3.1. Method Goals and Circumstances
The first step of any structured development of a chemical analysis protocol, and definitively when following a QbD approach, is to establish the requirements of the final method and the circumstances under which it will be applied, and then to assess the chemical properties of the analyte and the other species in the sample. After that, a review of prior knowledge in the scientific literature, regarding the quantification of similar analytes in comparable samples, may assist in the selection of a suitable analytical technique and provide a starting point of conditions for method development and optimization. This knowledge is also used for risk assessment of the selected analysis technique and its conditions and for planning of strategies for risk mitigation.
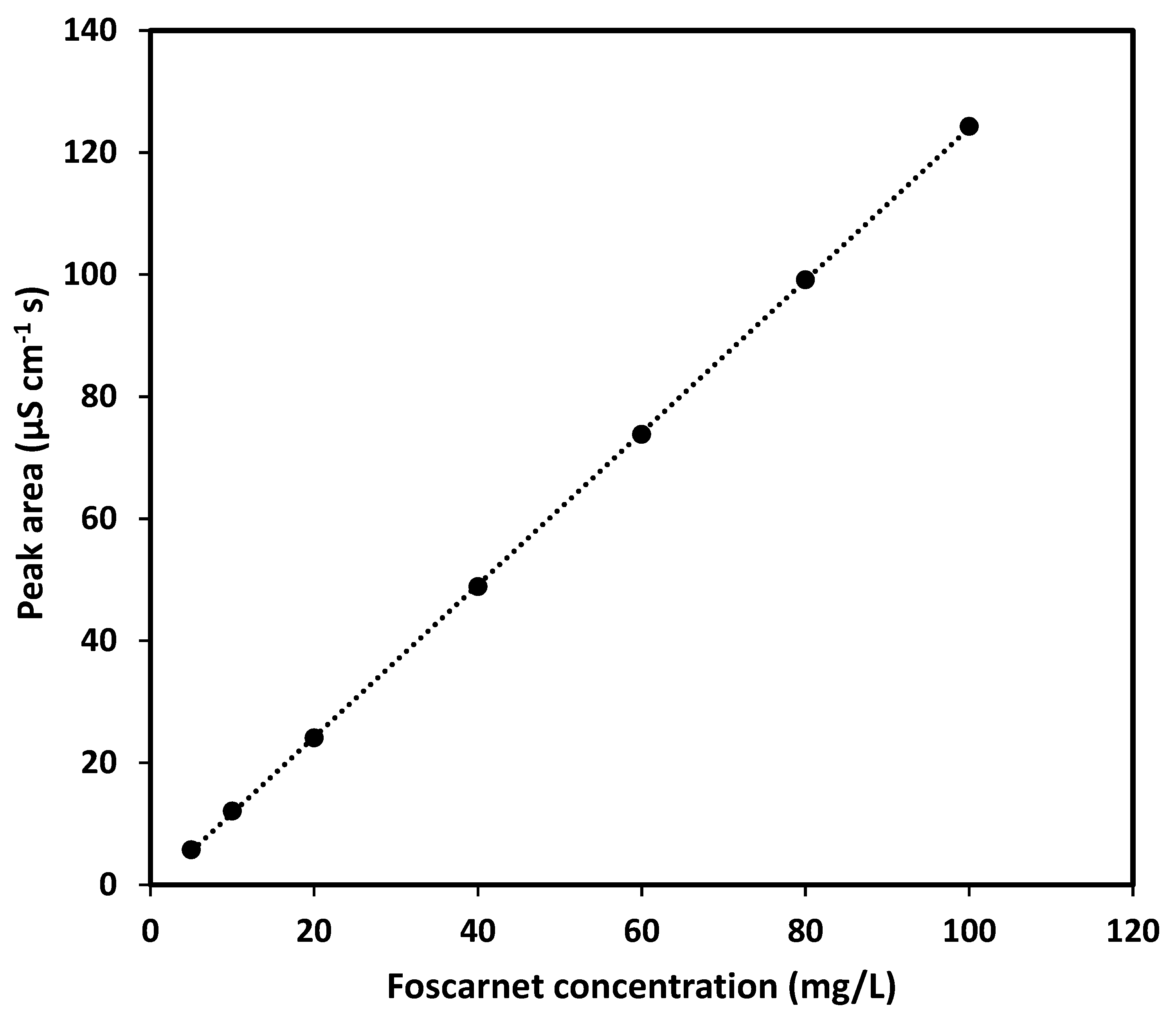
Since the aim of this study was to develop an assay for foscarnet sodium injection solution, the method must be able to quantify foscarnet in the range of 24 mg/mL (or lower if dilution is applied before analysis) while simultaneously being able to discriminate between other species in the pharmaceutical formulation to ensure specificity for foscarnet. These requirements call for a separation step in the analytical procedure, and according to FDA guidelines [
22], the chromatographic resolution (Rs) from the closest peak must then be at least 2, the analyte peak should have a retention factor (k’) above 2, a theoretical plate number (N) not less than 2000, and the tailing should not exceed 2. Naturally, the assay must also be reproducible and easily transferable between different instruments, analysts, and laboratories. It is further desirable that an assay method is quick and simple to perform and does not require expensive new equipment or complicated procedures.
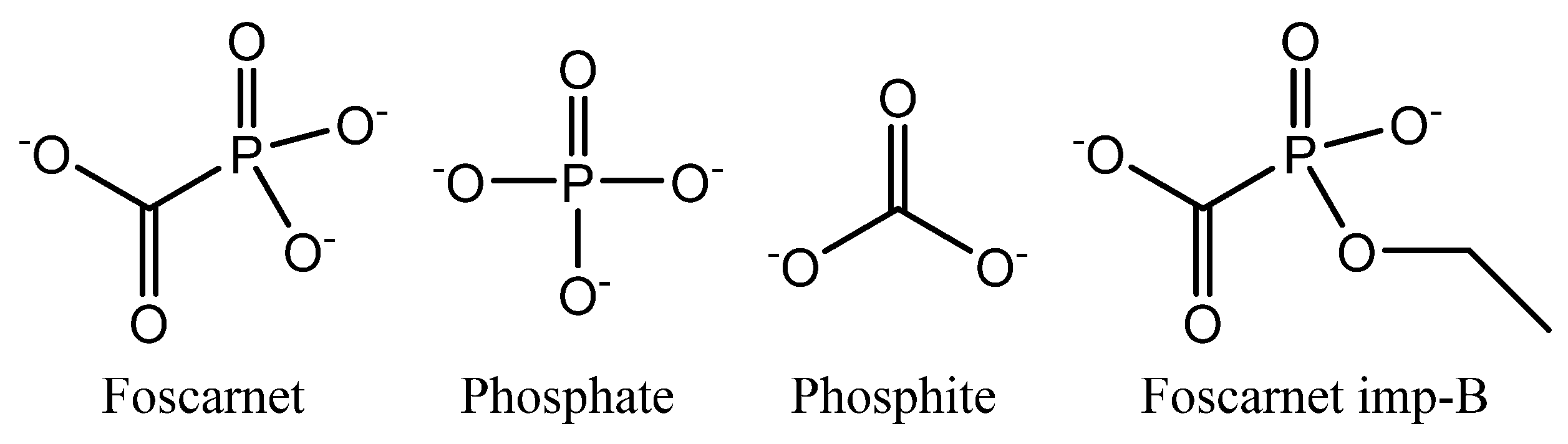
Foscarnet (
Figure 1) is a triprotic acid with pKa values of 0.49, 3.41, and 7.27 [
28,
29], and thus, it is multiply negatively charged at neutral pH and within most of the practical pH range. In the sodium hexahydrate salt form, foscarnet has a molecular weight of 300.0 g/mol [
18], and the molecule is remarkably hydrophilic, as depicted by its predicted [
30] negative logD value of −7.1 at neutral pH and spanning from −1.2 to −9.0 in the 0–14 pH range. Thus, the solubility in organic solvents is very low [
29,
31] but amounts to 5 wt % in water at pH 7 [
32]. Its intrinsic light absorption capabilities are poor, and it has been concluded that wavelengths of 205 nm or less would be required to achieve sufficient analytical detection sensitivity [
29,
31]. Foscarnet has been reported to form complexes with several metal ions [
33], and therefore, it often shows poor peak shapes during liquid chromatographic separations [
29,
31,
33,
34,
35,
36] and may adsorb to surfaces during sample preparation [
35] unless precautions have been taken. Foscarnet is known to degrade into phosphate and phosphite (see
Figure 1) while releasing carbon dioxide [
28,
37], especially in acidic aqueous solutions. A potential by-product from foscarnet synthesis is 1-ethoxy-1-hydroxyphosphinecarboxylic acid 1-oxide, which is registered as impurity B (see
Figure 1) in pharmacopoeia methods [
18,
19].
The scientific literature does contain several published methods for the separation and analysis of foscarnet and other phosphonates [
29,
31]. The first example employed ion exchange separation, using a silica-based anion exchange column, an acetic acid eluent, and post-column reaction detection [
34]. A later example of anion exchange separation used a polymethacrylate-based column using nitric acid eluent and conductivity detection [
36]. Both these procedures utilized multi-acidic eluent additives such as citric acid [
34] and succinic acid [
36] in millimolar concentrations to mask strong interactions and achieve acceptable peak shapes. Other analysis protocols have been based on ion-pair reversed-phase liquid chromatographic separations with electrochemical detection (coulometry and amperometry) [
33,
35] or with ultra-violet light absorption detection [
37,
38,
39] (presumably of the ion-pair complex), utilizing low amounts of sulfuric acid or pyrophosphoric acid as eluent additives to accomplish the needed peak shape improvement. More recent examples have exploited high pH suppressed ion chromatography with hydroxide gradient elution [
40] and avoided the need for peak shape improving eluent additives by utilizing IC instruments where the pump heads, flow path, and column body were constructed from PEEK rather than being based on stainless steel.
The most attractive separation mode for our target analyte foscarnet that has such a hydrophilic character and multiple anionic properties was concluded to be suppressed IC. In this mode, we could fully benefit from its triple negative charge at higher pH versus the fewer charges of all the related compounds, including the degradation products phosphate and phosphite, the impurity B, and the chloride present in the pharmaceutical formulation. We further selected an eluent suppressor setup that can be combined with any instrument system for LC or IC and then passivated the flow path [
26,
41,
42] if the instrument was based on stainless steel. As separation material, a rather low-capacity hydrophilic polyvinylalcohol-based anion exchanger in a PEEK column hardware (250 × 4 mm, 37 µeq/column) was selected to avoid the need for particularly strong eluents and limit the risk of peak deformation. The rather large particle size (9 µm) of this material would not necessarily be a drawback in the present context despite resulting in lower peak efficiency. It would give limited column pressure drop, thus possibly allowing faster flow rates to cut run times, and the larger peak volumes would make the method less sensitive to system dead volume and thus allow smooth method transfer between different instrument setups. An aqueous eluent system with a combination of carbonate and bicarbonate salts (with hydroxide as pH modifier if required) was favored, since such eluents are more resistant to sample pH effects due to their inherent buffer capacity, and they are typically easy to prepare and use routinely in isocratic mode with any type of LC instrument. To ensure perfect retention stability and avoid increasing noise and background levels, the eluent was prepared and stored in polypropylene plastic bottles and protected from ambient carbon dioxide by forcing all air entering the eluent bottle to pass through a cartridge containing a color-indicating carbon dioxide adsorption material. For details of the instrument setup and procedures, we refer to the Materials and Methods section.
3.2. Screening of Eluent Conditions
The initial screening of eluent conditions for separation of foscarnet from its related compounds phosphate, phosphite, and impurity B was accomplished with eluents with equal concentrations of sodium carbonate and sodium bicarbonate ranging from 5 mM to 10 mM of each salt. In addition, we screened higher pH conditions by employing a pure carbonate eluent and a pure hydroxide eluent. Although being present in the pharmaceutical formulation, chloride was not injected explicitly during this screening, since its single charge will force it to elute well before the other multiple charged related compounds and thus not constitute a risk of interference if foscarnet already is separated from phosphate, phosphite, and impurity B. Similarly, not all of the related compounds were injected with each eluent if some of the stipulated criteria already had failed.
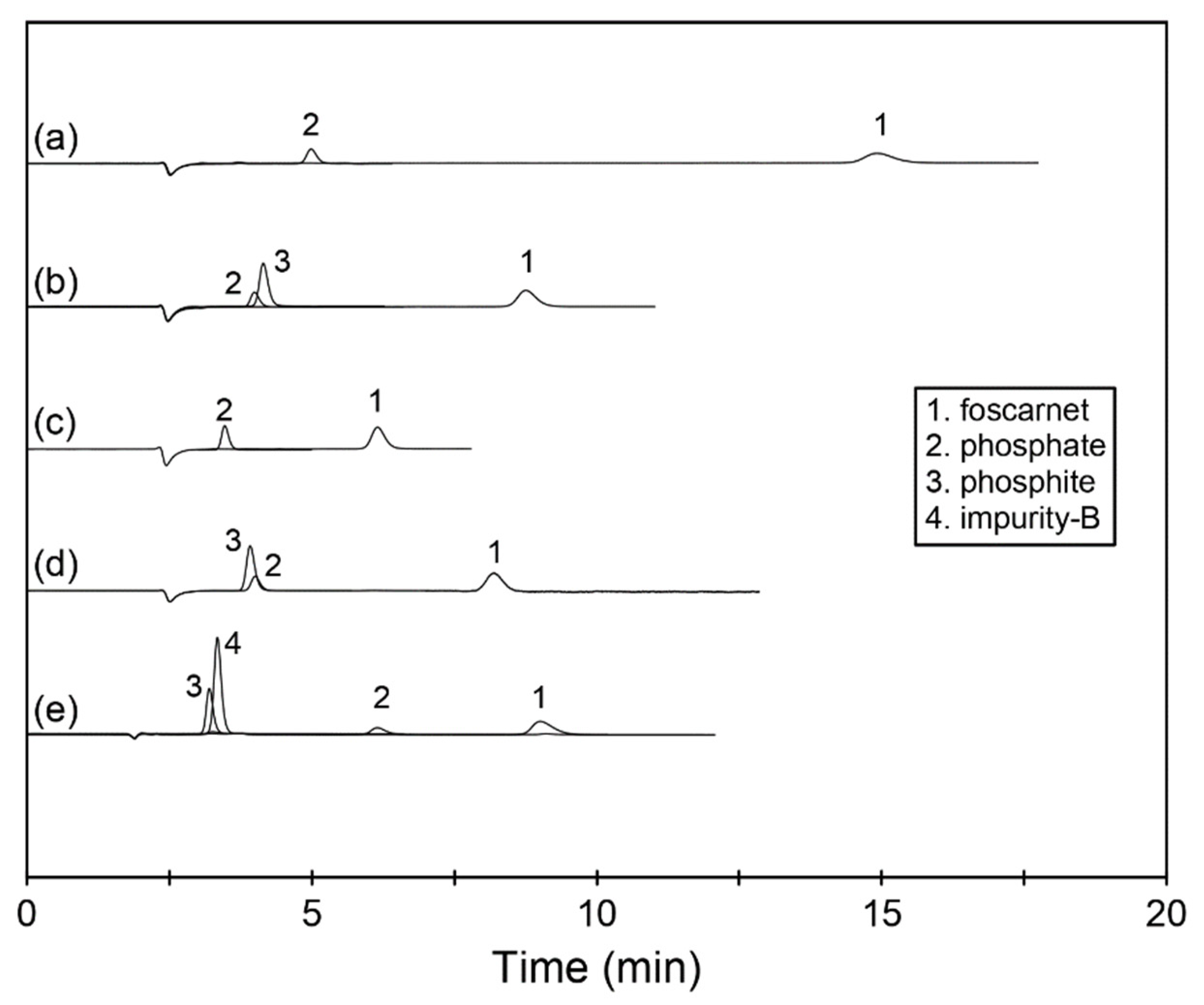
With all tested eluents, the foscarnet peak was well separated from all related compounds and displayed very good peak shape with acceptable efficiency; see
Figure 2 and
Table 1. However, the resolution criteria of >2 [
22] was not fully met with the pure hydroxide eluent. The weakest eluent with 5 mM Na
2CO
3 and 5 mM NaHCO
3 resulted in a retention time close to 15 min, which was deemed too long for an assay. The eluent containing 10 mM Na
2CO
3 and 10 mM NaHCO
3 was too strong, as it resulted in a retention factor well below 2.0, which is a recommended minimum criterion [
22]. However, the eluent with 7.5 mM Na
2CO
3 and 7.5 mM NaHCO
3 did result in a separation that tentatively seemed to meet all the stipulated method criteria.
Since we wanted to better understand the possibilities of separation selectivity manipulation for foscarnet and for its related compounds that largely co-eluted with the first three eluents, two additional conditions with higher pH were explored. The eluent with 10 mM Na
2CO
3 resulted in a retention for foscarnet that was comparable to the bicarbonate–carbonate eluent containing 7.5 mM of each component. The selectivity between phosphate and phosphite changed slightly though, and the two species incompletely changed their elution order; see
Figure 2. The reason for this behavior was concluded to be the further partial ionization among the population of phosphate ions.
Although the third pKa value for phosphate is 12.32 [
43], and the dominating ionization form in both eluents (at pH 10.3 and 11.1) therefore is the double charged HPO
42− ion, the increase in the proportion of triply charged molecules in the eluent with higher pH is probably responsible for this slight selectivity shift. Since neither phosphite nor impurity B can be ionized further than to a double negative charge, their selectivity appeared not to change with the modification of eluent pH.
The effect of additional ionization of phosphate from the double-charged HPO42− to the triply charged PO43− ion was even more evident when the eluent pH was increased even further to pH 12.6. With the 40 mM NaOH used to establish this pH, the retention for foscarnet, phosphite, and impurity B changed somewhat due to a change in the dominating eluting ion from carbonate to hydroxide, whereas the retention for phosphate changed dramatically, and it now eluted well separated from both foscarnet and the coeluted pair of phosphite plus impurity B. However, an unfortunate consequence of this change in pH was that the foscarnet peak started to show more tailing and thus broadened and lost separation efficiency.
Although not a focus of this study, these experiments also made it evident that to separate all the impurities with suppressed IC using carbonate–bicarbonate eluents, another column with additional selectivity features would be necessary. Our preliminary multivariate selectivity model for columns in suppressed IC [
44,
45] suggests that such a column should be a bit more hydrophobic to manage that type of separation.
3.3. Studying the Effect of Method Conditions Using Design of Experiments
As found in the screening experiments above, the mobile phase containing 7.5 mM Na
2CO
3 and 7.5 mM NaHCO
3 provisionally appeared to meet the target criteria of the method. However, to better understand how the different method parameters contributed to the quality attributes of the method, an experimental design was performed around these experimental conditions. In addition to concentration of the two mobile phase components, the eluent flow rate and column temperature were included as method parameters (factors) in the design, as detailed in
Table 2.
The eight quality attributes (responses) that were monitored included foscarnet retention time and retention factor, foscarnet peak efficiency and tailing, column back-pressure, and resolution between foscarnet and its related compounds phosphite, phosphate, and impurity B, respectively. The resulting full factorial design with four factors at two levels consisted of 19 experimental conditions in total, including three center point experiments. After performing these experiments in a fully randomized manner, the resulting data were subjected to multiple linear regression modeling to calculate the coefficients of each factor (method parameter) and their interactions to generate each type of response (the different quality parameters). The summary of fit for the model is outlined in
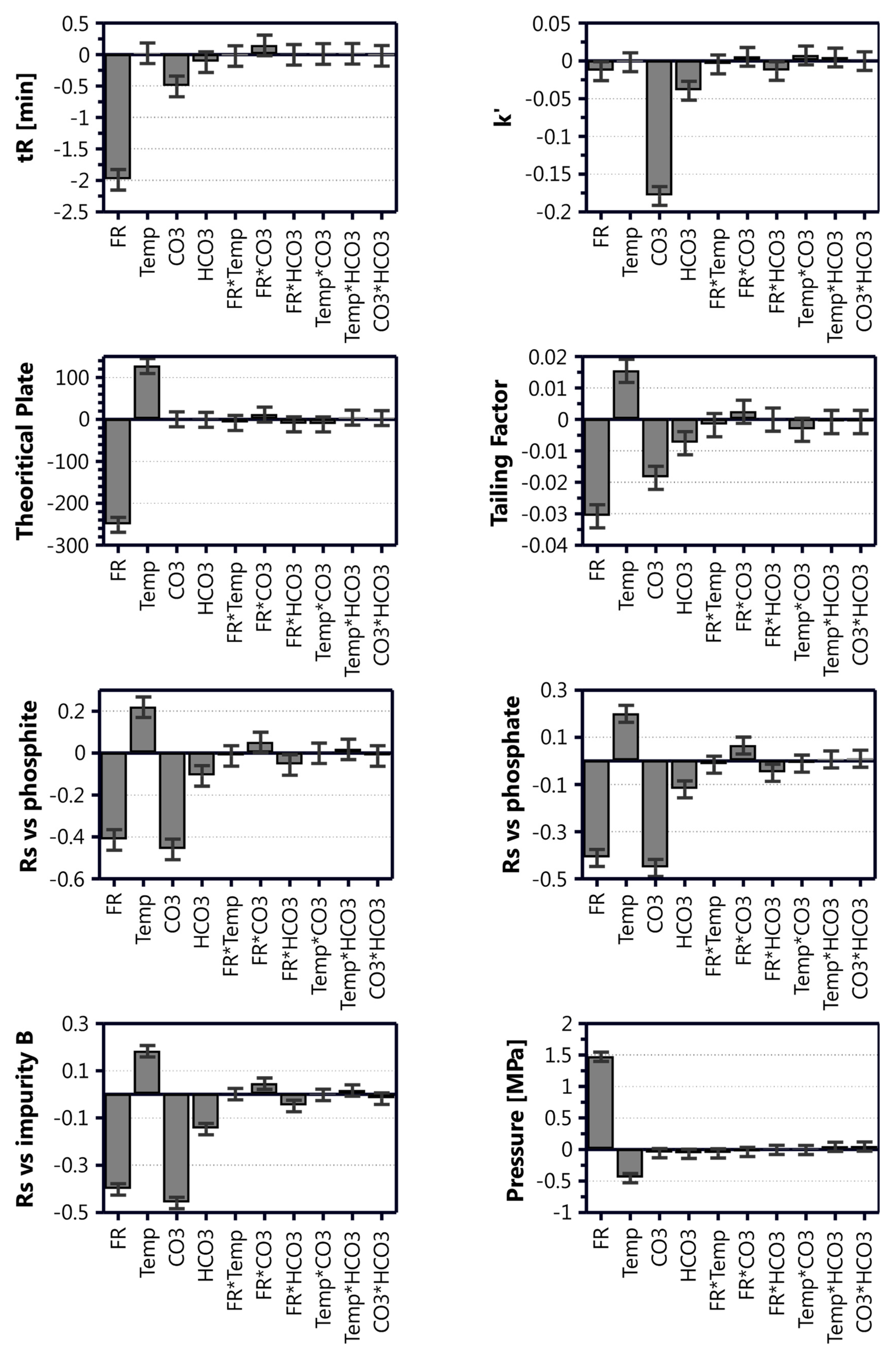
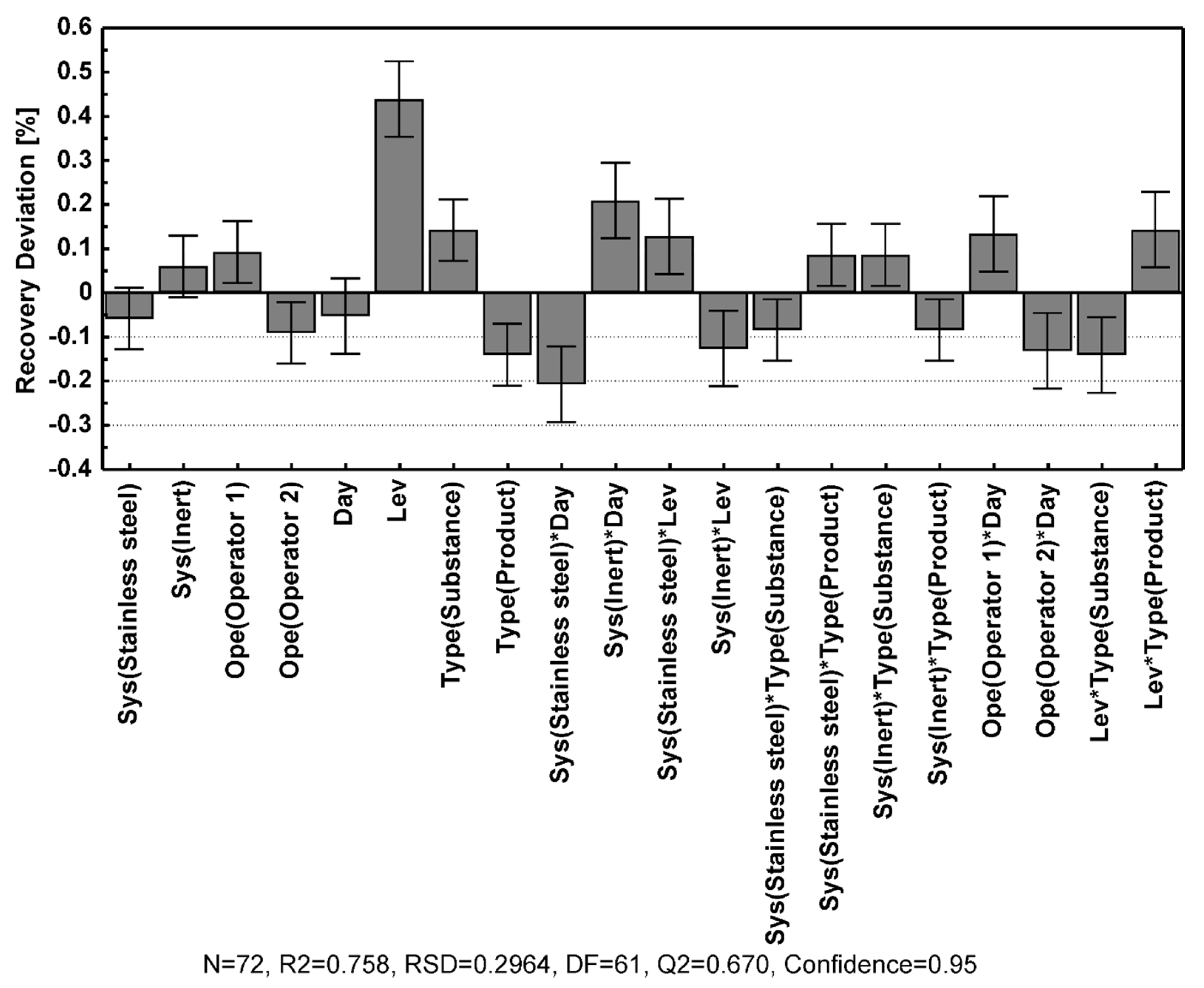
Table 3, and the contributions of the different model coefficients and their interactions are illustrated in
Figure 3.
In general, the data were highly reproducible, and most of the modeled quality control attributes showed excellent fit to the data and very high predictability, as manifested by the R2 and Q2 values displayed in
Table 3. The tailing factor showed a slightly inferior model fit compared to the other quality control attributes, although it still displayed a good data fit with R2 just below 0.99 and an adequate predictability with Q2 above 0.88.
Inspection of the model coefficient plots in
Figure 3 disclose the relative impact of the different parameters (factors) on the model attributes (responses). From this plot, it was clear that the interaction terms for all responses either were insignificant or very small in comparison to the single factor terms. The retention time was strongly affected by the flow rate and mobile phase composition, and the concentration of Na
2CO
3 had about four times stronger effect than NaHCO
3. Higher concentrations of Na
2CO
3 and NaHCO
3 reduced the retention time and retention factor, whereas the effect of temperature was insignificant on these attributes. The foscarnet peak was less tailing at higher flowrate and higher carbonate concentration, whereas higher temperature led to increased peak tailing. Low flow rate and high temperature within the studied range led to reduced peak efficiency with an increase in the number of theoretical plates, which is in good agreement with chromatographic theory [
46]. The effect of the method parameters on the resolution of foscarnet to the related substances was quite similar across the different species. Better resolution was achieved when using a higher temperature, lower flow rate, and lower eluent salt concentrations, where the flow rate and Na
2CO
3 concentration had the most significant effect.
3.4. Method Optimization and Design Space Evaluation
The limiting criteria for method optimization was set according to the FDA [
22]; i.e., the retention factor and resolution should be more than 2, the tailing factor should be less than 2, and the number of theoretical plates should be more than 2000. The back-pressure limit was set according to the manufacturer specification for the applied column, which states below 10 MPa. The desired retention time was set to below 10 min, since the aim was to arrive at a quick assay method. Following the criteria set above, as summarized in
Table 4, the design space was calculated utilizing multiple probability Monte Carlo simulations within the employed software [
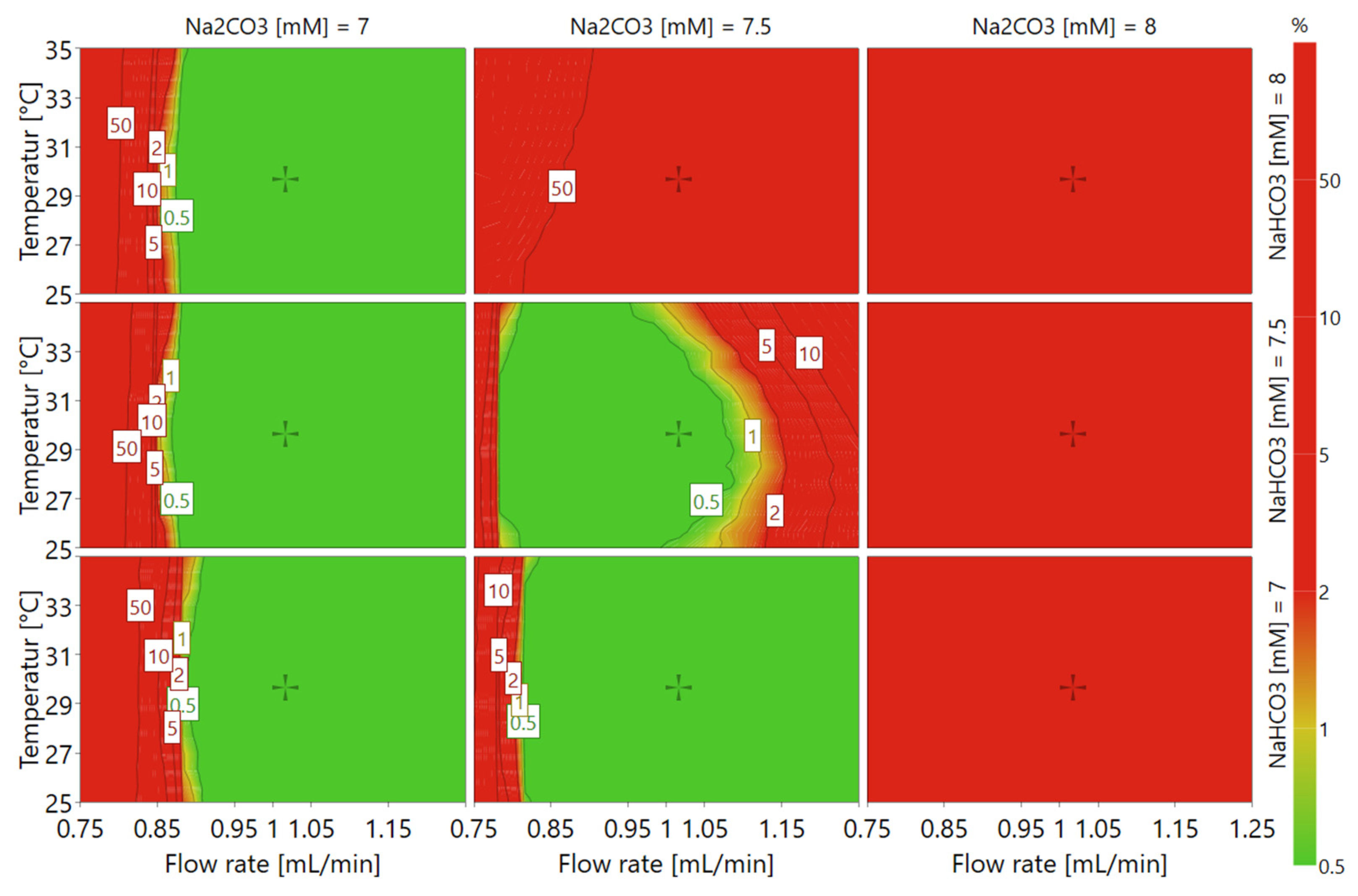
21] based on data from the experimental design. As illustrated by the resulting design space plot (
Figure 4), conditions with high Na
2CO
3 concentration or low flow rate would likely result in the method failing to meet the set criteria and should thus be avoided, as represented by the red colored area. On the contrary, low eluent buffer salt concentrations and moderate flow rate assure that the method criteria would be met within most of the temperature range, as illustrated by the green colored area.
Since a short analysis time and good peak shape are highly desired in an assay, a method optimization was performed with the target to minimize the retention and tailing factor while maximizing the retention factor by utilizing the “find robust setpoint” calculation within the commissioned multivariate software [
21]. The other attributes were not considered during this optimization, since the experimental results for these attributes always met the method criteria. The resulting optimum and robust setpoint values, plus the factor contributions and the predicted quality control attributes, are all listed in
Table 5. In order to simplify the assay setup and make it easy to transfer between different operators and laboratories, we tweaked the operating conditions slightly by rounding the value of flowrate to 1.0 mL/min, the concentrations of Na
2CO
3 and NaHCO
3 to 7.3 mM, and the temperature to 30 °C, which thus constitutes the conditions at which the method was validated. This condition did also show a very high probability of success within the design space (cf.
Figure 4).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}