
Expanding Antineoplastic Drugs Surface Monitoring Profiles: Enhancing of Zwitterionic Hydrophilic Interaction Methods

 ,
,  ,
,  ,
,  , , ,
, , ,  , and
, and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Instruments
2.3. Standard Solutions and Calibration Levels
2.4. Sample Preparation
2.5. Chromatography and Instrument Parameters
2.6. MS/MS Experiments
2.7. Performance Evaluation of LC-MS/MS Methods
2.8. Mitoxantrone Stability Evaluation
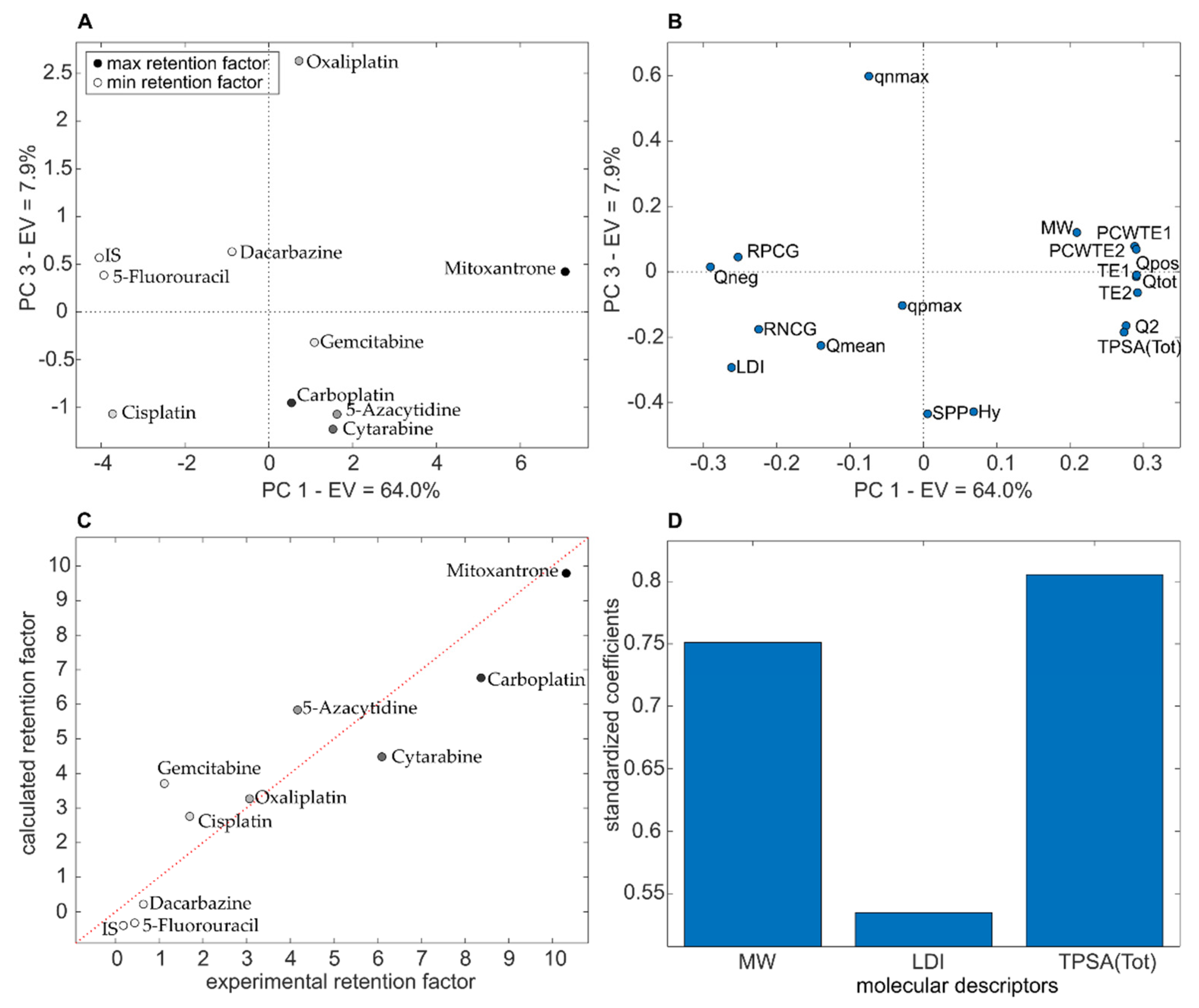
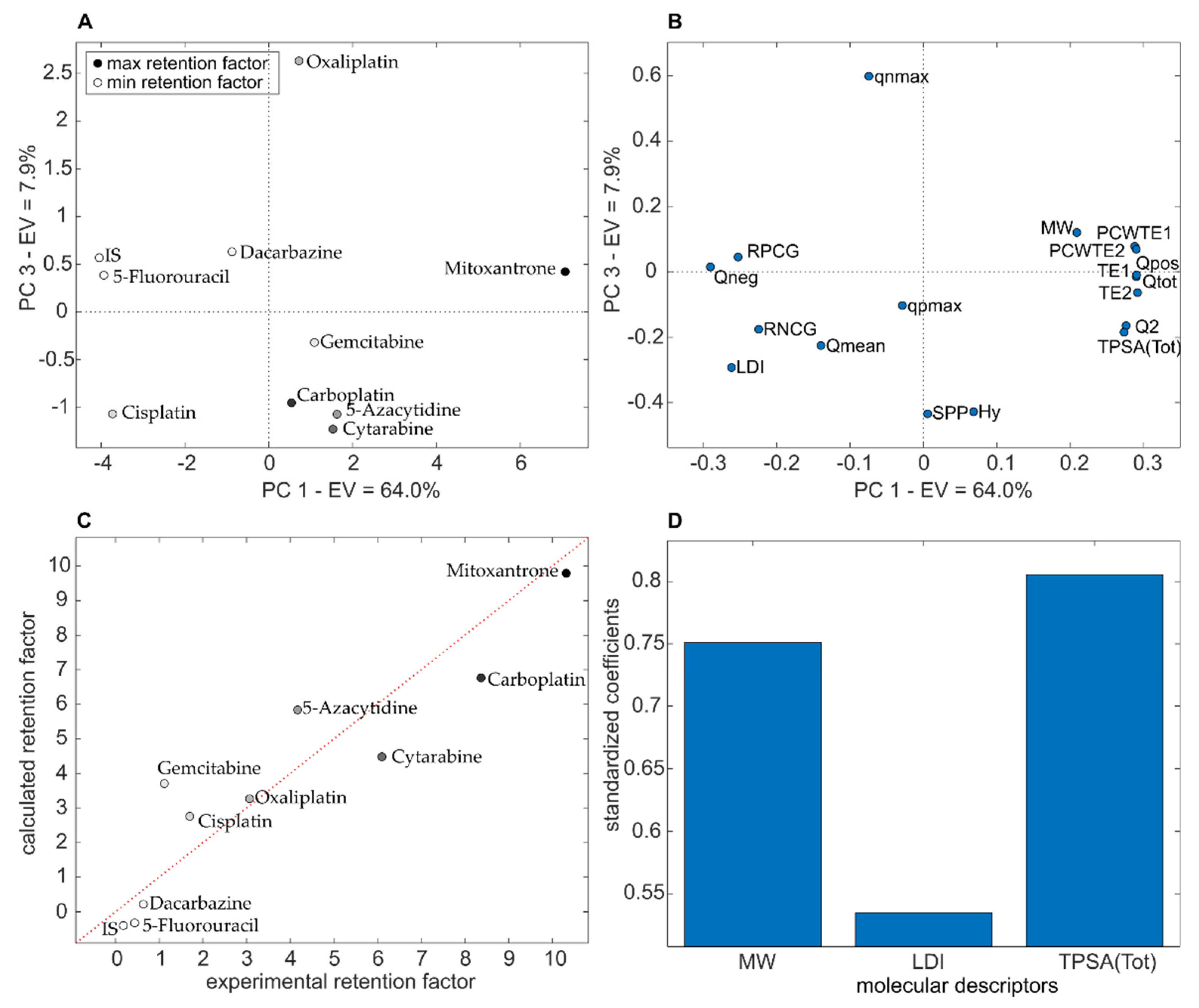
2.9. Quantitative Structure-Retention Relationships (QSRRs)
3. Results
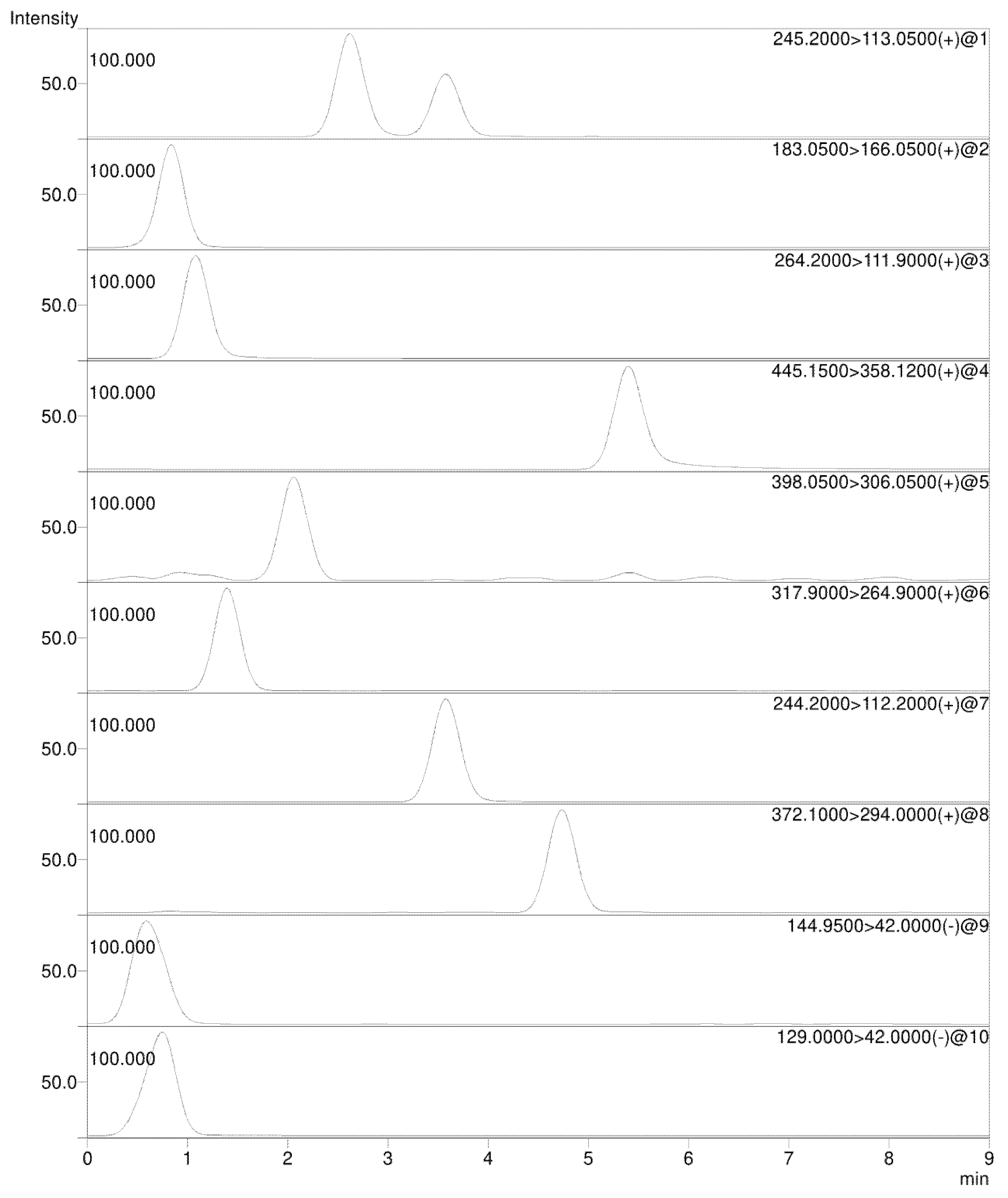
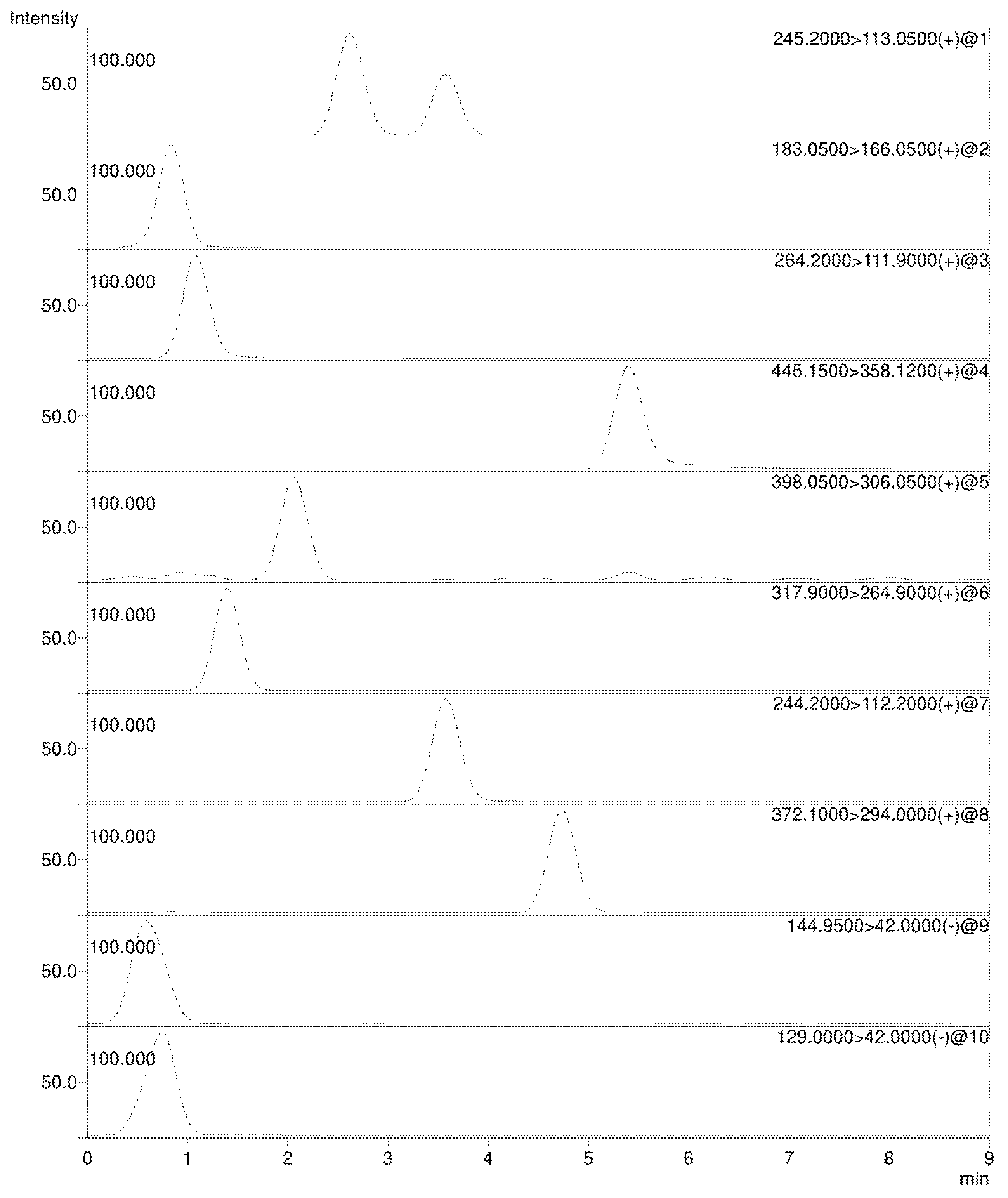
3.1. Chromatographic Conditions
3.2. Mass Spectrometry
3.3. Method Validation
3.3.1. Calibration Curves
3.3.2. Matrix Effect and Recovery
3.3.3. Accuracy and Precision
3.4. QSRR Results
3.5. Real Sample Analysis
3.6. Mitoxantrone Stability
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- Global Anticancer Drugs Market to Record 6.7% CAGR through 2027. Available online: https://www.biospace.com/article/global-anticancer-drugs-market-to-record-6-7-percent-cagr-through-2027/ (accessed on 14 November 2021).
- Dranitsaris, G.; Johnston, M.; Poirier, S.; Schueller, T.; Milliken, D.; Green, E.; Zanke, B. Are health care providers who work with cancer drugs at an increased risk for toxic events? A systematic review and meta-analysis of the literature. J. Oncol. Pharm. Pract. 2005, 11, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Mahmoodi, M.; Soleyman-Jahi, S.; Zendehdel, K.; Mozdarani, H.; Azimi, C.; Farzanfar, F.; Safari, Z.; Mohagheghi, M.-A.; Khaleghian, M.; Divsalar, K.; et al. Chromosomal aberrations, sister chromatid exchanges, and micronuclei in lymphocytes of oncology department personnel handling anti-neoplastic drugs. Drug Chem. Toxicol. 2016, 40, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Villarini, M.; Gianfredi, V.; Levorato, S.; Vannini, S.; Salvatori, T.; Moretti, M. Occupational exposure to cytostatic/antineoplastic drugs and cytogenetic damage measured using the lymphocyte cytokinesis-block micronucleus assay: A systematic review of the literature and meta-analysis. Mutat. Res. 2016, 770, 35–45. [Google Scholar] [CrossRef] [PubMed]
- Directive 2004/37/EC of the European Parliament and of the Council of 29 April 2004 on the Protection of Workers from the Risks Related to Exposure to Carcinogens or Muta-Gens at Work (Sixth Individual Directive within the Meaning of Article 16(1) of Council Directive 89/391/EEC). Available online: https://eur-lex.europa.eu/LexUriServ/LexUriServ.do?uri=OJ:L:2004:229:0023:0034:EN:PDF (accessed on 9 December 2021).
- European BioSafety Network. 2016. Preventing Occupational Exposure to Cytotoxic and Other Hazardous Drugs European Policy Recommendations. Available online: https://www.europeanbiosafetynetwork.eu/wp-content/uploads/2016/05/Exposureto-Cytotoxic-Drugs_Recommendation_DINA4_10-03-16.pdf (accessed on 9 December 2021).
- Kibby, T. A review of surface wipe sampling compared to biologic monitoring for occupational exposure to antineoplastic drugs. J. Occup. Environ. Hyg. 2017, 14, 159–174. [Google Scholar] [CrossRef] [PubMed]
- Jeronimo, M.; Colombo, M.; Astrakianakis, G.; Hon, C.-H. A surface wipe sampling and LC–MS/MS method for the simultaneous detection of six antineoplastic drugs commonly handled by healthcare workers. Anal. Bioanal. Chem. 2015, 407, 7083–7092. [Google Scholar] [CrossRef] [PubMed]
- Dugheri, S.; Bonari, A.; Pompilio, I.; Boccalon, P.; Tognoni, D.; Cecchi, M.; Ughi, M.; Mucci, N.; Arcangeli, G. Analytical strategies for assessing occupational exposure to antineoplastic drugs in healthcare workplaces. Med. Pract. 2018, 69, 589–604. [Google Scholar] [CrossRef] [PubMed]
- Lee Walton, A.M.; Bush, M.A.; Douglas, C.E.; Allen, D.H.; Polovich, M.; Spasojevic, I. Surface Contamination With Antineoplastic Drugs on Two Inpatient Oncology Units. Oncol. Nurs. Soc. Forum 2020, 47, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Chabut, C.; Bussières, J.-F. Characteristics of wipe sampling methods for antineoplastic drugs in North America: Comparison of six providers. Pharm. Technol. Hosp. Pharm. 2020, 5. [Google Scholar] [CrossRef]
- Mucci, N.; Dugheri, S.; Farioli, A.; Garzaro, G.; Rapisarda, V.; Campagna, M.; Bonari, A.; Arcangeli, G. Occupational exposure to antineoplastic drugs in hospital environments: Potential risk associated with contact with cyclophosphamide- and ifosfamide contaminated surfaces. Med. Pract. 2020, 71, 519–529. [Google Scholar] [CrossRef]
- Dugheri, S.; Mucci, N.; Squillaci, D.; Marrubini, G.; Bartolucci, G.; Melzi, C.; Bucaletti, E.; Cappelli, G.; Trevisani, L.; Arcangeli, G. Developing a Fast Ultra-High-Performance Liquid Chromatography–Tandem Mass Spectrometry Method for High-Throughput Surface Contamination Monitoring of 26 Antineoplastic Drugs. Separations 2021, 8, 150. [Google Scholar] [CrossRef]
- Dugheri, S.; Mucci, N.; Mini, E.; Squillaci, D.; Marrubini, G.; Bartolucci, G.; Bucaletti, E.; Cappelli, G.; Trevisani, L.; Arcangeli, G. Characterization and Separation of Platinum-Based Antineoplastic Drugs by Zwitterionic Hydrophilic Interaction Liquid Chromatography (HILIC)-Tandem Mass Spectrometry, and Its Application in Surface Wipe Sampling. Separations 2021, 8, 69. [Google Scholar] [CrossRef]
- Alpert, A.J. Hydrophilic-interaction chromatography for the separation of peptides, nucleic acids and other polar compounds. J. Chromatogr. A 1990, 499, 177–196. [Google Scholar] [CrossRef]
- Huber, J.F.K.; Pawlowska, M.; Markl, P. Selection of the solid support-a crucial step for the realization of solvent generated liquid-liquid chromatography. J. Chromatogr. A 1990, 500, 257–280. [Google Scholar] [CrossRef]
- Horváth, C.; Lipsky, S.R. Column design in high pressure liquid chromatography. J. Chromatogr. Sci. 1969, 7, 109–116. [Google Scholar] [CrossRef]
- Kirkland, J.J. Controlled surface porosity supports for high-speed gas and liquid chromatography. Anal. Chem. 1969, 41, 218–220. [Google Scholar] [CrossRef]
- DeStefano, J.J.; Langlois, T.J.; Kirkland, J.J. Characteristics of superficially-porous silica particles for fast HPLC: Some performance comparisons with sub-2-µm particles. J. Chromatogr. Sci. 2008, 46, 254–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gritti, F.; Guiochon, G. Comparison between the efficiencies of columns packed with fully and partially porous C18-bonded silica materials. J. Chromatogr. A 2007, 1157, 289–303. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Wei, T.-C. Superficially Porous Particles and Methods of Making and Using Same. U.S. Patent 7,846,337 B2, 7 December 2010. [Google Scholar]
- Narduzzi, L.; Royer, A.-L.; Bichon, E.; Guitton, Y.; Buisson, C.; Le Bizec, B.; Dervilly-Pinel, G. Ammonium Fluoride as Suitable Additive for HILIC-Based LC-HRMS Metabolomics. Metabolites 2019, 9, 292. [Google Scholar] [CrossRef] [Green Version]
- Dugheri, S.; Bonari, A.; Pompilio, I.; Boccalon, P.; Mucci, N.; Arcangeli, G. A new approach to assessing occupational exposure to antineoplastic drugs in hospital environments. Arh. Hig. Rada Toksikol. 2018, 69, 226–237. [Google Scholar] [CrossRef] [Green Version]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the assessment of matrix effect in quantitative bioanalytical methods based on HPLC-MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef]
- Greco, G.; Grosse, S.; Letzel, T. Study of the retention behavior in zwitterionic hydrophilic interaction chromatography of isomeric hydroxy- and aminobenzoic acids. J. Chromatogr. A 2012, 1235, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Validation of Analytical Procedures: Text and Methodology Q2(R1). Available online: https://database.ich.org/sites/default/files/Q2%28R1%29%20Guideline.pdf (accessed on 8 December 2021).
- Weininger, D.; Weininger, A.; Weininger, J.L. SMILES. 2. Algorithm for generation of unique SMILES notation. J. Chem. Inf. Comput. Sci. 1989, 29, 97–101. [Google Scholar] [CrossRef]
- Riniker, S.; Landrum, G.A. Better Informed Distance Geometry: Using What We Know To Improve Conformation Generation. J. Chem. Inf. Comp. Sci. 2015, 55, 2562–2574. [Google Scholar] [CrossRef] [PubMed]
- RDKit: Open-Source Cheminformatics. Available online: http://www.rdkit.org (accessed on 22 December 2021).
- Halgren, T.A. Merck molecular force field. I. Basis, form, scope, parameterization, and performance of MMFF94. J. Comput. Chem. 1996, 17, 490–519. [Google Scholar] [CrossRef]
- Ionescu, C.-M.; Sehnal, D.; Falginella, F.L.; Pant, P.; Pravda, L.; Bouchal, T.; Vařeková, R.S.; Geidl, S.; Koča, J. AtomicChargeCalculator: Interactive Web-based calculation of atomic charges in large biomolecular complexes and drug like molecules. J. Cheminform. 2015, 7, 50. [Google Scholar] [CrossRef] [Green Version]
- Geidl, S.; Bouchal, T.; Raček, T.; Vařeková, R.S.; Hejret, V.; Křenek, A.; Abagyan, R.; Koča, J. High-quality and universal empirical atomic charges for chemoinformatics applications. J. Cheminform. 2015, 7, 59. [Google Scholar] [CrossRef] [Green Version]
- Todeschini, R.; Vighi, M.; Finizio, A.; Gramatica, P. SAR & QSAR. Environ.Res. 1997, 7, 173–193. [Google Scholar]
- Ertl, P.; Rohde, B.; Selzer, P. Fast Calculation of Molecular Polar Surface Area as a Sum of Fragment-Based Contributions and Its Application to the Prediction of Drug Transport Properties. J. Med. Chem. 2000, 43, 3714–3717. [Google Scholar] [CrossRef]
- Stanton, D.T.; Jurs, P.C. Development and Use of Charged Partial Surface Area Structural Descriptors in Computer Assissted Quantitative Structure Property Relationship Studies. Anal. Chem. 1990, 62, 2323–2329. [Google Scholar] [CrossRef]
- Kaliszan, R.; Osmialowski, K.; Tomellini, S.A.; Hsu, S.-H.; Fazio, S.D.; Hartwick, R.A. Non-empirical descriptors of sub-molecular polarity and dispersive interactions in reversed-phase HPLC. Chromatographia 1985, 20, 705–708. [Google Scholar] [CrossRef]
- Osmialowski, K.; Halkiewicz, J.; Radecki, A.; Kaliszan, R. Quantum chemical parameters in correlation analysis of gas—Liquid chromatographic retention indices of amines. J. Chromat. 1985, 346, 53–60. [Google Scholar] [CrossRef]
- Katritzky, A.R.; Gordeeva, E.V. Traditional topological indexes vs electronic, geometrical, and combined molecular descriptors in QSAR/QSPR research. J. Chem. Inf. Comput. Sci. 1993, 33, 835–857. [Google Scholar] [CrossRef] [PubMed]
- Osmialowski, K.; Halkiewicz, J.; Kaliszan, R. Quantum chemical parameters in correlation analysis of gas-liquid chromatographic retention indices of amines II. Topological electronic index. J. Chromat. 1986, 361, 63–69. [Google Scholar] [CrossRef]
- Clare, B.W.; Supuran, C.T. Carbonic anhydrase activators. 3: Structure-activity correlations for a series of isozyme II activators. J. Pharm. Sci. 1994, 83, 768–773. [Google Scholar] [CrossRef] [PubMed]
- Karelson, M.; Lobanov, V.S.; Katritzky, A.R. Quantum-Chemical Descriptors in QSAR/QSPR Studies. Chem. Rev. 1996, 96, 1027–1043. [Google Scholar] [CrossRef] [PubMed]
- Kode srl. Dragon Version 7.0.6. 2016. Available online: https://chm.kode-solutions.net (accessed on 22 December 2021).
- Bro, R.; Smilde, A.K. Principal Component Analysis. Anal. Methods 2014, 6, 2812–2831. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Canela, C.; Campos, B.; Barata, C.; Lacorte, S. Degradation and toxicity of mitoxantrone and chlorambucil in water. Int. J. Environ. Sci. Technol. 2015, 12, 633–640. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
| Compound | Precursor Ion Species | Precursor Ion (m/z) | Quantifier Ion (m/z) [CE (V)] | Qualifier Ion (m/z) [CE (V)] |
|---|---|---|---|---|
| ISTD | [M − H]- | 144.9 | 42.0 [17.0] | 65.8 [28.5] |
| 5-Azacytidine | [M + H]+ | 245.2 | 113.0 [−10.0] | 86.0 [−30.0] |
| Dacarbazine | [M + H]+ | 183.0 | 166.0 [−13.0] | 123.0 [−19.0] |
| Gemcitabine | [M + H]+ | 264.2 | 111.9 [−17.0] | 95.1 [−42.0] |
| Mitoxantrone | [M + H]+ | 445.1 | 88.0 [−26.0] | 358.1 [−20.0] |
| Cytarabine | [M + H]+ | 244.2 | 112.2 [−15.0] | 94.1 [−43.0] |
| 5-Fluorouracil | [M − H]- | 129.0 | 42.0 [19.0] | 59.2 [23.0] |
| Oxaliplatin | [M + H]+ | 398.0 | 306.0 [−28.0] | 96.0 [−25.0] |
| Cisplatin | [M + NH4]+ | 317.9 | 264.9 [−15.0] | 300.7 [−15.0] |
| Carboplatin | [M + H]+ | 372.1 | 294.0 [−18.0] | 248.0 [−35.0] |
| Interday | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Compound | RT (min) | RT RSD | Width1/2 (min) | Width RSD | Tf | Tf RSD | Af | Af RSD | N (Plates) | k |
| 5-Azacytidine | 2.726 | 0.3% | 0.326 | 1.8% | 1.0 | 0.8% | 1.08 | 1% | 387 | 4.4 |
| Dacarbazine | 0.842 | 0.2% | 0.293 | 0.5% | 0.9 | 1.0% | 0.90 | 2% | 46 | 0.7 |
| Gemcitabine | 1.089 | 0.2% | 0.298 | 0.3% | 1.0 | 1.0% | 1.06 | 1% | 74 | 1.1 |
| Mitoxantrone | 5.621 | 1.8% | 0.529 | 24.3% | 1.8 | 10.7% | 3.20 | 10% | 634 | 10.1 |
| Cisplatin | 1.386 | 0.2% | 0.292 | 0.4% | 1.0 | 0.1% | 1.03 | 0% | 125 | 1.7 |
| Cytarabine | 3.754 | 0.2% | 0.319 | 1.5% | 1.0 | 0.5% | 1.05 | 1% | 769 | 6.4 |
| Carboplatin | 4.918 | 0.2% | 0.329 | 1.3% | 1.0 | 0.3% | 1.05 | 1% | 1237 | 8.7 |
| Oxaliplatin | 2.110 | 0.2% | 0.314 | 0.8% | 1.0 | 0.2% | 1.04 | 0% | 251 | 3.1 |
| IS | 0.594 | 1.4% | 0.365 | 1.6% | 1.1 | 1.5% | 1.22 | 3% | 15 | 0.2 |
| 5-Fluorouracil | 0.732 | 0.6% | 0.369 | 2.1% | 0.9 | 1.9% | 0.85 | 4% | 22 | 0.4 |
| Intraday | ||||||||||
| 5-Azacytidine | 2.630 | 0.5% | 0.318 | 1.6% | 1.0 | 1.3% | 1.06 | 2% | 378 | 4.2 |
| Dacarbazine | 0.834 | 0.2% | 0.293 | 0.5% | 0.9 | 1.2% | 0.91 | 2% | 45 | 0.6 |
| Gemcitabine | 1.077 | 0.3% | 0.298 | 0.6% | 1.0 | 0.3% | 1.04 | 0% | 72 | 1.1 |
| Mitoxantrone | 5.757 | 2.9% | 0.484 | 24.0% | 2.0 | 61.3% | 2.88 | 44% | 785 | 10.3 |
| Cisplatin | 1.372 | 0.3% | 0.294 | 0.6% | 1.0 | 0.6% | 1.03 | 1% | 120 | 1.7 |
| Cytarabine | 3.612 | 0.4% | 0.313 | 1.5% | 1.0 | 0.8% | 1.03 | 1% | 738 | 6.1 |
| Carboplatin | 4.764 | 0.3% | 0.324 | 1.3% | 1.0 | 2.8% | 1.04 | 3% | 1194 | 8.4 |
| Oxaliplatin | 2.071 | 0.2% | 0.315 | 1.0% | 1.0 | 0.6% | 1.03 | 1% | 240 | 3.1 |
| IS | 0.602 | 2.0% | 0.369 | 1.5% | 1.1 | 2.1% | 1.18 | 4% | 15 | 0.2 |
| 5-Fluorouracil | 0.730 | 0.7% | 0.365 | 2.5% | 0.9 | 0.8% | 0.84 | 1% | 22 | 0.4 |
| Interday | ||||
|---|---|---|---|---|
| Compound | Slope (PAR/ng/mL) | Intercept (PAR) | R2 | r |
| 5-Azacytidine | 0.037 | 0.012 | 0.9970 | 0.9985 |
| Dacarbazine | 9.176 | 12.894 | 0.9960 | 0.9980 |
| Gemcitabine | 1.161 | 0.065 | 0.9977 | 0.9988 |
| Mitoxantrone | - | - | - | - |
| Cisplatin | 0.004 | 0.149 | 0.9981 | 0.9990 |
| Cytarabine | 4.559 | −11.284 | 0.9884 | 0.9942 |
| Carboplatin | 0.207 | 0.094 | 0.9979 | 0.9989 |
| Oxaliplatin | 0.059 | −0.012 | 0.9985 | 0.9992 |
| 5-Fluorouracil | 0.102 | 0.032 | 0.9980 | 0.9990 |
| Intraday | ||||
| 5-Azacytidine | 0.037 | −0.005 | 0.9996 | 0.9998 |
| Dacarbazine | 8.963 | 12.529 | 0.9950 | 0.9975 |
| Gemcitabine | 1.079 | −0.077 | 0.9995 | 0.9997 |
| Mitoxantrone | - | - | - | - |
| Cisplatin | 0.005 | 0.131 | 0.9985 | 0.9992 |
| Cytarabine | 5.942 | −15.064 | 0.9901 | 0.9950 |
| Carboplatin | 0.048 | 0.022 | 0.9996 | 0.9998 |
| Oxaliplatin | 0.063 | −0.007 | 0.9996 | 0.9998 |
| 5-Fluorouracil | 0.109 | 0.029 | 0.9996 | 0.9998 |
| Compound | LOD (ng/mL) | LOQ (ng/mL) |
|---|---|---|
| 5-Azacytidine | 0.23 | 0.70 |
| Dacarbazine | 0.03 | 0.10 |
| Gemcitabine | 0.09 | 0.28 |
| Mitoxantrone | - | - |
| Cisplatin | 9.86 | 29.58 |
| Cytarabine | 0.02 | 0.06 |
| Carboplatin | 0.91 | 2.73 |
| Oxaliplatin | 0.12 | 0.35 |
| 5-Fluorouracil | 0.05 | 0.15 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dugheri, S.; Mucci, N.; Squillaci, D.; Bucaletti, E.; Cappelli, G.; Trevisani, L.; Valsecchi, C.; Consonni, V.; Gosetti, F.; Ballabio, D.; et al. Expanding Antineoplastic Drugs Surface Monitoring Profiles: Enhancing of Zwitterionic Hydrophilic Interaction Methods. Separations 2022, 9, 34. https://doi.org/10.3390/separations9020034
Dugheri S, Mucci N, Squillaci D, Bucaletti E, Cappelli G, Trevisani L, Valsecchi C, Consonni V, Gosetti F, Ballabio D, et al. Expanding Antineoplastic Drugs Surface Monitoring Profiles: Enhancing of Zwitterionic Hydrophilic Interaction Methods. Separations. 2022; 9(2):34. https://doi.org/10.3390/separations9020034
Chicago/Turabian StyleDugheri, Stefano, Nicola Mucci, Donato Squillaci, Elisabetta Bucaletti, Giovanni Cappelli, Lucia Trevisani, Cecile Valsecchi, Viviana Consonni, Fabio Gosetti, Davide Ballabio, and et al. 2022. "Expanding Antineoplastic Drugs Surface Monitoring Profiles: Enhancing of Zwitterionic Hydrophilic Interaction Methods" Separations 9, no. 2: 34. https://doi.org/10.3390/separations9020034
APA StyleDugheri, S., Mucci, N., Squillaci, D., Bucaletti, E., Cappelli, G., Trevisani, L., Valsecchi, C., Consonni, V., Gosetti, F., Ballabio, D., & Arcangeli, G. (2022). Expanding Antineoplastic Drugs Surface Monitoring Profiles: Enhancing of Zwitterionic Hydrophilic Interaction Methods. Separations, 9(2), 34. https://doi.org/10.3390/separations9020034