Abstract
An ever-increasing need exists within the forensic laboratories to develop analytical processes for the qualitative and quantitative determination of a broad spectrum of new psychoactive substances. Phenylethylamine derivatives are among the major classes of psychoactive substances available on the global market and include both amphetamine analogues and synthetic cathinones. In this work, an ultra-high-performance liquid chromatography-positive ion electrospray ionization tandem mass spectrometric method (UHPLC-ESI-MS/MS) has been developed and fully validated for the determination of 19 psychoactive substances, including nine amphetamine-type stimulants and 10 synthetic cathinone derivatives, in premortem and postmortem whole blood. The assay was based on the use of 1 mL premortem or postmortem whole blood, following solid phase extraction prior to the analysis. The separation was achieved on a Poroshell 120 EC-C18 analytical column with a gradient mobile phase of 0.1% formic acid in acetonitrile and 0.1% formic acid in water in 9 min. The dynamic multiple reaction monitoring used in this work allowed for limit of detection (LOD) and lower limit of quantitation (LOQ) values of 0.5 and 2 ng mL−1, respectively, for all analytes both in premortem and postmortem whole blood samples. A quadratic calibration model was used for the 12 quantitative analytes over the concentration range of 20–2000 ng mL−1, and the method was shown to be precise and accurate both in premortem and postmortem whole blood. The method was applied to the analysis of real cases and proved to be a valuable tool in forensic and clinical toxicology.
1. Introduction
Over the past decade, the trend towards substance abuse has changed, and many new psychoactive substances have emerged as “legal high” alternatives to traditional illicit drugs [1]. This phenomenon was accompanied by increased incidences of people in intensive care units of hospitals after substance use with unknown pharmacological and/or toxicological action [2]. The acute and chronic effects of new psychoactive substances (NPSs) are not always known, and safety data on their toxicity are usually not available [3]. By the end of 2018, the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) recorded more than 730 NPSs, 55 of which were detected for the first time on the drug market in Europe [4]. Phenylethylamine derivatives are serotoninergic receptor agonists leading to psychedelic effects and in some cases also inhibiting monoamine reuptake [5]. These compounds are among the major classes of psychoactive substances available on the global market and include both amphetamine and β-keto amphetamine analogues (synthetic cathinones). Synthetic cathinones are sold online as ‘‘plant food,’’ ‘‘bath salts,’’ or ‘‘research chemicals,’’ labeled as “not for human consumption’’ to avoid potential regulation [6], and according to the United Nations report, are one of the two largest categories of substances along with synthetic cannabinoids [7]. The forensic analysis of phenylethylamine derivatives is important because they can be involved in drug-related deaths. Fatal intoxication due to the recreational use of 3,4-methylenedioxymethamphetamine (MDMA) and 3,4-methylenedioxy-N-ethylamphetamine (MDEA) has been reported in Italy in 1996 [8]. MDMA overdosage can cause hyperthermia, cardiac arrhythmias, and renal failure, leading to death. More recently, 77 deaths where MDMA was detected in the body were reviewed and of these cases, 59 deaths had MDMA present in whole blood. [9]. In one case study, death was attributable to the toxic effects of 3,4-methylenedioxypyrovalerone (MDPV) [10] and in other nine postmortem cases, ethylone was confirmed [11]. According to the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA 2020 report), some drugs were more associated than others with admission to critical care, such as gamma-hydroxybutyrate (GHB)/gamma butyrolactone (GBL) (16%), methamphetamine (8%) and MDMA (8%) [4]. The Centers for Disease Control and Prevention (CDC) warns that overdose deaths in the USA from psychostimulants, including methamphetamine, have steadily increased from 1999 to 2019 [12].
A variety of analytical methods are employed by the forensic laboratories to cover the analysis of a great number of psychoactive substances in various biological samples [13,14,15]. Typically, immunoassay tests are used for the preliminary screening of psychoactive substances [16,17] followed by using hyphenated mass spectrometric methods interfaced either with gas chromatography (GC-MS/MS) [18,19,20,21,22] or liquid chromatography (LC-MS/MS) for confirmation and quantitation purposes [23,24,25,26,27,28,29]. Only a few screening procedures based on high resolution mass spectrometry have been published in the literature [30,31,32,33,34,35,36]. Whole human blood (premortem and/or postmortem) [37] and urine [38,39] are the biological samples of choice in forensic toxicological analyses [40,41] for several reasons. For postmortem cases, the pathologists try to collect whole blood during autopsies and inspections. Per recommended specimen collection procedures [42], they will typically collect blood from two different sources such as the heart and peripheral blood from the femoral vein. Femoral blood is the postmortem sample of choice for quantification of drugs and interpretation as it is less subject to contamination from trauma and/or postmortem redistribution. As for premortem samples from living subjects, many Driving Under the Influence of Drugs (DUID) laws, including those in the state of Delaware, are written specifically in reference to blood rather than serum or plasma, again making blood the preferred specimen for analysis [43]. Additionally, drug concentrations in blood are frequently available in the scientific literature for comparison of results and for purposes of interpretation. Considering the ever-increasing number of psychoactive substances, the increasing demands in the time of issuing the results, and the high cost of analyses, the development of analytical processes for the qualitative and quantitative determination of a broad spectrum of psychoactive substances is of great importance. An ever-increasing need exists within forensic laboratories to improve the sensitivity and reduce the total analysis time. The main drawback of GC–MS methods is that a derivatization step during sample preparation is usually required to improve the chromatography of amphetamine-type stimulants [44]. On the contrary, UHPLC, enables ultra-high-speed analysis and provides the full benefits of chromatography for the separations using shorter analytical columns and mobile phases at high linear velocities with greater resolution and sensitivity for analytical determinations, particularly when coupled to mass spectrometry [45,46,47].
The aim of this study was to develop a new ultra-high-performance liquid chromatography-positive ion electrospray ionization tandem mass spectrometry (UHPLC-ESI-MS/MS) method for the identification and confirmation of 19 psychoactive substances from two categories, including nine amphetamine-type stimulants, namely amphetamine (A), methamphetamine (MA), phenylpropanolamine (PHPR), ephedrine, pseudoephedrine (PSE), 3,4-methylenedioxyamphetamine (MDA), MDMA, MDEA, phentermine (PHRM), and ten synthetic cathinone derivatives, namely methylphenidate (MPHD), methcathinone (ephedrone), methedrone (PMMC), mephedrone (4-MMC), ethylone (MDEC), methylone (MDMC), MDPV, alpha-pyrrolidinopentiophenone (alpha-PVP), bupropion, and hydroxybupropion in premortem and postmortem whole blood samples. A quantitation method was also developed for the 12 psychoactive substances, including the nine amphetamine-type stimulants and three synthetic cathinone derivatives MPHD, bupropion, which is used for both therapeutic and recreational purposes, and its metabolite. The dynamic multiple reaction monitoring (dynamic MRM) method used in this work requires fewer ion transitions to be monitored concurrently in a chromatogram, and it allows the collection of many data points across narrow peaks, exhibiting excellent quantitative results. To the extent of our knowledge, this is the first reported UHPLC-ESI-MS/MS method targeting the determination of 19 psychoactive substances both in premortem and postmortem whole blood samples. UHPLC coupled to triple quadrupole tandem MS is an important tool for targeted analysis and brings further advantages in terms of selectivity, sensitivity, and high throughput for the analysis of complex samples. The proposed UHPLC-ESI-MS/MS method was validated based on the guidelines of the Scientific Working Group of Forensic Toxicology (SWGTOX) [47,48]. The assay was based on 1 mL premortem or postmortem whole blood, following solid phase extraction prior to the analysis. The method was successfully applied to the analysis of real premortem and postmortem whole blood samples obtained from various toxicological cases and proved to be a valuable tool in forensic and clinical toxicology.
2. Materials and Methods
2.1. Chemical and Reagents
Acetonitrile (ACN) of Optima LC-MS grade, methanol (MeOH), 2-propanol (C3H7OH), and methylene chloride (CH2Cl2) of HPLC grade were obtained by Thermo Fisher Scientific (Pittsburgh, PA, USA). Hydrochloric acid (HCl), ammonium hydroxide (NH4OH) of Certified ACS plus grade, glacial acetic acid (CH3COOH) of HPLC grade and sodium hydroxide (NaOH) of analytical purity grade were also supplied from Thermo Fisher Scientific (Pittsburgh, PA, USA). Formic acid (HCOOH) of LC-MS grade, sodium phosphate dibasic (Na2HPO4), and sodium phosphate monobasic monohydrate (NaH2PO4∙H2O) were obtained from Sigma Aldrich (St. Louis, MO, USA). HPLC-grade water was purchased by Aurora Corporation (Wilmington, DE, USA). The solid phase extraction (SPE) experiments were performed by using a positive pressure manifold of 48 positions purchased from UCT (Bristol, PA, USA) and Clean Screen® DAU SPE Cartridges (200 mg, 10 mL). Blank whole blood and premortem and postmortem whole blood samples from real toxicological cases were obtained according to the rules, ethics, and procedures of the Laboratory of Toxicology, Division of Forensic Science, Department of Safety and Homeland Security, State of Delaware, Wilmington, DE, USA.
2.2. Stock Standard Solutions
The stock standard solutions of amphetamine (A), methamphetamine (MA), phenylpropanolamine (PHPR), ephedrine, pseudoephedrine (PSE), 3,4-methylenedioxyamphetamine (MDA), 3,4-methylenedioxymethamphetamine (MDMA), 3,4-methylenedioxy-N-ethylamphetamine (MDEA), phentermine (PHRM), methylphenidate (MPHD), bupropion, hydroxybupropion, methcathinone (ephedrone), methedrone (PMMC), mephedrone (4-MMC), methylone (MDMC), ethylone (MDEC), 3,4-methylenedioxypyrovalerone (MDPV), and alpha-pyrrolidinopentiophenone (alpha-PVP) at a concentration of 1.0 mg mL−1 for each analyte in methanol were purchased from Cerilliant (Round Rock, Texas, USA). Stock standard solutions of the internal standards (ISTDs), namely amphetamine-d11, methamphetamine-d11, ephedrine-d3, pseudoephedrine-d3, norpseudoephedrine-d3, MDA-d5, MDMA-d5, MDEA-d5, phentermine-d5, methylphenidate-d9, bupropion-d9, hydroxybupropion-d6, mephedrone-d3, and methylone-d3, were also purchased from Cerilliant (Round Rock, TX, USA) at a concentration of 100 µg mL−1 in methanol.
2.3. Working Standard Solutions and Calibration Whole Blank Samples
A mixed working standard solution of the analytes was prepared at 10 μg mL−1 by appropriate dilutions of the stock standard solutions in methanol. A mixed working standard solution of the internal standards (ISTDs) was prepared at 1 μg mL−1 by appropriate dilutions of the stock standard solutions in methanol. The mixed working standard solutions were stored at −20 °C in dark amber vials to protect the analytes and the ISTDs from light degradation.
Calibration standards spiked in whole blood were freshly prepared by using the mixed working standard solution at eight different concentration levels, 20, 50, 100, 300, 500, 1000, 1500, and 2000 ng mL−1. Control whole blood spiked samples were also prepared in an analogous manner at three concentration levels 60, 800, and 1600 ng mL−1. Quality control whole blood spiked samples were prepared at two concentration levels 60 and 1000 ng mL−1, by using different mixed working solutions. Internal standard (ISTD) concentration was 100 ng mL−1 for each of the ISTDs used in all the spiked whole blood samples.
2.4. Sample Preparation Procedure
Cleanup of biological samples is carried out by solid phase extraction. On the day of extraction, the whole blood samples are vortex-mixed to ensure homogeneity. Consequently, a 1 mL aliquot is transferred to a glass test tube followed by addition of 100 μL of the mixed working solution of the ISTDs, 4 mL of LC-grade water and 2 mL of 0.1 M phosphate buffer at pH 6. The mixture is vortexed for 10 sec, remained at rest for 5 min, and centrifuged at 3000 rpm for 10 min at room temperature. Then the sample is subjected to solid phase extraction using a Clean Screen® DAU mixed mode SPE cartridge (200 mg, 10 mL). Conditioning of the cartridge is performed with 1 × 3 mL of methanol, 1 × 3 mL of LC-grade water, and 1 × 1 mL of 0.1 M phosphate buffer at pH 6. After loading of the sample and subsequent washing with 1 × 3 mL of LC-grade water, 1 × 1.25 mL of 0.1 M acetic acid, and 1 × 3 mL of methanol, the cartridge is dried under vacuum for 5 min. Elution of the analytes is then achieved with 1 × 3 mL of CH2Cl2:C3H7OH:NH4OH (78:20:2, v/v/v). An aliquot of 100 μL of 0.2% hydrochloric acid solution in 2-propanol is then added to the extract before the evaporation to dryness with nitrogen at 40 °C. The sample is reconstituted in 0.1% aqueous formic acid solution to a final volume of 0.2 mL prior to the UHPLC-ESI-MS/MS analysis.
2.5. UHPLC-ESI-MS/MS
The experiments were performed using a UHPLC system model 1260 Infinity (Agilent Technologies, CA, USA) coupled to an ESI ion source model G1948B and a triple quadrupole mass spectrometer model Agilent 6410B (Agilent Technologies, CA, USA). Highly pure nitrogen was produced by a Parker Balston nitrogen generator, model N2-14 (RJM Sales Inc., Somerset, NJ, USA). Electrospray ionization was performed in positive ion mode, and the ionization was optimized using MassHunter® MS Optimizer software (Agilent Technologies, CA, USA). The ionization source temperature was set to 350 °C, and the capillary potential was adjusted at 3.0 kV. Nitrogen was used as drying and nebulizing gas. The decomposition gas flow rate was set at 10 L min−1 and the gas pressure in the nebulization needle at 45 psi. A Poroshell® 120 EC-C18 analytical column (2.1 × 75 mm, 2.7 µm particle size) was used for the chromatography, which was performed at 45 ± 0.8 °C with an injection volume of 1 µL. The mobile phase consisted of 0.1% formic acid aqueous solution (eluent A) and 0.1% formic acid in acetonitrile (eluent B) and pumped at a 0.4 mL min−1 flow rate. The gradient elution program starts at 2% B for 2 min, increases to 10% B at 4 min, then to 30% B at 7 min, and finally to 90% B at 9 min, which is maintained for 1.5 min, and then equilibrates at the initial condition for 2 min. MassHunter® software ver. B.07.01, build 7.1.524.0 and ver. B.08.00, build 8.0.8023.0 for QQQ (Agilent Technologies, CA, USA) were used for data acquisition and analysis.
2.6. Method Validation
The method was validated separately in premortem and postmortem whole blood, according to the guidelines of the Scientific Working Group on Forensic Toxicology (SWGTOX) and the Commission Decision 2002/657/EC [48,49,50]. The validation parameters for the quantitation include limits of detection (LODs), lower limits of quantification (LLOQs), selectivity and specificity, calibration model, accuracy, precision, matrix effect, carryover, and extraction efficiency. For those analytes validated for qualitative purposes, only selectivity and specificity, LODs, matrix effect, carryover, and extraction efficiency were evaluated.
The selectivity of the current method was evaluated for any endogenous or exogenous interferences. To evaluate endogenous interferences, 70 different blank premortem and postmortem whole blood samples, which were negative for the analytes based on the preliminary screening Enzyme-linked Immunosorbent Assay (ELISA) method (cutoff level of 20 ng mL−1), have been analyzed by the proposed method. To evaluate any interferences originating from the deuterated internal standards, premortem whole blood blank samples spiked with the ISTDs (zero-blank samples) were also analyzed to observe possible cross fragmentation from the isotopic labeled ISTDs. The exogenous interferences were evaluated by the analysis of standard solutions prepared in 0.1% formic acid aqueous solution and spiked with 77 commonly used medicines and psychoactive substances, namely methadone and its metabolite at 4 μg mL−1; phencyclidine at 2 μg mL−1; tramadol at 4 μg mL−1; codeine, morphine, hydrocodone, hydromorphone, oxycodone, and oxymorphone at 4 μg mL−1; 6-acetylmorphine at 2 μg mL−1, fentanyl at 100 ng mL−1; norfentanyl, novel synthetic opioids (non-fentanyl analogues) such as U47700, AH-7921, MT-45, U-4990, and U-50488, 4-aminophenyl-1-phenethylpiperidine, sufentanil, carfentanil, butyryl fentanyl, o-fluoro-fentanyl, p-fluoro-fentanyl, isobutyricfentanyl, 3-methylfentanyl, acetyl fentanyl, 4-methyl-acetylfentanyl, p-methoxy-butyrfentanyl, β-hydroxythiofentanyl, p-fluor butyryl-fentanyl, acryl fentanyl, valeryl fentanyl, and 4-fluoro-iso-butyryl fentanyl at 0.5 ng mL−1; Δ9-tetrahydrocannabinol at 200 ng mL−1, 11-hydroxy-Δ9-tetrahydrocannabinol, and 11-nor-9-carboxy-Δ9-tetrahydrocannabinol at 500 ng mL−1; cyclobenzaprine at 4 μg mL−1; diphenhydramine at 4 μg mL−1; diazepam, nordiazepam, and alprazolam at 4 μg mL−1; mirtazapine, venlafaxine, desmethylvenlafaxine, citalopram, desmethylcitalopram, doxepin, nor-doxepin, imipramine, paroxetine, amitriptyline, nortriptyline, duloxetine, fluoxetine, norfluoxetine, and sertraline at 4 μg mL−1; trazodone at 40 μg mL−1; primidone, N-(1-phenylethyl) maleimide, levetiracetam, butalbital, pentobarbital, phenobarbital, and phenytoin at 4 ng mL−1; acetaminophen at 10 ng mL−1; topiramate at 2 ng mL−1; valproic acid at 20 ng mL−1; ibuprofen at 10 ng mL−1; butalbital, meprobamate, carisoprodol, glutethimide, theophylline, and carbamazepine at 4 ng mL−1; amobarbital at 2 ng mL−1, and secobarbital at 1 ng mL−1.
The carryover was evaluated by analyzing blank samples after the analysis of mixed standard solutions of the analytes at high concentration (4000 ng mL−1) prepared in 0.1% formic acid aqueous solution, and the analysis was performed in triplicate. The accepted criterion for carryover is that the peak area signals of the targeted analytes in the blank samples must be less than 5% of the peak area signals at the lower limit of quantitation (LLOQ).
Five calibration curves were constructed on five different days at eight different concentration levels, 20, 50, 100, 300, 500, 1000, 1500, and 2000 ng mL−1 for the nine amphetamine-type stimulants and the three synthetic cathinone derivatives including MPHD, bupropion, and its metabolite, hydroxy bupropion. The GraphPad Prism software program ver. 6.0 (GraphPad Software, Inc., California, USA) enabled automated comparison between the results of the linear and polynomial models, and it was used to select the optimal regression model for each one of the targeted analytes.
Precision and accuracy were assessed by the analysis of premortem and postmortem whole blood samples spiked with the targeted analytes at 60, 800, and 1600 ng mL−1 in three replicates on five different days (n = 15). One-way ANOVA was used to calculate the intra-assay precision, the total precision, and the total accuracy.
To assess the ionization suppression or enhancement, the matrix effect (%) was calculated both in premortem and postmortem whole blood samples by comparing the mean peak area signals of ten different blank whole blood samples spiked with the analytes after the sample preparation procedure, with the peak area signals of mixed standard solutions of the analytes diluted in 0.1% formic acid aqueous solution at equivalent concentration. The experiments were performed at two concentration levels, 60 and 1600 ng mL−1 for the analytes, and at 100 ng mL−1 for the internal standards, in ten replicates. The matrix effect (%) was then calculated based on the following equation:
To evaluate the extraction efficiency, the absolute recovery (%) was calculated both in premortem and postmortem whole blood samples and at two concentration levels, 60 and 1600 ng mL−1, for the targeted analytes and at 100 ng mL−1 for the ISTDs. The recovery (%) was calculated by comparing the mean peak area signals of ten individual whole blood samples spiked with the targeted analytes before the sample preparation with the mean peak area signals of ten whole blood samples spiked after the sample preparation.
2.7. Analysis of Real Samples
The applicability of the current method was evaluated by the analysis of one Proficiency Testing Scheme sample (PTS sample) provided by the CAP accreditation program (College of American Pathologists, Northfield, IL, USA), two pre-prepared samples provided by UTAK Laboratories, Inc. (Valencia, CA, USA), and two real cases samples, one premortem and one postmortem whole blood sample, that arrived for examination at the Laboratory of Toxicology, Division of Forensic Science, State of Delaware, USA. The PTS sample was evaluated by comparing the results of the current method to the mean values of the results obtained with methods of other toxicological laboratories enrolled in a proficiency testing program. The pre-prepared sample was evaluated based on the nominal concentrations given by UTAK. The real case samples were evaluated by comparison of the results obtained by the current method to the results obtained with other in-house methods or methods of an external laboratory.
3. Results and Discussion
3.1. Method Development
3.1.1. Mass Spectrometry
Analytes were detected in ESI positive ion mode by dynamic multiple reaction monitoring (dynamic MRM). The dynamic MRM is a relatively new and versatile technique where MRM transition lists are built dynamically throughout each UHPLC-MS run, based on the retention time windows of the analytes. This way the analytes are only monitored at their elution time and valuable MS duty cycle is not wasted by monitoring them when they are not expected. Identification of all the 19 psychoactive substances has been performed according to the optimum MRM parameters presented in Table 1 and by using at least four identification points [41]. These identification points include the retention time, the precursor ion, two fragment ions, and the relative fragment ions’ intensity, which is expressed as a percentage of the intensity of the most intense ion (precursor ion). The maximum permitted tolerance for the relative fragment ions’ intensity was set at ± 20% for a positive result of all the analytes in accordance with the standard for mass spectral data acceptance in forensic toxicology originally conceived by the SWGTOX and further developed by the toxicology subcommittee of the Organizational Scientific Area Committee (OSAC) [51]. A quantitation method was also developed for the 12 psychoactive substances, including the nine amphetamine-type stimulants and the three synthetic cathinone derivatives, including MPHD, bupropion, and its metabolite, hydroxybupropion. Fourteen isotopically labeled compounds were selected as internal standards and their MRM parameters are summarized in Table 1. For the quantitation of the targeted analytes, the ion transition with the highest sensitivity was used as a quantifier transition (Table 2).

Table 1.
Retention times (tR) and selected reaction monitoring conditions of the ISTDs used in the analysis of the targeted analytes.
Table 2.
Retention times (tR) and multiple reaction monitoring conditions for the analysis of the targeted analytes in whole blood samples.
3.1.2. Chromatography
Preliminary chromatographic experiments were conducted by using a Zorbax Eclipse Plus-C8 analytical column (50 × 2.1 mm, 1.8 μm particle size) and a Poroshell 120 EC-C18 analytical column (75 × 2.1 mm, 2.7 μm particle size). The packing material of the Poroshell 120 analytical column is based on superficially porous particle technology and features a solid silica core and a porous outer layer with the benefits of higher efficiencies and fast, high-resolution separations. The exhaustive end-capping of the Poroshell 120 packing material makes this column ideal for use with basic compounds like the targeted analytes and especially those that produce poor peak shapes on other columns. For all these reasons, the Poroshell 120 UHPLC analytical column allowed for better peak shapes and increased separation efficiency of the analytes, and it was therefore selected for the chromatography. In the present work, the selection of the organic solvent and the mobile phase composition was critical mainly for the separation of ephedrine and pseudoephedrine and the elution of phenylpropanolamine. Both methanol and acetonitrile were tested as organic modifiers, and acetonitrile provided the best results, and it was therefore selected for the chromatography. Under the chromatographic conditions described in Section 2.5, adequate separation of the targeted analytes from matrix interferences was achieved, and the analytes are eluted at retention times ranging from 2.81 to 7.71 min. Under these chromatographic conditions, ephedrine (tR 3.91 min) was adequately separated from its diastereomer, pseudoephedrine (tR 4.25 min). The retention times of the analytes are presented in Table 1 and the chemical structures of the analytes as drawn by ChemBioDraw ver. 13.0 are presented in Figure 1. Dynamic MRM chromatograms of a postmortem zero-blank sample spiked with the ISTDs at 100 ng mL−1, a premortem whole blood sample at 10 ng mL−1, and a postmortem whole blood sample at 10 ng mL−1, are presented in Figure 2.
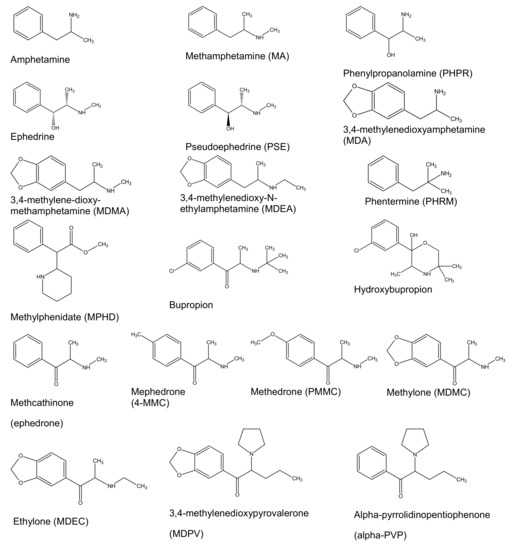
Figure 1.
Chemical structures of the targeted analytes.
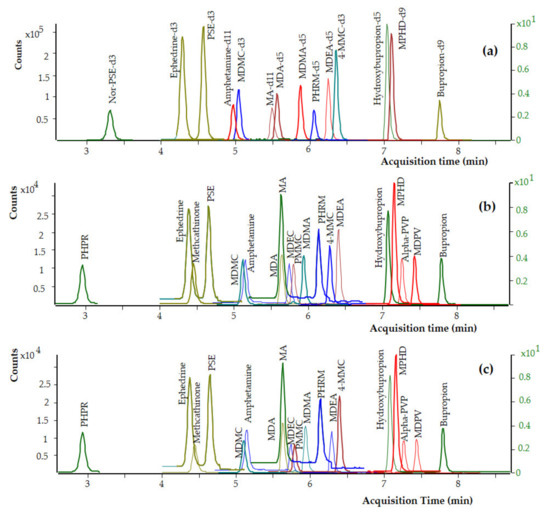
Figure 2.
Dynamic MRM chromatograms of (a) a postmortem whole blood zero-blank sample spiked with the ISTDs at 100 ng mL−1, (b) a premortem whole blood sample spiked with the analytes at 10 ng mL−1, and (c) a postmortem whole blood sample spiked with the analytes at 10 ng mL−1.
3.1.3. Optimization of the Sample Preparation Procedure
Preliminary experiments for the optimization of the sample preparation procedure were performed in premortem whole blood samples spiked with the analytes at 500 ng mL−1 using the Clean Screen® DAU mixed mode SPE cartridges (200 mg, 10 mL). These SPE cartridges are widely used for sample preparation in the Laboratory of Toxicology of the Division of Forensic Science, DE, USA. The Clean Screen® DAU SPE cartridge packing material is composed of two types of functional groups, a benzene sulphonic acid cation exchanger combined with a hydrophobic carbon chain, attached to a silica backbone. The unique copolymeric chemistry of this packing material allows for the controlled use of a mixed mode separation mechanism. For the optimization of the washing step, water, and methanol alone or in various mixtures were tested, including the use of 0.1% acetic acid aqueous solution. The optimum washing was achieved by using 1 × 3 mL of LC-grade water, 1 × 1.25 mL of 0.1 M acetic acid, and 1 × 3 mL of methanol. The type and the volume of the elution solvent are critical parameters in SPE procedure, and they were also optimized to deliver the maximum percentage recovery for each analyte. In this regard, various solvents such as ethyl-acetate, hexane, dichloromethane (CH2Cl2), and isopropanol (C3H7OH) have been tested and the best percentage recovery of the analytes was achieved using 1 × 3 mL CH2Cl2:C3H7OH:NH4OH (78:20:2, v/v/v). A 100 μL aliquot of a 0.2% hydrochloric acid solution in 2-propanol was then added to the SPE organic extract before the evaporation. This addition was deemed necessary and solved the problem of the partial loss of the volatile nitrogen-containing analytes such as amphetamine, MA, MDA, MDMA, and MDEA during the evaporation of the eluent. The optimum conditions for sample preparation are presented in detail in Section 2.4.
3.2. Statistical Analysis of Data
3.2.1. Limits of Detection (LODs) and Limits of Quantitation (LOQs)
Blank whole blood specimens spiked with decreasing quantities of each analyte have been analyzed, and the limit of detection (LOD) and the lower limit of quantitation (LLOQ) were evaluated on the basis of the signal-to-noise ratio (SNR). The LODs and LLOQs were identified as the concentrations with SNR 3:1 and 10:1, respectively. LOD and LLOQ values of 0.5 and 2 ng mL−1, respectively, were achieved for all the analytes in premortem and postmortem whole blood samples. Dynamic MRM chromatograms of a blank premortem whole blood sample, a premortem whole blank sample spiked with the analytes at LOD level (0.5 ng mL−1), and a premortem whole blank sample spiked with the analytes at LOQ level (2 ng mL−1) are presented in Figure 3. No interferences from coeluting peaks greater than 10% of the peak area of the targeted analytes at 2 ng mL−1 were observed after the analysis of 12 whole blood samples. The intra-day percentage CVs ranged from 4.9 to 11.7%, the total precision (inter-day % CVs) ranged from 4.1 to 9.7%, and the total accuracy ranged from 99.2 to 106.6% in whole blood samples spiked with the analytes at 2 ng mL−1.
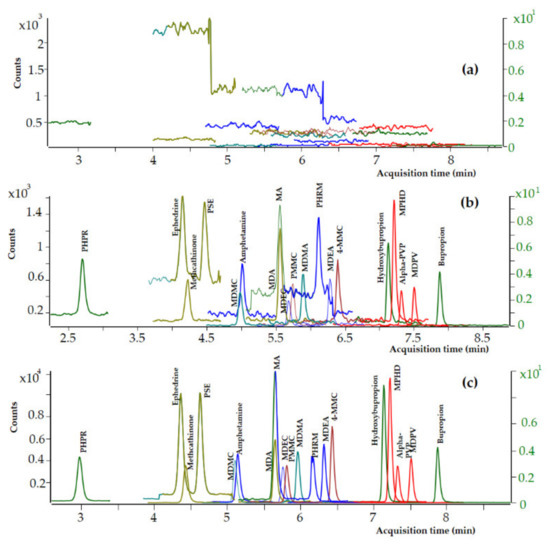
Figure 3.
Dynamic MRM chromatograms of (a) a premortem whole blood blank sample, (b) a premortem whole blood sample at LOD level (0.5 ng mL−1), and (c) a premortem whole blood sample at LOQ level (2 ng mL−1).
In the Laboratory of Forensic Science of the State of Delaware, any positive sample after the preliminary screening by Enzyme-linked Immunosorbent Assay (ELISA) (cutoff level of 20 ng mL−1) will be sent for confirmation by the proposed UHPLC-MS/MS method. Therefore, the limit of decision that will be used to discriminate between positive and negative results in the current method was administratively defined as 20 ng mL−1.
Based on the above, the UHPLC-MS/MS method was further validated to confirm the identity of all the 19 targeted analytes at 20 ng mL−1 and to quantitate 12 psychoactive substances including the nine amphetamine-type stimulants and MPHD, bupropion, and its metabolite, at 20 ng mL−1. Dynamic MRM chromatograms of a premortem whole blood blank sample, a premortem whole blood sample spiked with the analytes at LOD level (0.5 ng mL−1), and a premortem whole blood sample spiked with the analytes at LOQ level (2 ng mL−1) are presented in Figure 3.
3.2.2. Selectivity and Specificity
No interferences from coeluting peaks greater than 10% of the peak area of the targeted analytes at 20 ng mL−1 were observed after the analysis of 70 premortem and postmortem whole blood samples but with some exceptions. Interferences were detected for two premortem whole blood samples at the retention time of amphetamine, one premortem, and two postmortem blood samples at the retention time of methamphetamine, one premortem whole blood sample at the retention time of pseudoephedrine, and one postmortem whole blood sample at the retention times of bupropion, and hydroxybupropion. Due to these interferences, these samples were not used any further for the method validation. No interferences from exogenous substances were observed after the analysis of mixed standard solutions of the analytes spiked with 77 commonly used medicines and psychoactive substances. No cross fragmentation from the isotopic labeled ISTDs was observed in the analyzed zero-blank sample at the retention time of the analytes and ISTDs.
The carryover test met the predefined requirements; no interfering peaks with responses greater than 10% of the peak areas of the targeted analytes at 20 ng mL−1 were detected in blank whole blood samples analyzed after the analysis of mixed standard solutions of the analytes at 4000 ng mL−1.
The selectivity of the current method is further demonstrated in Figure 3, where dynamic MRM chromatograms of a postmortem zero-blank sample spiked with the ISTDs at 100 ng mL−1 (Figure 3a) and a postmortem (Figure 3b) and a premortem (Figure 3c) whole blood sample spiked with the analytes at 10 ng mL−1 are presented.
3.2.3. Calibration Model, Precision, and Accuracy
Calibration data were interpreted using linear and polynomial models and weighting factors 1/x and 1/x2. The results indicated that the polynomial model gave the best fit for all the analytes with a weighting factor of 1/x except for phenylpropanolamine, pseudoephedrine, and hydroxybupropion where a weighting factor of 1/x2 gave the optimum results. The coefficient of determination was higher than 0.994 for all the analytes and the back-calculated concentrations ranged from −6.6 to 8.9% for amphetamine, −9.0 to 11.9% for methamphetamine, −5.1 to 9.4% for MDA, −4.4 to 7.6% for MDMA, −9.0 to 12.8% for phentermine, −7.7 to 6.7% for pseudoephedrine, −6.9 to 10.7% for bupropion, −11.8 to 10.2% for hydroxybupropion, −7.9 to 10.7% for ephedrine, −10.1 to 7.8% for MDEA, −8.6 to 11, 4% for phenylpropanolamine, and −8.3 to 17.9% for methylphenidate. Autosampler stability was also evaluated both in premortem and postmortem samples, and the targeted analytes remained constant for at least four days.
The results for precision and accuracy evaluation are shown in Table 3 and in Table 4 for premortem and postmortem whole blood, respectively, and the precision and accuracy tests met the predefined requirements. In the premortem whole blood samples, the repeatability (intra-day % CVs) ranged from 7.1 to 1.4%, the total precision (inter-day % CVs) ranged from 1.8 to 7.5%, and the total accuracy ranged from 96.2 to 107.2%.
Table 3.
Accuracy and precision in premortem whole blood samples at low, medium, and high QC levels for the 12 psychoactive substances validated for quantitation. (n = 5 runs; 3 replicates per run).
Table 4.
Accuracy and precision in postmortem whole blood samples at low, medium, and high QC levels for the psychoactive substances validated for quantitation. (n = 5 runs; 3 replicates per run).
In the postmortem whole blood samples, the intra-day % CVs ranged from 2.1 to 4.2%, the total precision (inter-day % CVs) ranged from 1.9 to 6.8%, and the total accuracy ranged from 96.5 to 108.7%, as indicated by the results presented in Table 4.
3.2.4. Matrix Effect and Extraction Efficiency
The results of the matrix effect (%) are illustrated in Figure 4, and most of the analytes showed signal suppression. The matrix effect (%) of the analytes ranged from −31% to +3% in premortem whole blood and from −32.8 to 10.9% in postmortem whole blood.
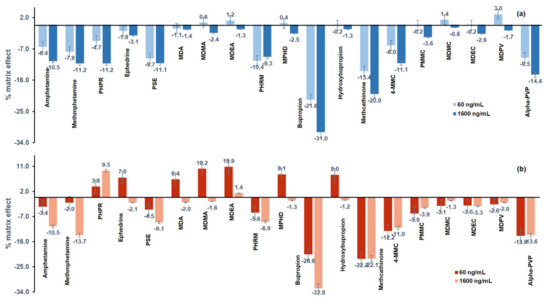
Figure 4.
Matrix effect (%) in (a) premortem (blue) and (b) postmortem (red) whole blood samples, spiked with the psychoactive substances at 60 ng mL−1 and at 1600 ng mL−1.
The results of the matrix effect (%) for the ISTDs are illustrated in Figure 5. Ion suppression was also observed for most of the ISTDs used in the current method, and the matrix effect (%) ranged from −21.2 to 0.9% and from −23.5 to 5.4% in premortem and postmortem whole blood, respectively.
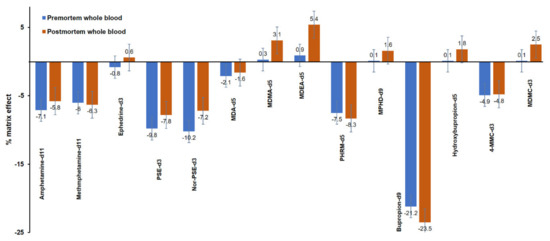
Figure 5.
Matrix effect (%) in premortem (blue) and postmortem (red) whole blood samples spiked with the internal standards at 100 ng mL−1.
Extraction efficiency (%) for the analytes ranged from 79.9 to 98.5% in premortem whole blood and from 86.2 to 95.0% in postmortem whole blood, and the results are presented in Table 5. For the ISTDs, the extraction efficiency (%) ranged from 78.9 to 92.7% and from 81.8 to 91.9% in premortem and postmortem whole blood, respectively.
Table 5.
Extraction efficiency (%) of the psychoactive substances in premortem and postmortem whole blood (n = 10).
3.3. Applicability of the Method to the Analysis of Real Cases
The applicability of the current method was evaluated by the analysis of one PTS sample, two pre-prepared samples, and two real case samples, one premortem and one postmortem whole blood sample.
The PTS sample, provided by the CAP accreditation program, was collected from a 34-year-old male with a history of drug use who was found dead at his home. Results of the in-house ELISA immunoassay preliminary screening method showed that the sample was positive for amphetamine, methamphetamine, and fentanyl. Both an in-house GC-MS method and the current UHPLC-ESI-MS/MS method were used for confirmation. The GC-MS analysis confirmed that the sample was positive for fentanyl, and the UHPLC-ESI-MS/MS analysis confirmed that the sample was positive for amphetamine and methamphetamine at concentration levels of 157 ng mL−1 and 761 ng mL−1, respectively. The analysis of this PTS sample by an external laboratory confirmed that the sample was positive for amphetamine and methamphetamine at 154 and 785 ng mL−1, respectively.
The two pre-prepared samples were obtained from UTAK. The first pre-prepared whole blood sample was found positive for amphetamine, methamphetamine, MDMA, MDA, and MDEA at concentrations of 103, 109, 99, 100, and 102 ng mL−1, respectively. The second pre-prepared whole blood sample was found positive for amphetamine, methamphetamine, MDMA, MDA, and MDEA at concentrations of 470, 480, 475, 466, and 484 ng mL−1, respectively. The pre-prepared samples were evaluated based on the nominal concentrations provided by UTAK, which were 100 and 500 ng mL−1, for all the analytes in the first and the second pre-prepared samples, respectively.
A premortem whole blood sample was collected from a 50-year-old male with a history of a car accident with lethargic symptoms, sluggish and slow speech, loss of coordination, confusion, and systolic pupil constriction. Results of the in-house ELISA immunoassay preliminary screening method showed that the sample was positive for amphetamine and phencyclidine. The GC-MS analysis confirmed that the sample was positive for phentermine and phencyclidine. The current method confirmed the positive result for phentermine at 256 ng mL−1. For the identification of phentermine, the retention time of this compound was at 6.19 min, and the two ion transitions m/z 150.1 > 91.1 (MRM1) and m/z 150.1 > 65.1 (MRM2) were also found within the acceptable limit of ±20%. However, the current method did not confirm the presence of amphetamine; most probably the ELISA immunoassay gave a false positive result for amphetamine due to the high percentage of the cross-reactivity of phentermine with amphetamine in this assay. The analysis of this sample by another validated method confirmed the presence of phentermine.
A postmortem whole blood sample was collected from a 46-year-old female. Results of the ELISA immunoassay preliminary screening method showed that the sample was positive for methamphetamine, cannabinoids, benzodiazepines, and buprenorphine. Analysis with the proposed UHPLC-ESI-MS/MS method showed that the sample was positive for pseudoephedrine at 1508 ng mL−1. For the identification of pseudoephedrine, the retention time of this compound was 4.77 min, and the ion transitions m/z 166.1 > 148.1 (MRM1) and m/z 166.1 > 91.1 (MRM2) were found within the acceptable limit of ±20%. The analysis of this postmortem whole blood sample by an external laboratory confirmed that the sample was positive for pseudoephedrine at 1400 ng mL−1.
3.4. Comparison with Other LC-MS/MS Analytical Methods
The proposed UHPLC-MS/MS method has been compared with other LC-MS/MS methods that have been reported in the literature and were dedicated to the analysis of psychoactive substances in whole blood. The results of this literature survey are presented in Table 6. Among the reported methods, only the current method allows for the analysis of the specific combination of psychoactive substances both in premortem and postmortem whole blood samples including the analysis of both ephedrine and pseudoephedrine (isobaric compounds). The method allows for the determination of the targeted analytes at adequately low LOQ and LOD values in relation to those reported in open literature (Table 6).
Table 6.
Comparison of the proposed method with LC-MS/MS methods published in literature for the determination of psychoactive substances in whole blood samples.
4. Conclusions
In this work, a UHPLC-MS/MS method was developed and validated for the determination of 19 psychoactive substances both in premortem and postmortem whole blood. By using the proposed method, the chromatographic analysis of each sample is completed in less than 9 min. The method was evaluated according to the SWGTOX guidelines and allows for the confirmation and identification of the 19 analytes at 20 ng mL−1 and for the quantitation of 12 psychoactive substances at 20 ng mL−1. Considering the problems that occur during the analysis of postmortem whole blood due to the decomposition of the body, the use of a common extraction method is important. Up to now, there is no method published in the literature to include the simultaneous determination of the 19 phenethylamine derivatives both in premortem and postmortem whole blood. Most of the published methods refer to the analysis of premortem whole blood, and only one publication [25] refers to the determination of eight psychoactive substances both in premortem and postmortem whole blood out of the 19 compounds analyzed by the current method. The efficiency of the method has been proven with the analysis of real premortem and postmortem whole blood samples, PTS samples, and pre-prepared samples obtained from UTAK.
Author Contributions
Conceptualization, S.K. and J.S.; methodology, S.K.; validation, S.K. and J.S.; formal analysis, S.K.; investigation, S.K. and J.S.; resources, J.S.; writing—original draft preparation, I.P.; writing—review and editing, I.P., S.K., and J.S.; supervision, J.S. and I.P. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
This is an observational, non-interventional, retrospective study on non-identifiable human material contained in laboratoty of Toxicology banks (Forensic Laboratory), in samples from which no genetic material could be extracted or/and reproduced due to the sample preparation. The study is not a clinical trial and it conforms with the guidelines set by the Declaration of Helsinki as the research is on non-identifiable, non-reproducible human material and there is no need to obtain informed consent and submit a study protocol for consid-eration and approval of a research ethics committee in our country.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data presented in this study are available on request from the corresponding author.
Acknowledgments
The authors acknowledge the Laboratory of Toxicology, Division of Forensic Science, Department of Safety and Homeland Security, State of Delaware, the USA for providing the materials and the equipment used for this research.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Vandrey, R.; Johnson, M.V.; Johnson, P.S.; Khali, M.A. Novel Drugs of Abuse: A Snapshot of an Evolving Marketplace. Adolesc. Psychiatry 2013, 3, 123–134. [Google Scholar] [CrossRef]
- Calpe-López, C.; García-Pardo, M.P.; Aguilar, M.A. Cannabidiol Treatment Might Promote Resilience to Cocaine and Methamphetamine Use Disorders: A Review of Possible Mechanisms. Molecules 2019, 24, 2583. [Google Scholar] [CrossRef]
- Drasch, G.; Dahlmann, F.; von Meyer, L.; Roider, G.; Eisenmenger, W. Frequency of different anti-depressants associated with suicides and drug deaths. Int. J. Legal Med. 2008, 122, 115–121. [Google Scholar] [CrossRef]
- European Monitoring Centre for Drugs and Drug Addiction. European Drug Report 2019: Trends and Developments; Publications Office of the European Union: Luxemburg, 2020; Available online: https://www.emcdda.europa.eu/ (accessed on 7 June 2020).
- Langman, L.J.; Snozek, C.L.H. Introduction to Drugs of Abuse. In Critical Issues in Alcohol and Drugs of Abuse Testing, 2nd ed.; Mercolini, L., Ed.; Elsevier: Amsterdam, The Netherlands, 2019; Volume 6, pp. 71–78. [Google Scholar]
- Ellefsen, K.N.; Concheiro, M.; Huestis, M.A. Synthetic cathinone pharmacokinetics, analytical methods, and toxicological findings from human performance and postmortem cases. Drug Metab. Rev. 2016, 48, 237–265. [Google Scholar] [CrossRef]
- United Nations Office on Drugs and Crime. World Drug Report 2015; United Nations Publications: New York, NY, USA, 2015. [Google Scholar]
- Fineschi, M.; Masti, A. Fatal poisoning by MDMA (ecstasy) and MDEA: A case report. Int. J. Legal Med. 1996, 108, 272–275. [Google Scholar] [CrossRef]
- Milroy, C.M. “Ecstasy” associated deaths: What is a fatal concentration? Analysis of a case series. Forensic Sci. Med. Pathol. 2011, 7, 248–252. [Google Scholar] [CrossRef] [PubMed]
- Wyman, J.F.; Lavins, E.S.; Engelhart, D.; Armstrong, E.J.; Snell, K.D.; Boggs, P.D.; Taylor, S.M.; Norris, R.N.; Miller, F.P. Postmortem Tissue Distribution of MDPV Following Lethal Intoxication by “Bath Salts”. J. Anal. Toxicol. 2013, 37, 182–185. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Chronister, C.W.; Hoyer, J.; Goldberger, B.A. Ethylone-Related Deaths: Toxicological Findings. J. Anal. Toxicol. 2015, 39, 567–571. [Google Scholar] [CrossRef][Green Version]
- National Institute on Drug Abuse. Advancing Addiction Science, Center for Health Statistics, Overdose Death Rates. Available online: https://www.drugabuse.gov/drug-topics/trends-statistics/overdose-death-rates (accessed on 25 May 2021).
- Kraemer, T.; Paul, L. Bioanalytical procedures for determination of drugs of abuse in blood. Anal. Bioanal. Chem. 2004, 388, 1415–1435. [Google Scholar] [CrossRef]
- Meyer, M.R. New psychoactive substances: An overview on recent publications on their toxicodynamics and toxicokinetics. Arch. Toxicol. 2016, 90, 2421–2444. [Google Scholar] [CrossRef] [PubMed]
- Peters, F.T.; Wissenbach, D.K.; Busardo, F.P.; Marchei, E.; Pichini, S. Method development in forensic toxicology. Curr. Pharm. Des. 2018, 23, 5455–5467. [Google Scholar] [CrossRef]
- Nieddu, M.; Burrai, L.; Baralla, E.; Pasciu, V.; Varoni, M.V.; Briguglio, I.; Demontis, M.P.; Boatto, G. ELISA detection of 30 new amphetamine designer drugs in whole blood, urine and oral fluid using Neogen® “amphetamine” and “methamphetamine/MDMA” kits. J. Anal. Toxicol. 2016, 40, 492–497. [Google Scholar] [CrossRef] [PubMed]
- Roda, E.; Buscaglia, E.; Papa, P.; Rocchi, L.; Locatelli, C.A.; Coccini, T. Evaluation of two different screening ELISA assays for synthetic cathinones (mephedrone/methcathinone and MDPV) with LC-MS method in intoxicated patients. J. Clin. Toxicol. 2016, 6. [Google Scholar] [CrossRef]
- Marquet, P.; Lacassie, E.; Battu, C.; Faubert, H.; Lachâtre, G. Simultaneous determination of amphetamine and its analogs in human whole blood by gas chromatography–mass spectrometry. J. Chromatogr. B Biomed. Sci. Appl. 1997, 24, 77–82. [Google Scholar] [CrossRef]
- Pelição, F.S.; Peres, M.D.; Pissinate, J.F.; Martinis, B.S. A one-step extraction procedure for the screening of cocaine, amphetamines and cannabinoids in postmortem blood samples. J. Anal. Toxicol. 2014, 38, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Mercieca, G.; Odoardi, S.; Cassar, M.; Rossi, S.S. Rapid and simple procedure for the determination of cathinones, amphetamine-like stimulants and other new psychoactive substances in blood and urine by GC-MS. J. Pharm. Biomed. Anal. 2018, 149, 494–501. [Google Scholar] [CrossRef] [PubMed]
- Woźniak, M.K.; Banaszkiewicz, L.; Wiergowski, M.; Tomczak, E.; Kata, M.; Szpiech, B.; Namieśnik, J.; Biziuk, M. Development and validation of a GC-MS/MS method for the determination of 11 amphetamines and 34 synthetic cathinones in whole blood. Forensic Toxicol. 2020, 38, 42–58. [Google Scholar] [CrossRef]
- Alexandridou, A.; Mouskeftara, T.; Raikos, N.; Gika, H.G. GC-MS analysis of underivatised new psychoactive substances in whole blood and urine. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2020. [Google Scholar] [CrossRef]
- Maurer, H.H. Advances in analytical toxicology: The current role of liquid chromatography–mass spectrometry in drug quantification in blood and oral fluid. Anal. Bioanal. Chem. 2005, 381, 110–118. [Google Scholar] [CrossRef]
- Moretti, M.; Freni, F.; Valentini, B.; Vignali, C.; Groppi, A.; Visonà, S.D.; Osculati, A.M.M.; Morini, L. Determination of Antidepressants and Antipsychotics in Dried Blood Spots (DBSs) Collected from Post-Mortem Samples and Evaluation of the Stability over a Three-Month Period. Molecules 2019, 24, 3636. [Google Scholar] [CrossRef]
- Øiestad, E.L.; Johansen, U.; Øiestad, A.M.L.; Christophersen, A.S. Drug screening of whole blood by ultra-performance liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2011, 35, 280–293. [Google Scholar] [CrossRef]
- Wang, C.C.; Hartmann-Fischbach, P.; Krueger, T.R.; Lester, A.; Simonson, A.; Wells, T.L.; Wolk, M.O.; Hidlay, N.J. Fast and Sensitive Chiral Analysis of Amphetamines and Cathinones in Equine Urine and Plasma Using Liquid Chromatography Tandem Mass Spectrometry. Am. J. Anal. Chem. 2015, 6, 995–1003. [Google Scholar] [CrossRef]
- Bjørk, M.K.; Nielsen, M.K.K.; Markussen, L.Ø.; Klinke, H.B.; Linnet, K. Determination of 19 drugs of abuse and metabolites in whole blood by high-performance liquid chromatography-tandem mass spectrometry. Anal. Bioanal. Chem. 2010, 396, 2393–2401. [Google Scholar] [CrossRef]
- Adamowicz, P.; Tokarczyk, B. Simple and rapid screening procedure for 143 new psychoactive substances by liquid chromatography-tandem mass spectrometry. Drug Test. Anal. 2016, 8, 652–667. [Google Scholar] [CrossRef] [PubMed]
- Concheiro, M.; Castaneto, M.; Kronstrand, R.; Huestis, M.A. Simultaneous determination of 40 novel psychoactive stimulants in urine by liquid chromatography-high resolution mass spectrometry and library matching. J. Chromatogr. A 2015, 1397, 32–42. [Google Scholar] [CrossRef]
- Sørensen, L.K. Determination of cathinones and related ephedrines in forensic whole-blood samples by liquid-chromatography-electrospray tandem mass spectrometry. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2011, 879, 727–736. [Google Scholar] [CrossRef] [PubMed]
- Middleberg, R.A.; Homan, J. Quantitation of amphetaminetype stimulants by LC-MS/MS. Methods Mol. Biol. 2012, 902, 105–114. [Google Scholar]
- Montesano, C.; Vannutelli, G.; Gregori, A.; Ripani, L.; Compagnone, D.; Curini, R.; Sergi, M. Broad screening and identification of novel psychoactive substances in plasma by high-performance liquid chromatography-high-resolution mass spectrometry and post-run library matching. J. Anal. Toxicol. 2016, 40, 519–528. [Google Scholar] [CrossRef][Green Version]
- Montesano, C.; Vannutelli, G.; Massa, M.; Simeoni, M.C.; Gregori, A.; Ripani, L.; Compagnone, D.; Curini, R.; Sergi, M. Multi-class analysis of new psychoactive substances and metabolites in hair by pressurized liquid extraction coupled to HPLC-HRMS. Drug Test. Anal. 2017, 9, 798–807. [Google Scholar] [CrossRef]
- Stephanson, N.N.; Signell, P.; Helander, A.; Beck, O. Use of LC-HRMS in full scan-XIC mode for multi-analyte urine drug testing—A step towards a ‘black-box’ solution? J. Mass Spectrom. 2017, 52, 497–506. [Google Scholar] [CrossRef] [PubMed]
- Meyer, M.R.; Maurer, H.H. Review: LC coupled to low- and high-resolution mass spectrometry for new psychoactive substance screening in biological matrices—where do we stand today? Anal. Chim. Acta 2016, 927, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Concheiro, M.; Anizan, S.; Ellefsen, K.; Huestis, M.A. Simultaneous quantification of 28 synthetic cathinones and metabolites in urine by liquid chromatography-high resolution mass spectrometry. Anal. Bioanal. Chem. 2013, 405, 9437–9448. [Google Scholar] [CrossRef]
- Vaiano, F.; Busardo, F.P.; Palumbo, D.; Kyriakou, C.; Fioravanti, A.; Catalani, V.; Mari, F.; Bertol, E. A novel screening method for 64 new psychoactive substances and 5 amphetamines in blood by LC-MS/MS and application to real cases. J. Pharm. Biomed. Anal. 2016, 129, 441–449. [Google Scholar] [CrossRef] [PubMed]
- Al-Saffar, Y.; Stephanson, N.N.; Beck, O. Multicomponent LC-MS/MS screening method for detection of new psychoactive drugs, legal highs, in urine-experience from the Swedish population. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2013, 930, 112–120. [Google Scholar] [CrossRef]
- Olesti, E.; Pascual, J.A.; Ventura, M.; Papaseit, E.; Farré, M.; Torre, R.; Pozo, O.J. LC-MS/MS method for the quantification of new psychoactive substances and evaluation of their urinary detection in humans for doping control analysis. Drug Test. Anal. 2020, 12, 785–797. [Google Scholar] [CrossRef]
- Maurer, H.H.; Brandt, S.D. New Psychoactive Substances, Pharmacology, Clinical, Forensic and Analytical Toxicology in Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2018; Volume 252, pp. 413–439. [Google Scholar] [CrossRef]
- Levine, B. Principles of Forensic Toxicology, 4th ed.; AACC: Washington, DC, USA, 2013; p. 5. [Google Scholar]
- The International Association of Forensic Toxicologists Committee of Systematic Toxicological Analysis. Recommendations on Sample Collection. TIAFT-Bulletin XXIX, Number 1. Available online: http://www.tiaft.org/data/uploads/documents/tiaft-sta-recommendations-on-sample-collection.pdf (accessed on 27 May 2021).
- The Delaware Code Online. Available online: https://delcode.delaware.gov/title21/c041/sc09/ (accessed on 27 May 2021).
- Ondra, P.; Válka, I.; Knob, R.; Ginterová, G.; Maier, V. Analysis of Amphetamine-Derived Designer Drugs by Gas Chromatography with Mass Spectrometry. J. Anal. Toxicol. 2016, 40, 78–85. [Google Scholar] [CrossRef] [PubMed]
- Tang, M.H.; Ching, C.K.; Lee, C.Y.; Lam, Y.H.; Mak, T.W. Simultaneous detection of 93 conventional and emerging drugs of abuse and their metabolites in urine by UHPLC-MS/MS. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 969, 272–284. [Google Scholar] [CrossRef]
- Kalovidouris, M.; Michalea, S.; Robola, N.; Koutsopoulou, M.; Panderi, I. Ultra-performance liquid chromatography/tandem mass spectrometry method for the determination of lercanidipine in human plasma. Rapid Commun. Mass Spectrom. 2006, 20, 2939–2946. [Google Scholar] [CrossRef]
- Rathod, R.H.; Chaudhari, S.R.; Patil, A.S.; Shirkhedkar, A.A. Ultra-high-performance liquid chromatography-MS/MS (UHPLC-MS/MS) in practice: Analysis of drugs and pharmaceutical formulations. Future J. Pharm. Sci. 2019, 5, 6. [Google Scholar] [CrossRef]
- Scientific Working Group for Forensic Toxicology. Scientific Working Group for Forensic Toxicology (SWGTOX) standard practices for method validation in forensic toxicology. J. Anal. Toxicol. 2013, 37, 452–474. [Google Scholar] [CrossRef]
- Wille, S.M.R.; Coucke, W.; De Baere, T.; Peters, F.T. Update of Standard Practices for New Method Validation in Forensic Toxicology. Curr. Pharm. Des. 2017, 23, 5442–5454. [Google Scholar] [CrossRef] [PubMed]
- European Comission. Commission Decision No. 2002/657/EC of 12 August 2002 Implementing Council Directive 96/23/EC Concerning the Performance of Analytical Methods and the Interpretations of Results; EUROPEAN Comission: Brussel, Belgium, 2002; Volume 221, pp. 8–36. [Google Scholar]
- Organizational Scientific Area Committee. Standard for Mass Spectral Data Acceptance in Forensic Toxicology. Available online: https://www.nist.gov/system/files/documents/2019/03/20/standard_for_mass_spec_spectral_data_acceptance_-_asb.pdf (accessed on 27 May 2021).
- Cunha, R.L.; Olivieta, G.S.L.; Oliviera, A.L.; Maldaner, A.O.; Pereira, P.A.P. Fast determination of amphetamine-type stimulants and synthetic cathinones in whole blood samples using protein precipitation and LC-MS/MS. Microchem. J. 2021, 163, 105895. [Google Scholar] [CrossRef]
- Lau, T.; Concheiro, M.; Cooper, G. Determination of 30 synthetic cathinones in postmortem blood using LC–MS-MS. J. Anal. Toxicol. 2020, 44, 679–687. [Google Scholar] [CrossRef] [PubMed]
- Ong, R.S.; Kappatos, D.C.; Russell, S.G.G.; Poulsen, H.A.; Banister, S.D.; Gerona, R.R.; Glass, M.; Johnson, C.S.; McCarthy, M.J. Simultaneous analysis of 29 synthetic cannabinoids and metabolites, amphetamines, and cannabinoids in human whole blood by liquid chromatography-tandem mass spectrometry—A New Zealand perspective of use in 2018. Drug Test. Anal. 2020, 12, 195–214. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, Z.H.; Li, H.; Liu, Y.; Zhao, M.; Jiang, Y.; Zhao, W.S. Simultaneous determination of 12 illicit drugs in whole blood and urine by solid phase extraction and UPLC-MS/MS. J. Chromatogr. B Biomed. Appl. 2014, 995–996, 10–19. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).