitrogen atmosphere was maintained, and the solution was neutralized to pH 6 by the addition of concentrated HCl. The flask, still filled with nitrogen, was tightly stoppered, and was placed in the refrigerator overnight. The precipitate was dissolved in 150 mL of 2 N HCl, was treated with a small amount of darko at room temperature, and was filtered. The solution was slowly neutralized with 3 N NH
4OH. The crystalline precipitate is filtered and washed with water (36% yield) [
24].
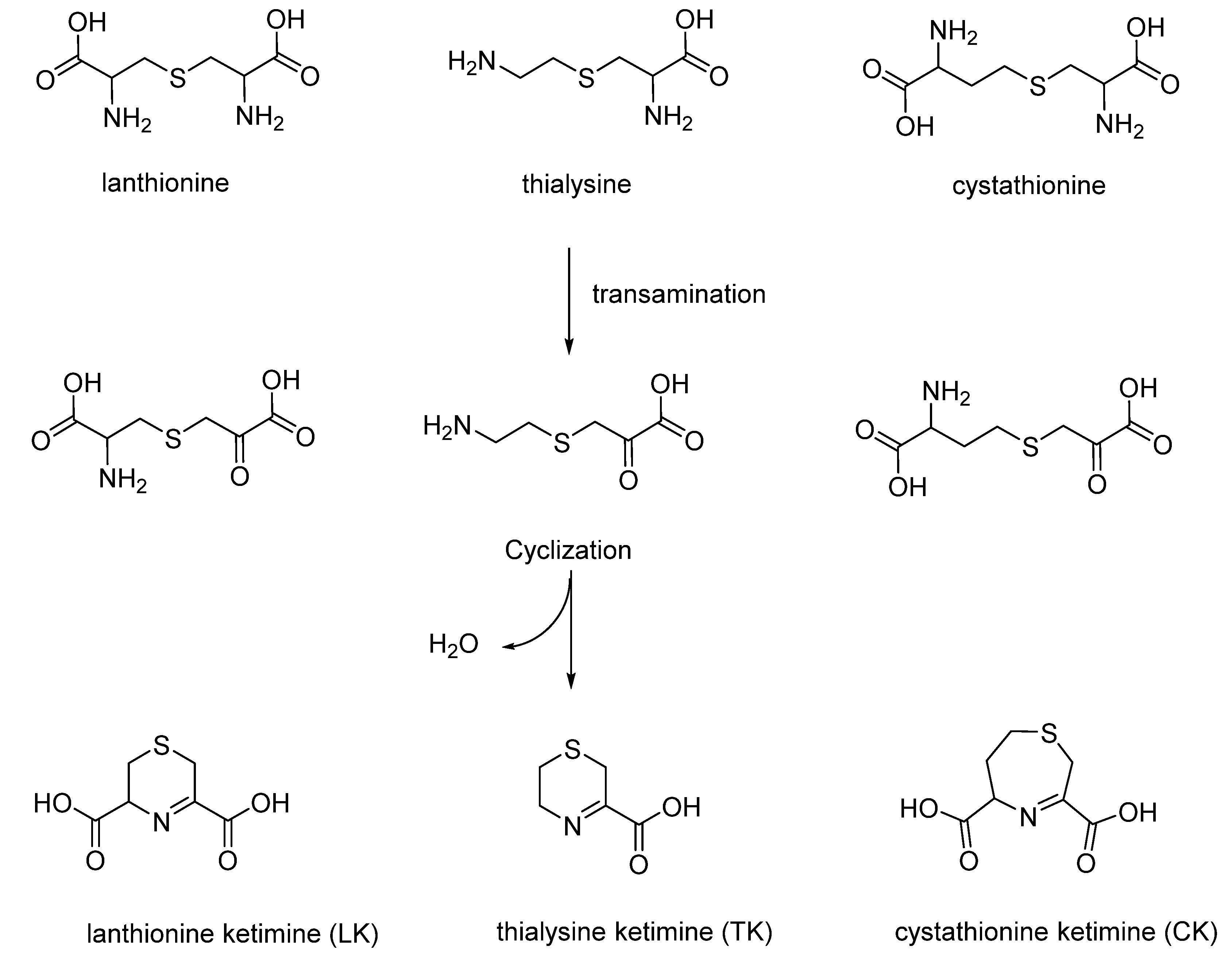
3. Cyclic Sulfur Ketimines (Lanthionine Ketimine, Cystathionine Ketimine and Thyalysine Ketimine)
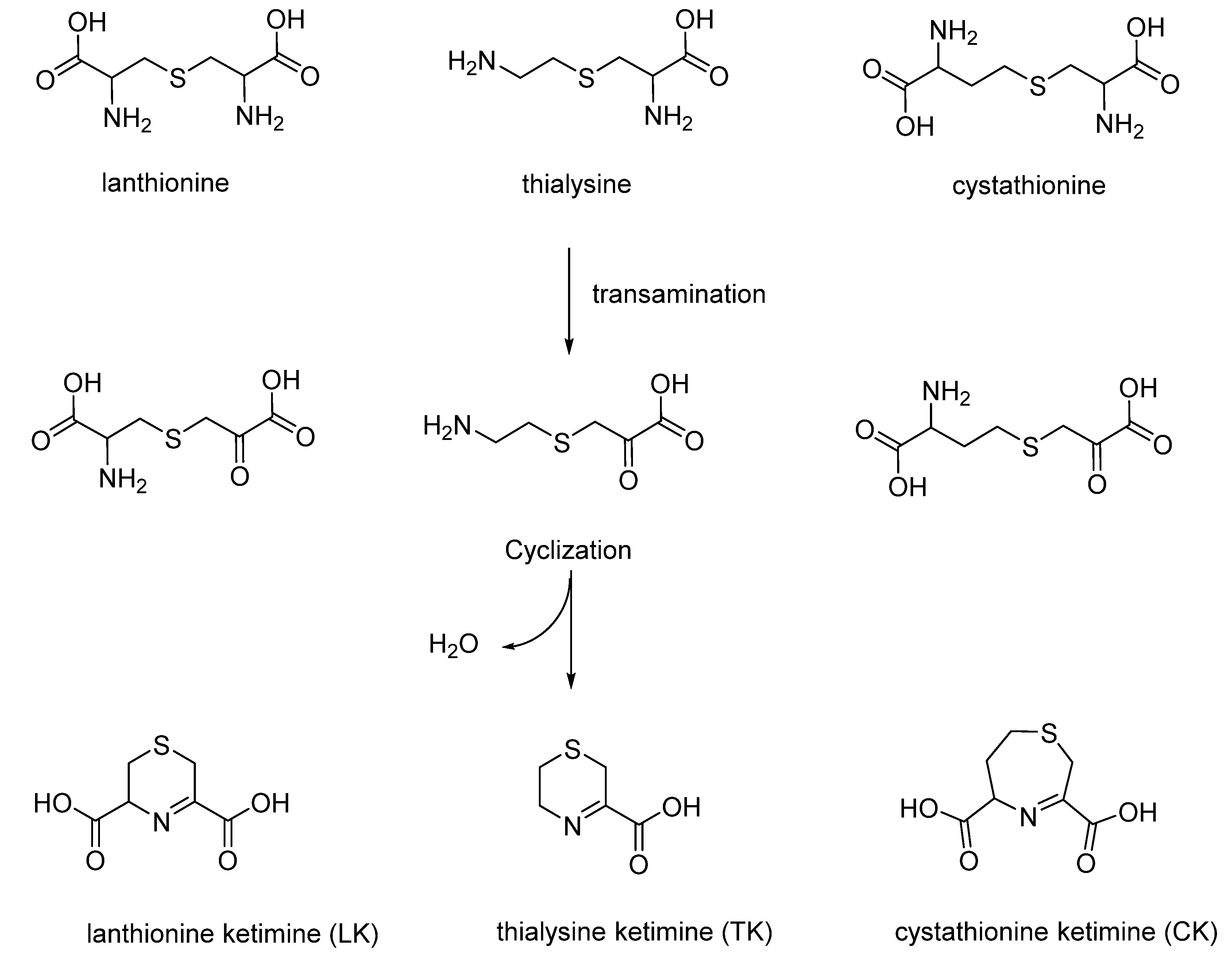
Sulfur cyclic compounds derived from thioether metabolism are very interesting class of products that were synthesized, studied in depth and found in mammals and humans by Prof. Cavallini during the past century (
Figure 4) [
7,
25,
26].
Cavallini and co-workers developed a totally aqueous synthetic strategy to produce lanthionine ketimine LK, cystathionine ketimine CK, thialysine ketimine TK and its decarboxylated dimer TK-DD. It is important to take into account that subsequent studies on these compounds, especially LK, underlined the unique biological properties of these natural products, in particular as neuroprotective molecules [
10,
27,
28,
29]. In
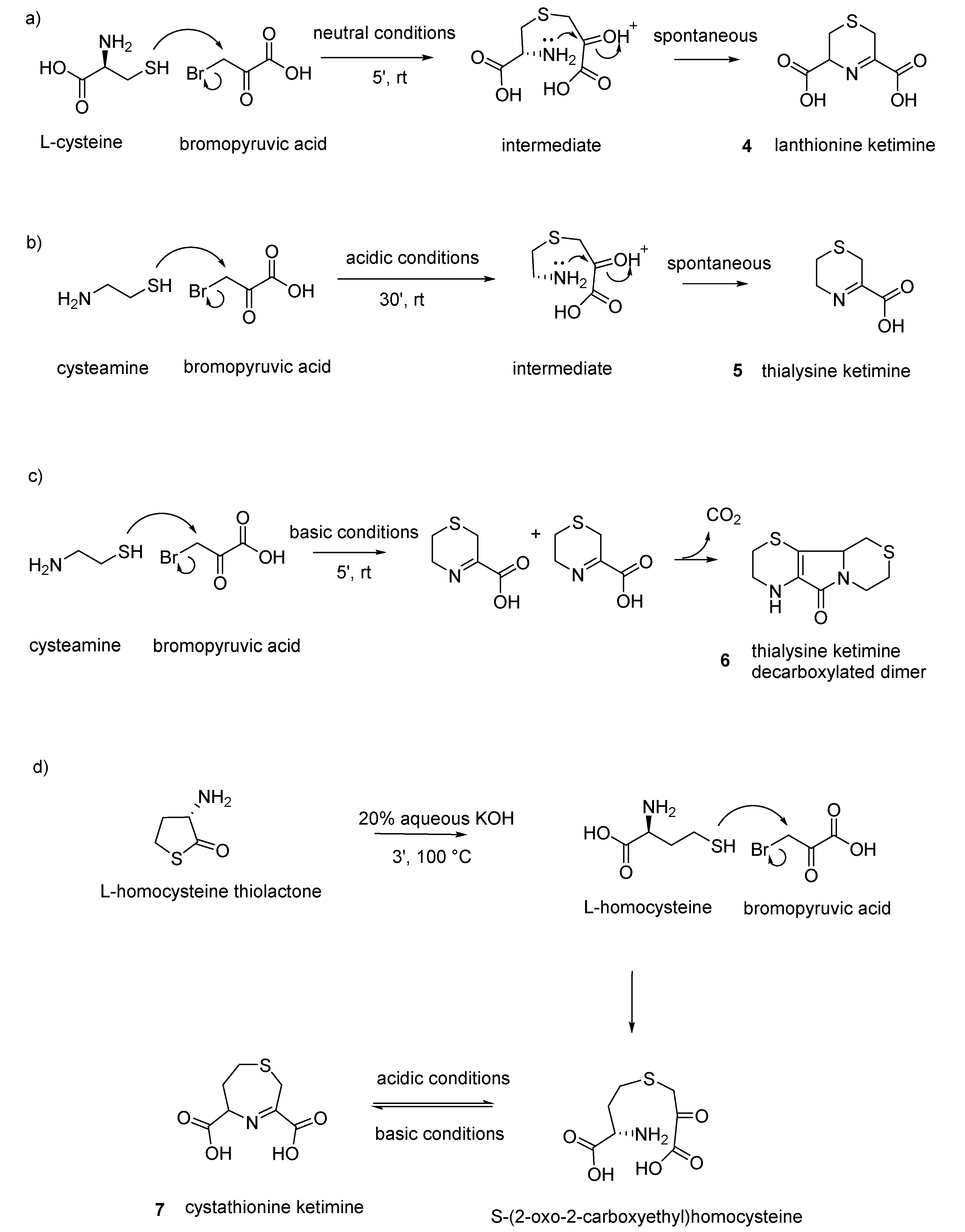
Figure 5, the synthetic strategies for the synthesis and purification of the four compounds are reported.
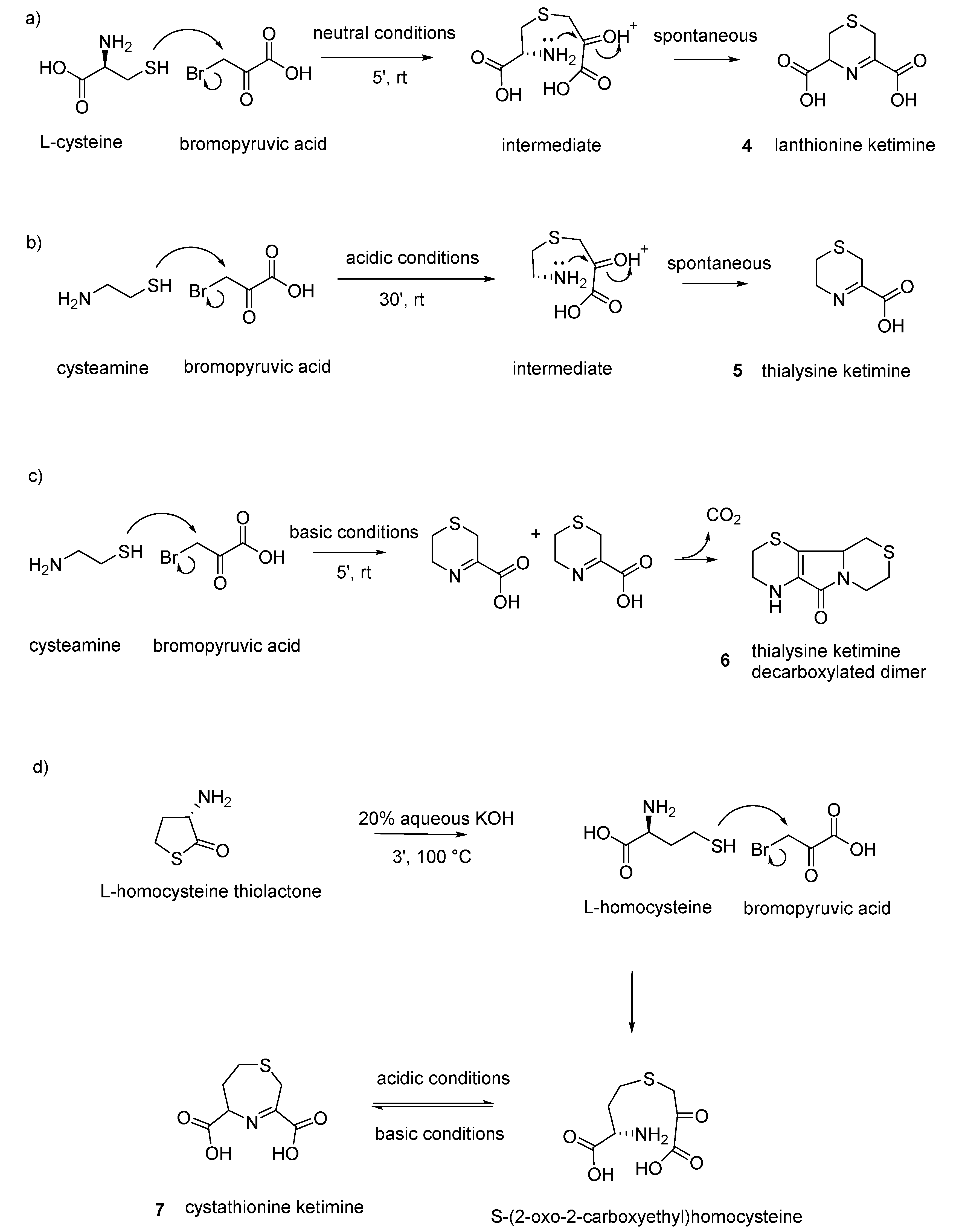
(
4)
Lanthionine ketimine (LK): 6 mmol (0.95 g) of L-cysteine is dissolved in 5 mL of water, and 6 mmol (1 g) of bromopyruvic acid in 2 mL of water is added. The condensation between brompyruvic acid and cysteine is followed-up by checking the disappearance of thiol group with the nitroprusside test. After few minutes at room temperature, the reaction is complete and the condensation product precipitates as white solid crystals. Crystals are filtrated and washed three times with cold acidic water and dried (50% yield) [
30].
(
5)
Thialysine ketimine (TK): 136 mg of cysteamine hydrochloride is added to a solution of 200 mg (1.2 mmol) of bromopyruvic acid dissolved in 2 mL of glacial acetic acid. After 30 min at room temperature, a white product is formed (sometimes preceded by the formation of a yellow sticky product that, after 2–3 h, becomes white). The product (36% yield) is washed with acetic acid and dried under vacuum [
30].
(
6)
Decarboxylated thialysine ketimine dimer (TK-DD): 3.4 g (20.0 mmol) of bromopyruvic acid dissolved in 5 mL of water is added to a solution of 2.27 g of cysteamine hydrochloride (20 mmol) dissolved in 1 mL of water and 10 mL of 6 N NaOH (room temperature). After 30 min standing, 4 mL 6 N HCl is added, and after 30 min in an ice bath the precipitate is collected in a sintered glass funnel. Next, 120 mL of water is added to the product and heated for 30 min until decarboxylation occurs. A yellow product crystallizes and precipitates after one night at 4 °C. The solid product is washed two times with cold water and dried (1.15 g: 50% yield). The product exhibits the spectral and chromatographic properties reported earlier [
31,
32]. Recrystallization may be done by dissolving 0.5 g in 50 mL boiling water and cooling [
33]. A procedure for thialysine ketimine decarboxylated dimer synthesis in gram amounts has been also presented [
34].
(
7)
Cystathionine ketimine (CK): with respect to the other ketimine, CK does not cyclize spontaneously after the reaction and needs two sequential steps to be prepared. The first step is the synthesis of the condensation product between bromopyruvic acid and cystathionine to give rise to the opened-ring keto-acid S-(2-oxo-2-carboxyethyl)homocysteine (OCEHC) in a solid form, and then there is a second step in which OCEHC is cyclized to CK. OCEHC is prepared as follows: 154 mg (1 mmol) of L-homocysteine thiolactone hydrochloride is dissolved in 2 mL 1 N NaOH and heated, under N
2 bubbling, in a boiling water bath for 3 min to open the thiolactone ring. Next, 1.2 mmol (0.2 g) of bromopyruvic acid in 5 mL water is added to this solution after cooling under N
2 atmosphere. After 10 min, the reaction is complete (negative to nitroprusside test) and the solution is passed through a 1 cm × 5 cm column of Dowex 50 × 8 (H
+ form, 200–400 mesh). The column is eluted with water, and the fractions between 70 and 120 mL (checked by the iodoplatinate test) are collected and dried with a rotary evaporator at 40 °C to obtain about 80 mg of OCEHC yellowish crystals. Then, 22 mg OCEHC (0.1 mmol) is dissolved in 1 mL degassed water under N
2 bubbling; 6 mL of 96% ethanol is added; and, under gentle N
2 bubbling, four portions of 25 µL of 2 N NaOH are added over a period of 20 min. After cyclization (15 min), the product is dried under vacuum with a rotator evaporator. The residue is dissolved in 0.25 mL water and 5 mL of absolute ethanol is added. The precipitate is removed by centrifugation, and the supernatant is dried under vacuum to obtain about 10 mg of white powder. Solid CK salt is stored in the dessiccator under vacuum [
35].
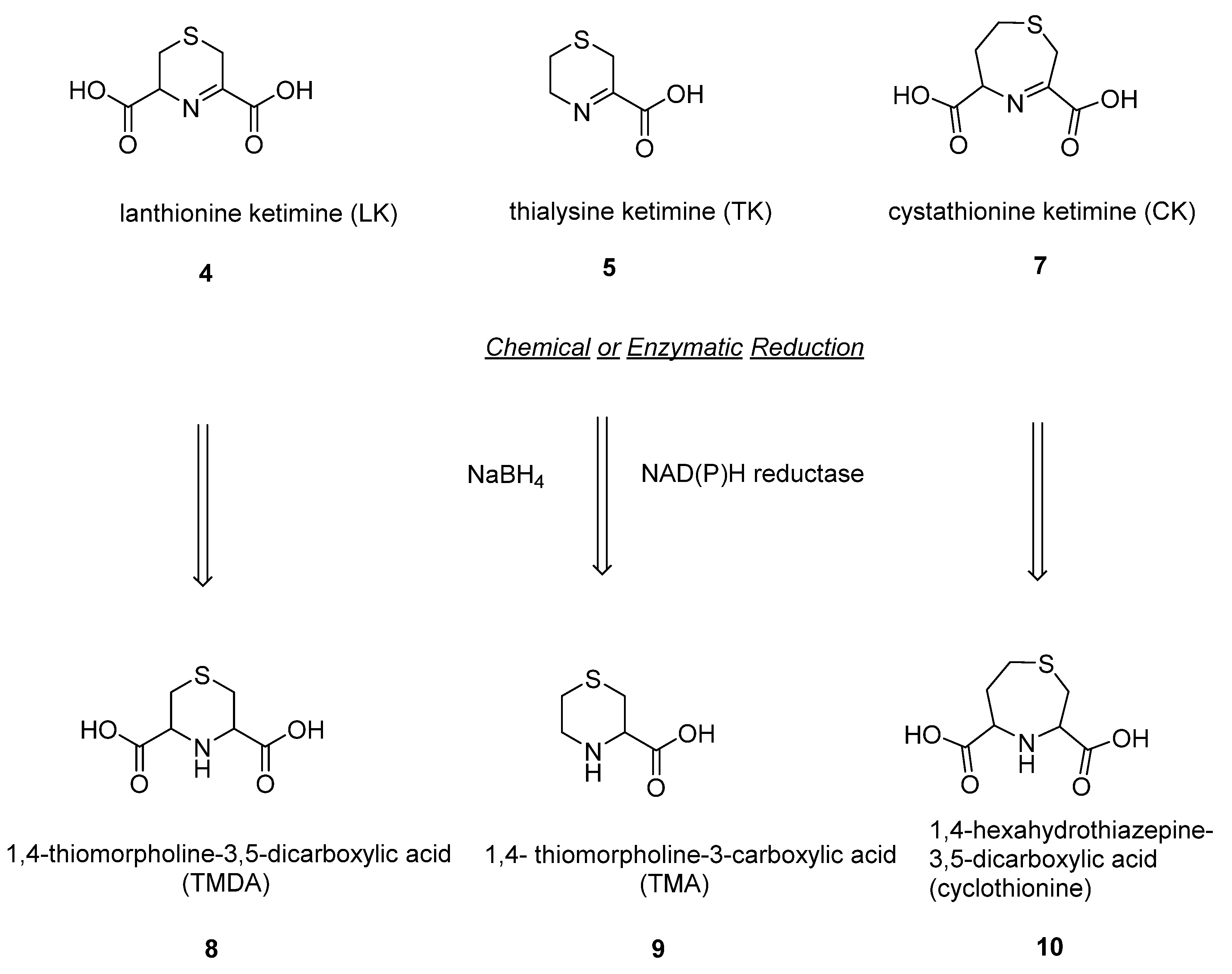
4. Cyclic Reduced Sulfur Ketimines (1,4-Thiomorpholine-3-carboxylic Acid; 1,4-Thiomorpholine-3,5-dicarboxylic Acid; 1,4-Hexahydrothiazepine-3,5-dicarboxylic Acid)
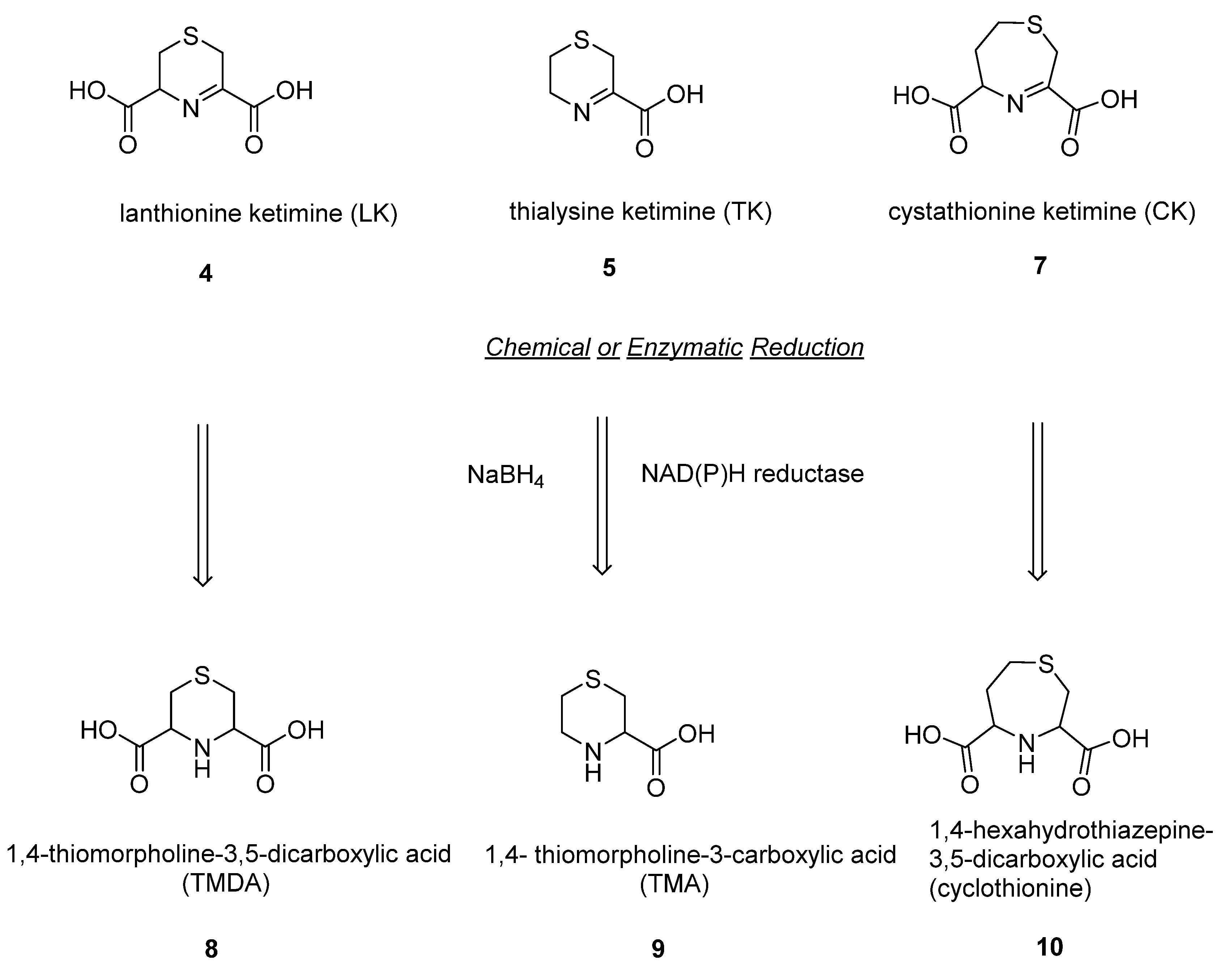
The enzymatic reduction of the sulfur-containing cyclic lanthionines (LK, CK and TK) produces novel saturated compounds detected in different mammalian districts, whose biological functions are still unknown [
36,
37,
38,
39,
40]. These compounds can be chemically generated by the reduction of their unsaturated precursors with NaBH
4; meanwhile, their biological synthesis seems to be mediated by an NAD(P)H-dependent enzyme found in animal tissues (
Figure 6) [
41,
42].
(
8)
1,4-thiomorpholine-3,5-dicarboxylic acid (
reduced lanthionine ketimine, TMDA): 1.5 mmol of LK dissolved in 50 mL of water 25 mmol of NaBH
4 is added by maintaining the pH of the solution at 7 with HCl. After the reduction (monitored by the disapparence of the UV absorbance at 296 nm), the reaction is acidified to pH 2 with 4 N HCl and then passed on a DOWEX 50 × 8 column (H
+ form, 200–400 mesh, 2 cm × 35 cm). After washing with water, the elution is conducted with 3 N of ammonia (eluted fractions are positive to iodoplatinate test for tioethers). Eluted fractions are dried under reduced pressure, dissolved in a minimum amount of water and passed through a second DOWEX 50 × 8 column (H
+ form, 200–400 mesh, 2 cm × 5 cm). Elution with water gives rise to two positive fractions regarding iodoplatinate test (180–320 mL and 380–420 mL eluates). The first fraction, after drying, produces 140 mg of a white solid [
43].
(
9)
1,4-thiomorpholine-3-carboxylic acid (
reduced thialysine ketimine, TMA): 600 mg of bromopyruvic acid (3.6 mmol) dissolved in 90 mL of water 408 mg (3.6 mmol) of cysteamine HCl is added, and the formation of a chromophore at 296 nm is checked in phosphate buffer pH 7.6 (epsilon 6200). The solution is then adjusted to pH 8 with NaOH, and 30 excesses of NaBH
4 are added. In a few minutes, the absorption at 296 nm disappears and the pH is brought to 3 with HCl. One third of the final volume (100 mL) is then loaded on a DOWEX 2 × 8 column (200–400 mesh, 3 cm × 15 cm, OH-form). After washing with 300 mL of water, the column is eluted with 1 N acetic acid. 150 mL of acidic eluate (yellow fractions positive to ioplatinate test) is dried and redissolved in a small amount of water. After acidification to pH 2 with acetic acid, the product is passed through a DOWEX 50 × 4 column (200–400 mesh, H
+ form, 2 cm × 35 cm). The column is washed with 300 mL of water and then eluted with 1 N HCl. Three fractions are positive with regard to iodoplatinate test (first, 20 mL, second 80 mL and third 150 mL). The third fraction is dried under reduced pressure, and 120 mg of crystals is obtained [
43].
(
10)
1,4-hexahydrothiazepine-3,5-dicarboxylic acid (
reduced cystathionine ketimine, cyclothionine): 1 mmol of L-homocysteine, obtained from homocysteine thiolactone hydrochloride as described above, is slowly added under N
2 bubbling with 0.2 g of bromopyruvic acid dissolved in 50 mL of water, and the pH is adjusted continuously at 12. The solution is first placed in a bath at 37 °C for 60 min; temperature is then raised to 50 °C, and 2 g of NaBH
4 is added in portions. Reduction is followed by the decrease of the absorbance at 296 nm. After 5 h, the absorbance almost disappears. The excess NaBH
4 is destroyed by dropwise addition of concentrated HCl up to pH 5. The solution is then introduced into a Dowex 50 column (H
+ form, 200–400 mesh, 2 cm × 30 cm), which is eluted with deionized water. The compound is located with the iodoplatinate reagent between 400 and 1200 mL of the effluent. The solution is dried under reduced pressure, yielding a white solid which is recrystallized twice from hot water (120 mg yield) [
35].
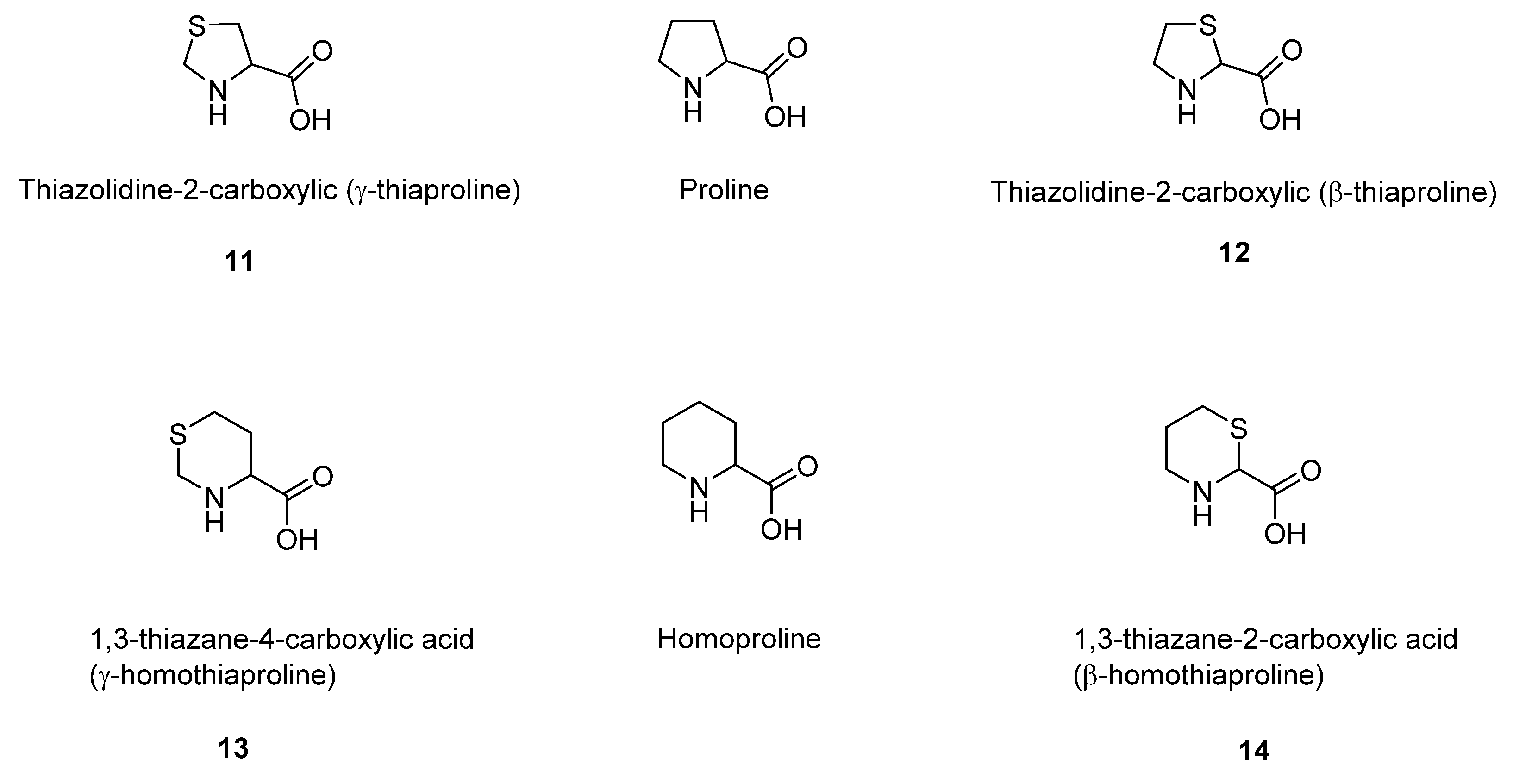
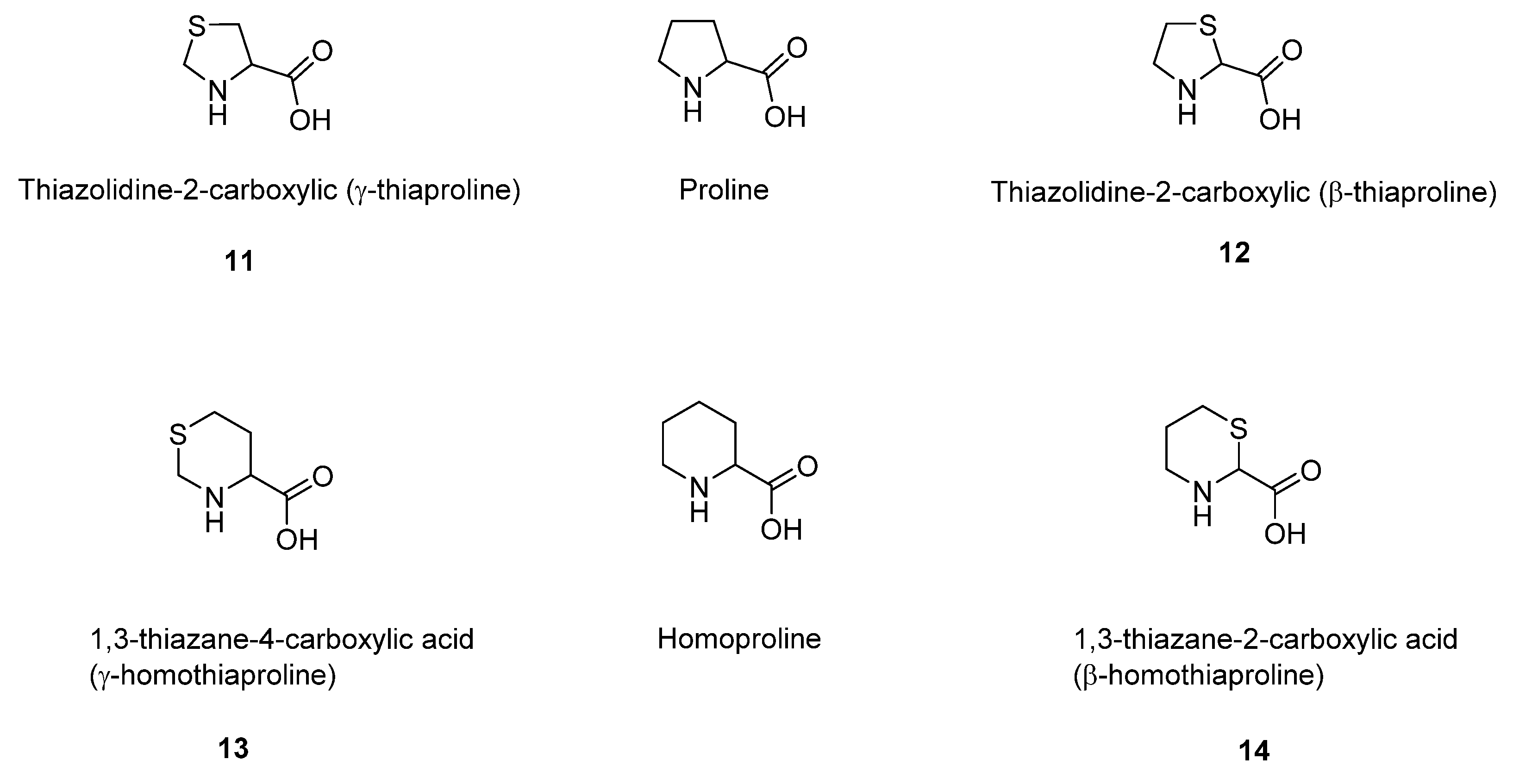
5. Sulfur-Containing Analogues of Proline and Homopropoline
Another important class of sulfur organic metabolites is represented by the sulfur-containing proline (and homoproline) analogues. During the last century, Prof. Doriano Cavallini and his research team studied the capacity of different enzymatic biological systems to metabolize thiaproline (thiazolidin-carboxylic acid) and homothiaproline (thiazane-carboxylic acid) [
44,
45,
46]. Cavallini and his co-workers were among the first ones able to synthesize and characterize some of these analogues. In more recent studies, thiaproline was found to be an effective nitrite-trapping agent in human body, to protect cells from oxidative stress and able to be incorporated into cellular proteins [
47,
48,
49]. Thiaproline is the analogue of proline in which a methylene group in the ring is substituted by a thioeter sulfur atom. Thiazolidine-2-carboxylic acid (β-thiaproline) differs from thiazolidine-4-carboxylic acid (γ-thiaproline) for the position of the sulfur atom in the thiazolidine ring (
Figure 7). Homoproline is the one carbon in-ring elongated analogue of thiaproline. Additionally, in this case one of the CH
2 cyclic groups can be substituted with a thioeter moiety to give rise to homothiaproline, which can exist as 1,3-thiazane-4-carboxylic acid (γ-homothiaproline) and 1,3-thiazane-2-carboxylic acid (β -homothiaproline) (
Figure 7).
(
11)
Thiazolidine-4-carboxylic (
γ-thiaproline): The cysteine hydrochloride from 30 g of cystine was dissolved in 100 mL of water; 22 mL of commercial 40% formaldehyde (1.1 mol) was added, and the mixture allowed to stand overnight at room temperature. On the addition of 25 mL of pyridine, crystals soon separated. The whole was diluted with 50 mL of alcohol and filtered. The product (28.2 g.) was recrystallized from hot water. The resulting long, colorless needles, which melt with decomposition at 196–197°, are insoluble in alcohol; somewhat soluble in cold water; readily soluble in hot water, in acid and in alkali [
50].
(
12)
Thiazolidine-2-carboxylic (β-thiaproline): a solution of 2-aminoethanethiol hydrochloride (cysteamine HCl) (19.2 g, 0.17 mmol) in MeOH was treated with NaOCH
3 (9.1 g, 0.17 mmol). After the solvent was removed in vacuo, the free base was treated with a solution of glyoxylic acid (12.5 g, 0.17 mmol) in absolute EtOH (100 mL). This mixture was shaken vigorously for several minutes and then cooled in an ice bath. The yellow precipitate was collected and recrystallized from 70% EtOH to afford 11.2 g of the products (50% yield) [
51].
(
13)
1,3-thiazane-2-carboxylic acid (
γ-homothiaproline): Homocysteine (18.4 mmol) was dissolved in 30 mL of hot, air-free, double distilled water and filtered to remove undissolved homocystine present as a contaminant. The solution was then transferred to an aeration vessel, the pH adjusted to approximately 8 with dilute NaOH, and the vessel flushed with nitrogen for 5 min. An equimolar amount of formaldehyde (552 mg in 22.2 mL of water) was added, and the vessel was again flushed with nitrogen and sealed. On the following day, the mixture was filtered and lyophilized. The powder was twice crystallized from methanol-ether, to which a little concentrated HCI was added and a crystalline hydrochloride was obtained. The yield of thiazane carboxylic acid hydrochloride, recrystallized once, was 2.5 g (74% yield) [
52].
(
14)
1,3-thiazane-2-carboxylic acid (
β-homothiaproline): homocysteamine (ca. 2.0 mmol) was obtained by reduction of homocystamine (250 mg) with dithioerythriol DTE (monitoring spectrofotometrically the oxidized DTE formation at 280 nm). After homocystamine reduction, 296 mg (2.3 mmol) of sodium glyoxate in alkali conditions was added, and the solution was kept in a water bath at 50 °C in a nitrogen atmosphere. After 30 min, the nitroprusside and the Folin–Marenzi reactions showed that thiol groups had practically disappeared, thus indicating that all homocysteamine had reacted. The solution was percolated through a 2 cm × 5 cm column od DOWEX 50-H
+ (200–400 mesh), and the column washed neutral with water and then eluted with 1 N NH
4OH, collecting fractions of 5 mL. Unreacted products eluted in the acidic fraction and in the washing process. 1,3-thiazane-2-carboxylic acid was present in the first alkali fractions (ninhydrin and iodoplatinate tests). Fractions were pooled together and dried by a rotary evaporator. The residue was taken up in EtOH and dried under vacuum again, 230 mg of yellow microcrystalline powder was obyained (78% yield), 2.5 mL of boiling EtOH was added, and the product dissolved by dropwise addition of water and allowed to crystallize over night at -20 °C [
53].
6. Sulfinates and Sulfonates (Cysteine/Homocysteine Sulfinic Acid, Hypotaurine/Homohypotaurine and Cysteic/Homocysteic Acid, and Taurine/Homotaurine)
The versality of sulfur compounds in cell metabolism lies in their ability to cycle through a variety of biologically relevant oxidation states. Disulfides and thiols can be easily oxidized to sulfinates and sulfonates, including copper ions (Cu
2+) as catalyst. Autoxidation of disulfides (RSSR) and disulfoxides (RSOSOR) in alkaline solutions (reaction 1 and 2), or dismutation of disulfoxides (reaction 3), gives rise to sulfinates according to these reactions [
54]:
Interestingly, Cavallini showed that cystine cleavage in alkaline medium in the presence of Cu
2+ ions is initiated by an α,β elimination with production of cysteine persulfide, called thiocysteine (RSSH) (reaction 4), as intermediate in the formation of cysteine sulfinic acid (RSO
2H) [
55].
Consequently, thiocysteine undergoes hydrolysis favored by copper ions, leading to cysteine sulfenic acid (RSOH) (reaction 5). Cysteine sulfenic acid is readily converted to cysteine sulfinic acid by dismutation (reaction 6).
Cysteine sulfinic acid: The sulfinate is prepared from cystine disulfoxide according to Lavine [
54]. Cystine disulfoxide: 1.2 g (4.4 mmol) of cystine is transferred in a 0.2 L flask with 25 mL of acetonitrile. To the suspension, 1.68 mL of 70% perchloric acid and 30 mL of acetonitrile are added. After shaking and cooling on ice, 3 mL of acetic anhydride is added to the solution (the solution must not become yellow). Then, 1.6 g of perbenzoic acid in about 15 mL of chloroform is added with shaking and slight cooling, along with 33 mL of acetonitrile, resulting in a a final volume of 100 mL. After standing for an hour at room temperature, the solution is transferred to a separatory funnel and extracted with 25 mL of 1 N HCl and after with 10 mL of 1 N HCl. The two aqueous solutions are combined and extracted four times with 5–10 mL portions of CHCl
3. Then, the aqueous solution is slowly neutralized, with active stirring and cooling, initially with about 8 N ammonia until precipitation starts and with the neutralization being completed with dilute NH
3 (1.0 N) up to pH 4–5. After standing about 10 min, the precipitate is filtered off by suction; washed with water, alcohol, and acetone; and dried in vacuum at room temperature. The yield is 1.1 g. Cysteine sulfinic acid: 1 g of cystine disulfoxide is dissolved in 2 N NH
3. After 1 h at room temperature, the reaction mixture is evaporated to dryness at 30 °C and 10 mL of water is added; this is repeated until no more ammonia is present. The precipitate is filtered off, the recipient is washed with 10 mL of water, and the filtrate is added to a Dowex-50 column (H
+ form, 200–400 mesh, 2.5 cm × 5 cm). The acid eluate is collected and dried under vacuum. The precipitate is suspended in absolute ethanol and dried again. Finally, the precipitate is broken in absolute ethanol with a steel spatula and crystallization of the product is obtained. The ethanol is withdrawn. The crystals are not hygroscopic and have a yield of 650 mg.
Homocysteine sulfinic acid: the sulfinate is prepared by copper-catalyzed alkaline autoxidation of homocystine [
56]. Next, 1.34 g (5.0 mmole) of homocystine is dissolved in 150 mL 0.1 N NaOH, and 5 mL 0.1 M CuCl
2 (0.5 mmole) is added. The mixture is incubated for 3 h at 38 °C in a thermostated water bath. Incubation is in a 1 L Erlenmeyer flask, with continuous mechanical shaking, to assure good oxygenation of the solution. Under these conditions, ca. 1.4 moles homocysteine sulfinic acid is produced per mole homocystine. At the end of the incubation, the reaction mixture is acidified to pH 2 with 5 mL 2 N HCl, filtered off from a little fine precipitate, and concentrated to about 20 mL in a flash-evaporator at 70 °C. Dowex 50 chromatography: the above concentrated solution is passed through a 2 cm × 28 cm column of Dowex 50, H
+. The column is washed with 100 mL H
2O to remove homocysteic acid. Homocysteine sulfinic acid is then eluted with 2 M NH
4OH, collecting 150 mL eluate before appearance of the alkaline front and 150 mL thereafter. The bulk of homocysteine sulfinic acid is contained in the alkaline fraction, together with traces of other aminated compounds and of homocysteic acid possibly originating from homocysteine sulfinic acid on the column. The eluate preceding the alkaline front still contains appreciable amounts of homocysteine sulfinic acid. All these compounds are easily detected by paper chromatography (ninhydrin reaction). The elution of homocysteine sulfinic acid, since it readily reduces KMnO
4, may be easily followed by dropping the eluate from the column directly into a small volume of a dilute acid solution of KMnO
4. The collected eluate is then reduced to about 20 mL in a flash-evaporator as above, eliminating the ammonia in the meantime. The slight yellow concentrated solution is decolorized with charcoal. Dowex-1 chromatography: the above solution is loaded on a 2 cm × 13 cm column of Dowex-1, HCOO
−-form. The column is washed with 100 mL H
2O, and the homocysteine sulfinic acid is eluted with 0.5 M HCl in fractions of 10 mL. The acid fractions reducing KMnO4 are collected (about 150 mL) and dried in a flash-evaporator at 70 °C. To the oily residue, 4–5 mL H
2O is added, and it is dried again; this step is repeated twice, to remove almost all of the HCI. Second Dowex-50 chromatography: the oily residue of the above step is diluted with 20 mL H
2O and loaded on a 2 cm × 4 cm column of Dowex 50, H
+-form. Owing to the small amount of resin, HCS is not retained on the column. The column is washed with water, collecting fractions of 10 mL. The first 5–6 fractions contain traces of an unidentified compound, which also reduces permanganate, and are discarded; HCS is eluted in the following 20 fractions, which readily reduce permanganate; these are collected and dried in a flash-evaporator at 70 °C. The white amorphous residue is dissolved in the minimum amount of 0.01 N HCl at 70 °C, 5 vol absolute ethanol is added, and it is allowed to stand overnight at −20 °C. A crystalline precipitate is formed that is collected on a sintered-glass funnel, washed with ethanol, and dried with ether. Yield: 674 mg. All of the above procedure was followed with either L- and DL-homocystine with the same results. It has been demonstrated, moreover, that homocysteine-thiolactone may be used as substrate instead of homocystine, with about the same results. In this case, a double molar amount of homocysteine-thiolactone (10 mmol) was used in the standard incubate.
Hypotaurine: in a 100 mL conical flask, 25 mL 2 N HCl and 4.55 mg of cysteamine hydrochloride are mixed. After dissolution and cooling, 6 mL of 30% hydrogen peroxide and 50 mg of solid potassium iodide (KI) are added while stirring. The solution heats up strongly and is colored. After 15 min at room temperature, the flask is placed in a boiling water bath for 2 min. It is allowed to cool at room temperature for 30–45 min, and then 20 mL 2 N NaOH is added. After 10 min, the solution is poured into a 2.5 cm × 30 cm Dowex-50, X4 200–400 Mesh H
+-form column. It is washed with water until neutral effluent (about 250 mL) and eluted with 1 N NH
3. The effluent is collected in 10 mL fractions up to 6–8 fractions beyond the alkaline front. Hypotaurine is localized with the permanganate test (generally the fraction before and 3–4 after the alkaline front). The positive fractions are poured into a column of Amberlite IRC-50 in the H
+-form (40–80 mesh 2.5 cm × 20 cm) to eliminate ammonia and traces of cystamine. The effluent and the subsequent washing are collected in 10 mL fractions (25 fractions). Hypotaurine is localized with the permanganate test, and the positive fractions (100–150 mL) are dried on a water bath at the water pump. The yield is 1–1.5 g of slightly yellow material [
57,
58].
Homohypotaurine: homohypotaurine is prepared by oxidation of homocystamine to the corresponding disulfoxide, which is then allowed to undergo dismutation in alkali [
59]. Homohypotaurine is extracted by ion-exchange chromatography. Ten mmoles homocystamine dihydrobromide (3.4 g) or dihydrochloride (2.5 g) are dissolved in 10 mL 0.1 N HCI; 20 mg KI are added, and then dropwise 2.3 mL 30% H
2O
2 (22 mmoles). The solution becomes yellow and slightly warms up. After a 30-min standing time, the solution is kept for 3 min in a boiling water bath, during which time it completely decolorizes; it is then allowed to cool at room temperature. The nitroprusside test for S-S groups is now negative. The solution is brought to pH 12–13 with 40% NaOH and left for 1 hr at room temperature. It is then loaded on a 25 cm × 2 cm column of Dowex 50, X8, H
+. The column is washed with water and then eluted with 1 N NH
4OH in fractions of 10 mL. Homohypotaurine is eluted in two fractions before the alkaline front and in two to three thereafter. It is easily localized since it readily decolorizes acidic KMnO
4. These fractions are collected and dried in a rotary evaporator. The oily residue is repeatedly added with a little absolute ethanol and again dried until a crystalline product is obtained. The yield is 1.2–1.3 g. Recrystallized from alcohol-ether homohypotaurine melts at 190–192 °C.
Cysteic acid: in a 500-mL round-bottomed flask, 24 g (99.8 mmol) of cystine is dissolved in a cold mixture of 150 mL of water and 50 mL of concentrated HCl. To the solution is added, dropwise, 80 g (25 mL, 0.5 mol) of commercial bromine, with occasional stirring for 40 min. The temperature of the mixture rises to about 60°. The resulting solution, which contains a little unreduced bromine, is then evaporated under reduced pressure on a steam bath. The dark-colored crystalline residue is dissolved in 100 mL of distilled water and filtered from a small quantity of amorphous insoluble matter. The filtrate is concentrated by evaporation on a water bath to 65 mL and allowed to crystallize by standing overnight in a refrigerator. The crystals are filtered with suction and washed well with about 100 mL of 95% ethanol in several portions, with the washings being collected separately. The crystals are dried under reduced pressure over phosphorus pentoxide. A second crop is obtained by diluting the washings with an equal volume of water, evaporating until free of ethanol, adding the residue to the mother liquor, and evaporating the combined solution to dryness on the water bath. The residue is dissolved in 30–40 mL of water, decolorized with 0.5–1.0 g of charcoal, concentrated to 15 mL and (when cold) treated with 30 mL of 95% ethanol. The crystals so formed are collected, washed with ethanol and dried as before. The total yield is 30.5–33.5 g of pure cysteic acid monohydrate (81–90%) [
60].
Homocysteic acid: L-Homocystine (268 mg, 1.0 mmol) is dissolved in 8 mL of 3 N HCI contained in a 25-mL round-bottomed flask. The solution is chilled on an ice bath and stirred magnetically as bromine (0.8 g, 260 μL, 10 mmol) is added in several portions over a 30-min period. The portions initially added decolorize spontaneously as homocystine is oxidized, but the solution soon develops and maintains a reddish-brown bromine color. After stirring an additional 15 min, the solution is dried with a rotary evaporator under reduced pressure (water aspirator). The residue is dissolved in 10 mL of water, and the resulting solution is passed through a small column (0.6 cm × 8 cm) of Dowex 50-X8 (H
+-form, 200–400 mesh). The product does not bind and is completely washed through with 10 mL of water. The combined eluents are evaporated to dryness, and the solid product is freed of most residual HC1 and HBr by twice adding and then evaporating small portions of water. Residual bromine is removed from the light tan product by adding and evaporating small portions of chloroform three times. The final white product is dried in a vacuum desiccator over P
20
5 [
61].
Taurine/Homotaurine: taurine and homotaurine are prepared by oxidizing, with an excess of 30% H
2O
2, in the presence of traces of ammonium molybdate, an aqueous solution of hypotaurine or homohypotaurine. The solution is dried in vacuo, and the residue is dissolved in the minimum amount of water and then crystallized by addition of ethanol [
59].
7. Thiosulfonates (Thiotaurine/Homothiotaurine, Alanine Thiosulfonate)
Thiosulfonates contain a highly reactive sulfur atom defined as sulfane sulfur. This sulfur atom can be released as elemental sulfur and transferred to another sulfur atom or reduced to hydrogen sulfide (H
2S) by thiols. Recent investigations suggest that thiosulfonate effect on biological cell responses can be attributed to its involvement in H
2S signaling, as H
2S donors and/or as reactive sulfane-sulfur species [
20]. Thiosulfonates are obtained by reacting sulfinates with elemental sulfur.
Thiotaurine: 220 mg of hypotaurine (2 mmol) was placed in a ground-cap tube topped with reflux coolant and dissolved in 1 mL 0.2 N NaOH, and 70 mg of finely dispersed elemental sulfur and 20 mL of ethanol were added. The reaction mixture was refluxed with ethanol. After 30 min boiling, the suspension was placed at 0 °C for 12 h. The obtained crystals were washed twice with 5 mL of carbon sulfide and once with 5 mL of ethanol, 1.5 mL of water was added and heated to dissolve the crystals, and 10 mL of absolute ethanol was added to the still cloudy solution. It was placed at 0 °C, and after 12 h the crystals were decanted and left to dry in the air. The yield was 200–250 mg of crystalline material [
62].
Homothiotaurine: To 0.7 g homohypotaurine (5 mmole) dissolved in 1 mL 0.2 N NaOH, 200 mg of finely powdered sulfur (ca. 6 matoms) and 15 mL ethanol were added. The suspension was refluxed for 30 min until most of the sulfur had dissolved and then cooled overnight at −20 °C. A crystalline precipitate formed. The alcohol was carefully decanted, and the crystals were washed twice with 4 mL carbon disulfide, twice with 4 mL absolute ethanol, and dried at 120 °C. The yield was 0.7 g, i.e., 92% [
59].
Alanine thiosulfonate: this thiosulfonate was obtained by reacting cysteinsulfinic acid with elemental sulfur and then precipitated as a barium salt. 612 mg (4 mmol) of cysteinsulfinic acid was dissolved in 4 mL of water, and 200 mg of elemental sulfur (approximately 6 mmol) and 20 mL of pyridine and boiled under reflux for 5–10 min until the sulfur completely dissolved. The suspension was dried on a boiling water bath at reduced pressure by means of a water pump, and 800 mg BaOH-8H
2O dissolved in 10 mL of water (H
2O) was added to the dry residue. This was filtered to eliminate the excess of sulfur and traces of barium carbonate, and it was precipitated with 10 volumes of alcohol at 95°. After 12–24 h at 0 °C, the precipitate was collected on Buchner, washed with absolute ethanol, and dried with ether. About 800 mg of a white, slightly hygroscopic, slightly pasty powder was obtained, which was kept in a dryer [
63].
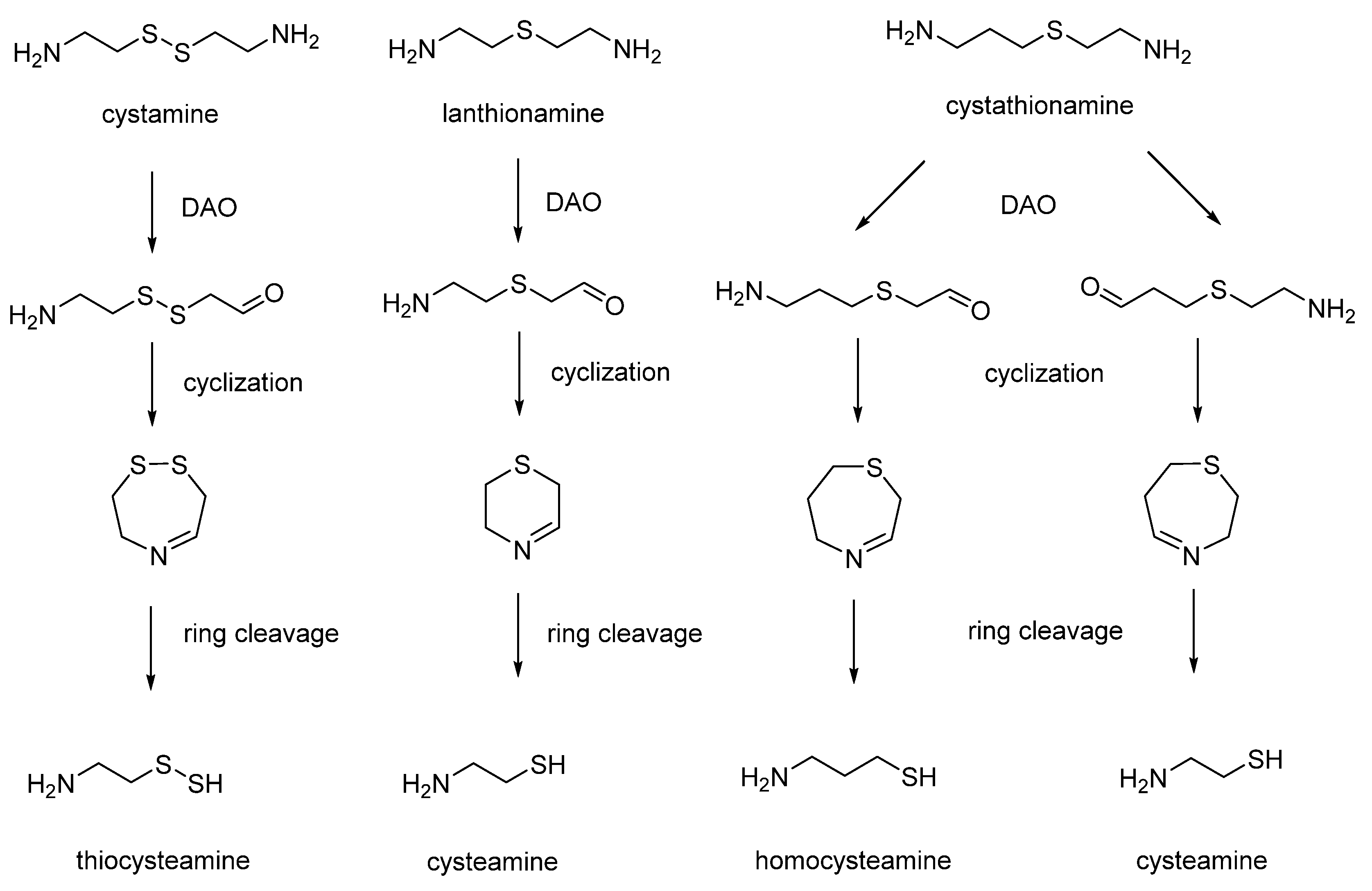
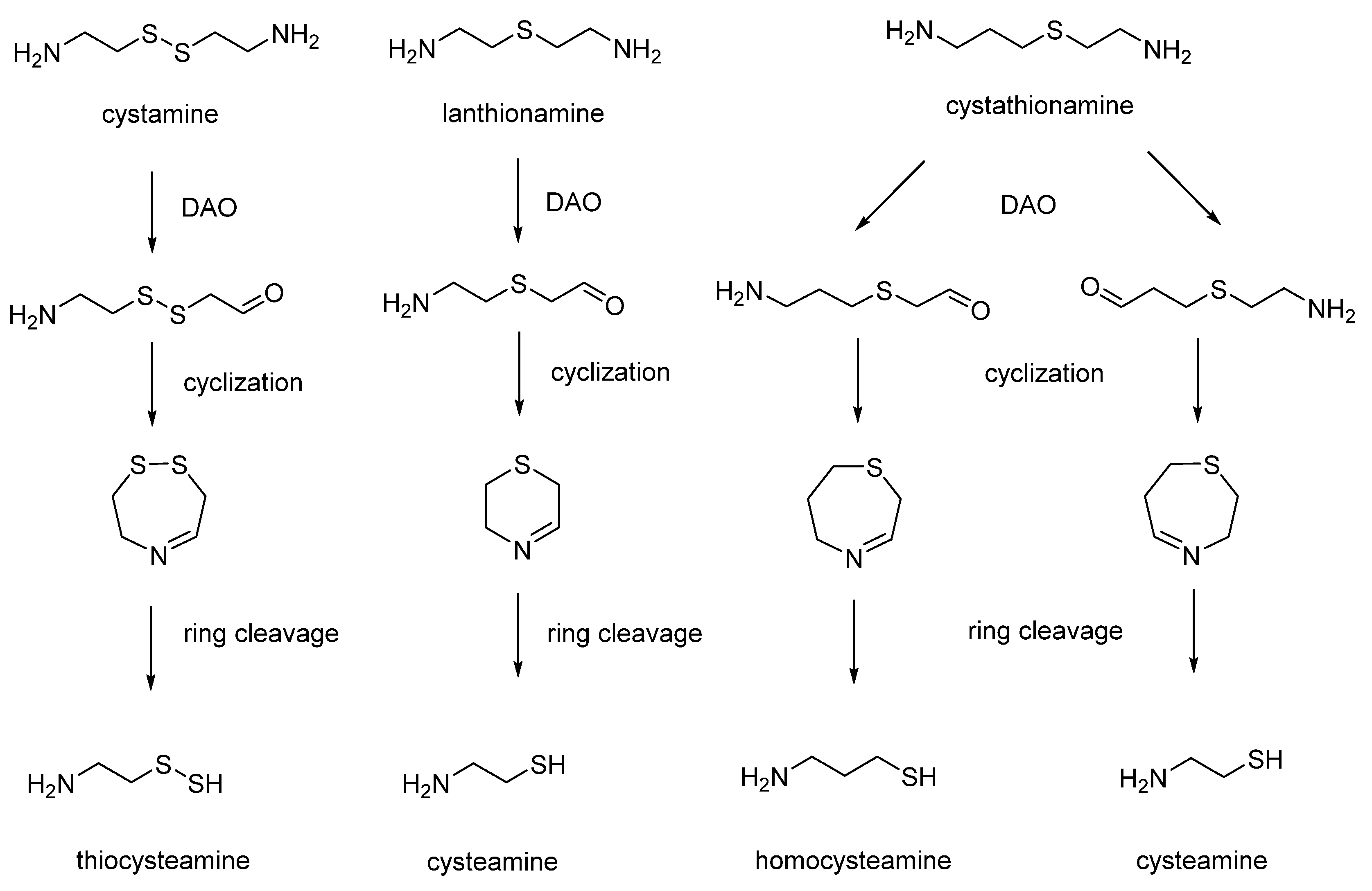
9. Thiodiamines (Cystamine, Homocystamine, Lanthionamine, Cystathionamine and Homolantionamine)
In 1956, Cavallini assayed sulfur-containing diamine analogues as substrates for diamine oxidase (also named histaminase) [
66]. Surprisingly, he found that diamino oxidase was unable to recognize the substitution of a methylene moiety of the substrates with one or two sulfur atoms. The enzymatic oxidation of thiodiamines was followed by the formation of number of cyclic intermediates, including cystaldimine [
67]. The cleavage of cyclic cystaldimine led to the production of interesting biomolecules, such as thiocysteamine, hypotaurine and thiotaurine (
Figure 8) [
68,
69,
70].
Cystamine: cystamine dihydrochloride is prepared by iodine oxidation of cysteamine hydrochloride. To completely free the product from HI formed in the reaction, the solution after oxidation was passed through a column of anionic resin (Dowex 2, OH
−-form), collecting eluates and washes with water positive for disulfide test. The liquid containing the cystamine acidified with concentrated HCl was dried on a boiling water bath, and the cystamine was recrystallized from the alcohol [
57].
Homocystamine: homocystamine was prepared by acid degradation of S-sulfo-homocysteamine (homocysteamine S-sulfonate) obtained from 3-bromopropylamine and thiosulfate [
53], according to the following reaction scheme:
2.6 g (10.5 mmol) of sodium thiosulfate was dissolved in 10 mL water. 2.2 g (10 mmol) of 3-bromopropylamine hydrobromide was then added, and the solution was allowed to stand at room temperature until almost complete consumption of thiosulfate, as checked by titration. The solution containing S-sulfo.homocysteamine was dried at 60 °C in a rotatory evaporator, and the residue dissolved in 2 mL 2 N HCl and dried again. The treatment with HCl was repeated three more times, and the residue was finally dissolved in 10 mL 2 N NaOH to convert the homocystamine dihydrochloride to the free base. The alkaline solution was repeatedly extracted with chloroform until no more homocystamine was taken out, as checked by nitroprussiate test. The pooled chloroform extracts were dehydrated with MgSO4, filtered, and dried in a rotatory evaporator. The residue was dissolved in 2 mL 2 N HCl, dried again and absolute ethanol was repeatedly added. Finally, the residue was suspended in 5 mL boiling ethanol, dissolved by dropwise addition of water, filtered and allowed to stand overnight at −20 °C. The crystalline precipitate was collected on a Buchner funnel, washed with absolute ethanol, and dried with ether. 750 mg of white crystal of homocystamine dihydrochloride was obtained. By reduction of homocystamine with dithithreitol, homocysteamine was obtained.
Lanthionamine: in a 50 mL flask, 2.2 g of bromoethylamine hydrobromide, 0.8 g of cysteamine base, 10 mL of boiled and N
2-saturated water were mixed. They were left under a current of nitrogen for 30 min. The reaction mixture was loaded on Dowex 2, OH
−-form, (200–400 mesh) 2.5 cm × 5 cm column. The effluent was concentrated on boiling water bath at reduced pressure. Then, 3 mL of concentrated HCl was added and dried. It was dissolved by boiling in 20 mL of ethanol. It was left at 0 °C and decanted from an oil that was then discarded, and 30 mL of acetone was added to the supernatant. It was left at −20 °C overnight. The next day, the flaky precipitate was centrifuged, washed with ether and dried. The preparation was slightly hygroscopic and contained some traces of cystamine [
71].
Cystathionamine: 1.15 g (5.2 mmol) of 3-bromopropylamine-HBr was dissolved in 2 mL H
2O bubbled with nitrogen for 10 min. The reaction was made alkaline with concentrated ammonia (pH 8–9) and placed in water-bath at 60 °C, and 0.55 g (7.1 mmol) cysteamine-HCl was added. The reaction was allowed to stand in the water-bath under continuous bubbling of nitrogen until the nitroprusside test for SH group proved negative (about 15 min). The reaction mixture was concentrated to 1–2 mL under reduced pressure in a boiling water-bath and loaded on a 2 × 10 cm Dowex 1, OH
−-form, column. The column was eluted with water, and the alkaline eluate was collected (about 50 mL), acidified with concentrated HCl to pH 3–4 and dried in a rotatory evaporator. The oily residue was dried by repeated additions of absolute ethanol, and finally 0.9 g white material was obtained. Traces of cystamine were eliminated by treating the reaction mixture before loading on Dowex column in the following manner: 0.3 mL of a 6% solution of sulfurous acid was added, followed by a few mgs of solid monochloroacetic acid [
72].
Homolanthionamine: homolanthionamine was prepared by allowing 3-bromopropylamine-HBr (5 mmol) to react in N2-saturated water with Na2S (2.5 mmol). The reaction was allowed to stand in the water-bath a 60 °C under continuous bubbling of nitrogen until the nitroprusside test for SH group was negative (about 10 min). Then, a few drops of concentrated ammonia were added, and the reaction mixture was degassed. Then, the suspension was acidified with HBr and dried. The precipitate was suspended in 5 mL of 96% ethanol, and the undissolved precipitate was Na2Br. The suspension was filtered and dried by repeated addition of 96% ethanol. The precipitate was suspended by boiling in 4 mL absolute ethanol, and 20 mL ether was added to obtain a precipitate corresponding to homolanthionamine (unpublished results).




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}