Highly Informative Fingerprinting of Extra-Virgin Olive Oil Volatiles: The Role of High Concentration-Capacity Sampling in Combination with Comprehensive Two-Dimensional Gas Chromatography





Abstract
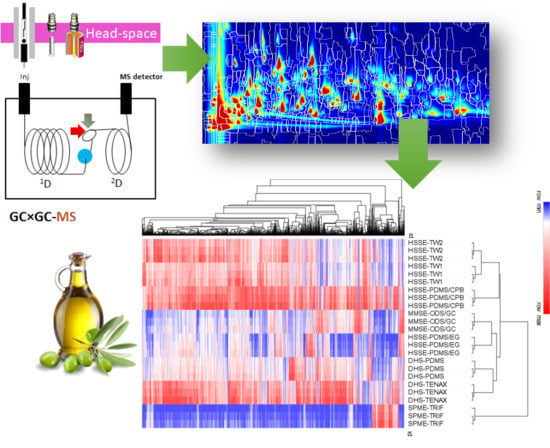
1. Introduction
2. Materials and Methods
2.1. Reference Compounds and Samples
2.2. Sample Preparation
2.2.1. Automated Headspace Solid Phase Microextraction
2.2.2. Headspace Sorptive Extraction
2.2.3. Monolithic Material Sorptive Extraction
2.2.4. Dynamic Headspace Sampling
2.3. GC × GC-MS Instrument Set-Up and Analytical Conditions
2.4. Analyte Identification
2.5. Method-Performance Parameters
2.6. Raw Data Acquisition and GC × GC Data Handling
3. Results and Discussion
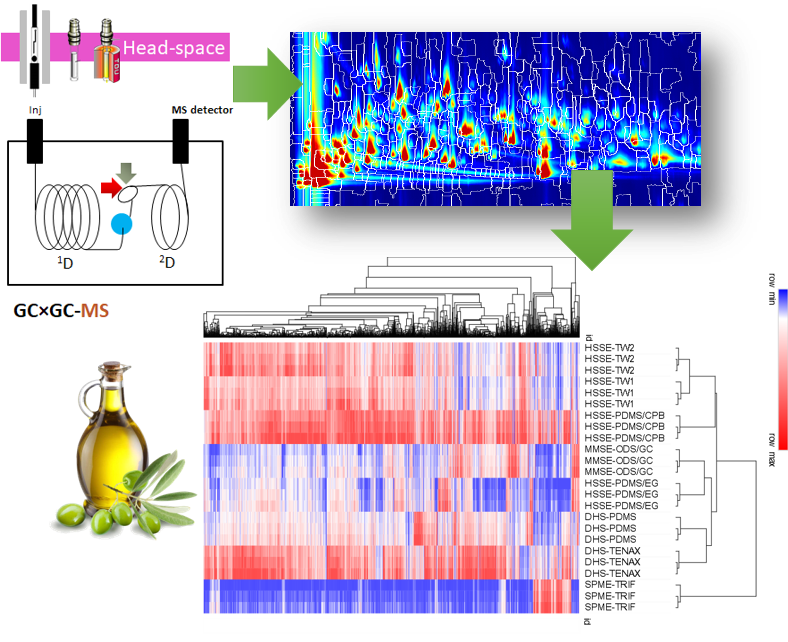
3.1. Extra-Virgin Olive Oil Complex Volatilome by GC × GC Fingerprinting
3.2. Sampling Information Potentials Based on Untargeted Data
3.3. Focus on Informative Targeted Analyte Signatures
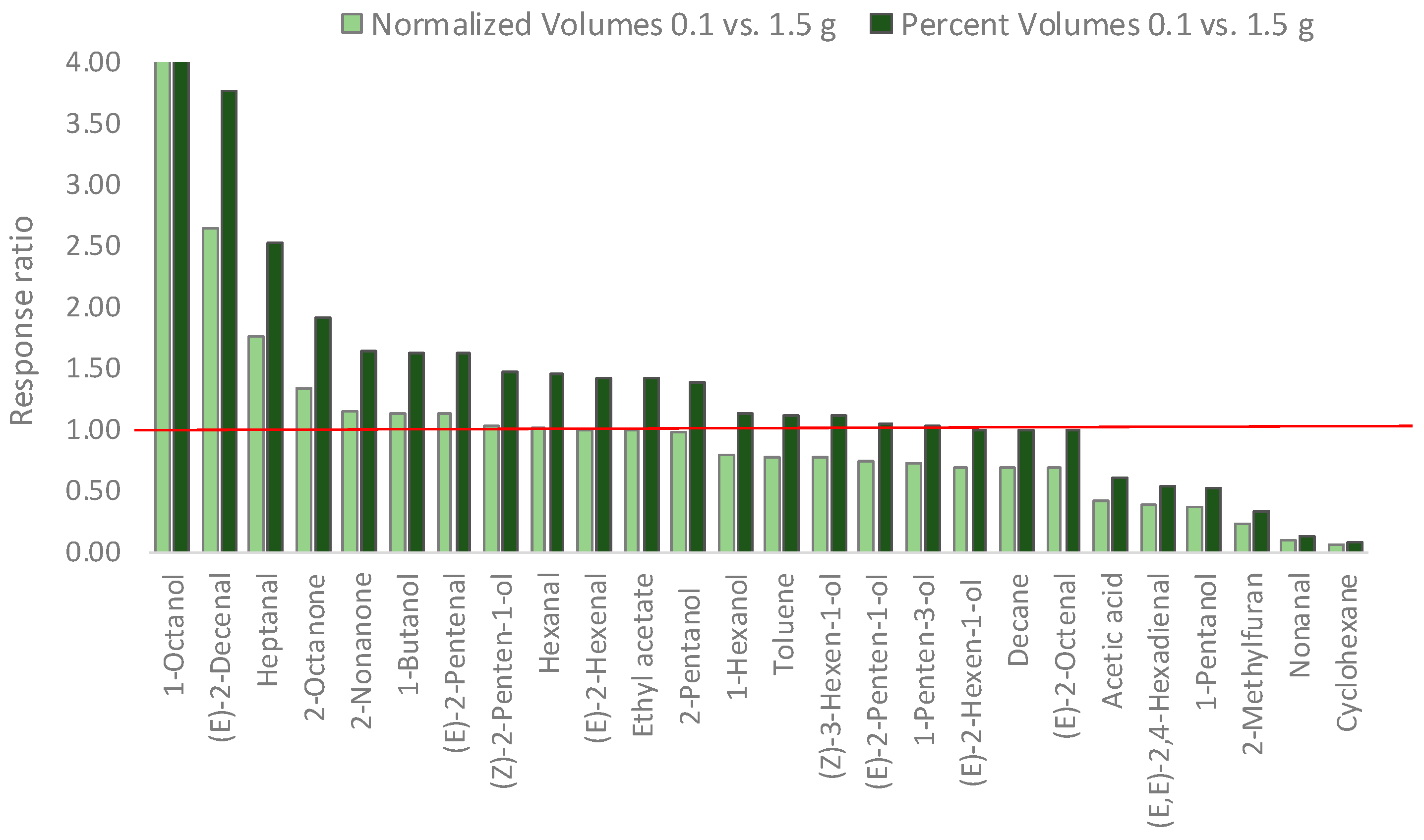
3.4. Headspace Linearity and Its Impact on Analyte Relative Distribution
- Step 1.
- Exhaustive analyte extraction from calibration solutions in a range that matches real-sample concentrations.
- Step 2.
- Exhaustive analyte extraction from selected samples that show comparable matrix effects in order to define HS linearity boundaries.
- Step 3.
- Use of MHE on samples of interest.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Cordero, C.; Kiefl, J.; Reichenbach, S.E.; Bicchi, C. Characterization of odorant patterns by comprehensive two-dimensional gas chromatography: A challenge in omic studies. Trends Anal. Chem. 2018, 364–378. [Google Scholar] [CrossRef]
- Purcaro, G.; Cordero, C.; Liberto, E.; Bicchi, C.; Conte, L.S. Toward a definition of blueprint of virgin olive oil by comprehensive two-dimensional gas chromatography. J. Chromatogr. A 2014, 1334, 101–111. [Google Scholar] [CrossRef]
- Romero, I.; García-González, D.L.; Aparicio-Ruiz, R.; Morales, M.T. Validation of SPME-GCMS method for the analysis of virgin olive oil volatiles responsible for sensory defects. Talanta 2015, 134, 394–401. [Google Scholar] [CrossRef]
- Aparicio, R.; Morales, M.T.; Aparicio-Ruiz, R.; Tena, N.; García-González, D.L. Authenticity of olive oil: Mapping and comparing official methods and promising alternatives. Food Res. Int. 2013, 54, 2025–2038. [Google Scholar] [CrossRef]
- Stilo, F.; Liberto, E.; Reichenbach, S.E.; Tao, Q.; Bicchi, C.; Cordero, C. Untargeted and Targeted Fingerprinting of Extra Virgin Olive Oil Volatiles by Comprehensive Two-Dimensional Gas Chromatography with Mass Spectrometry: Challenges in Long-Term Studies. J. Agric. Food Chem. 2019, 67, 5289–5302. [Google Scholar] [CrossRef]
- Commission of the European Communities. Commission Regulation (Eec) No 2568/91. Off. J. Eur. Comm. 1991, 5, 1–83. [Google Scholar]
- International Oil Council. COI/T.20/DOC.15/Rev.10 Sensory Analysis of Olive Oil—Method for the Organoleptic Assessment of Virgin Olive Oil, COI/T.20/DOC.15/Rev.10. 2018. Available online: www.internationaloliveoil.org/documents/viewfile/3685-orga6 (accessed on 12 July 2019).
- Stilo, F.; Liberto, E.; Bicchi, C.; Reichenbach, S.E.; Cordero, C. GC×GC–TOF-MS and Comprehensive Fingerprinting of Volatiles in Food: Capturing the Signature of Quality. LC-GC Eur. 2019, 32, 234–242. [Google Scholar]
- Vaz-Freire, L.T.; Da Silva, M.D.R.G.; Freitas, A.M.C. Comprehensive two-dimensional gas chromatography for fingerprint pattern recognition in olive oils produced by two different techniques in Portuguese olive varieties Galega Vulgar, Cobrançosa e Carrasquenha. Anal. Chim. Acta. 2009, 633, 263–270. [Google Scholar] [CrossRef]
- Cajka, T.; Riddellova, K.; Klimankova, E.; Cerna, M.; Pudil, F.; Hajslova, J. Traceability of olive oil based on volatiles pattern and multivariate analysis. Food Chem. 2010, 121, 282–289. [Google Scholar] [CrossRef]
- Lukić, I.; Carlin, S.; Horvat, I.; Vrhovsek, U. Combined targeted and untargeted profiling of volatile aroma compounds with comprehensive two-dimensional gas chromatography for differentiation of virgin olive oils according to variety and geographical origin. Food Chem. 2019, 270, 403–414. [Google Scholar] [CrossRef]
- Magagna, F.; Valverde-Som, L.; Ruíz-Samblás, C.; Cuadros-Rodríguez, L.; Reichenbach, S.E.; Bicchi, C.; Cordero, C. Combined untargeted and targeted fingerprinting with comprehensive two-dimensional chromatography for volatiles and ripening indicators in olive oil. Anal. Chim. Acta 2016, 936, 245–258. [Google Scholar] [CrossRef]
- Magagna, F.; Guglielmetti, A.; Liberto, E.; Reichenbach, S.E.; Allegrucci, E.; Gobino, G.; Bicchi, C.; Cordero, C. Comprehensive Chemical Fingerprinting of High-Quality Cocoa at Early Stages of Processing: Effectiveness of Combined Untargeted and Targeted Approaches for Classification and Discrimination. J. Agric. Food Chem. 2017, 65, 6329–6341. [Google Scholar] [CrossRef]
- Magagna, F.; Liberto, E.; Reichenbach, S.E.; Tao, Q.; Carretta, A.; Cobelli, L.; Cordero, C. Advanced fingerprinting of high-quality cocoa: Challenges in transferring methods from thermal to differential-flow modulated comprehensive two dimensional gas chromatography. J. Chromatogr. A 2018, 1536, 122–136. [Google Scholar] [CrossRef]
- Reichenbach, S.E.; Zini, C.A.; Nicolli, K.P.; Welke, J.E.; Cordero, C.; Tao, Q. Benchmarking Machine Learning Methods for Comprehensive Chemical Fingerprinting and Pattern Recognition. J. Chromatogr. A 2019, 1595, 158–167. [Google Scholar] [CrossRef]
- Bressanello, D.; Liberto, E.; Collino, M.; Chiazza, F.; Mastrocola, R.; Reichenbach, S.E.; Bicchi, C.; Cordero, C. Combined untargeted and targeted fingerprinting by comprehensive two-dimensional gas chromatography: Revealing fructose-induced changes in mice urinary metabolic signatures. Anal. Bioanal. Chem. 2018, 410, 2723–2737. [Google Scholar] [CrossRef]
- Bicchi, C.; Cordero, C.; Rubiolo, P. A survey on high-concentration-capability headspace sampling techniques in the analysis of flavors and fragrances. J. Chromatogr. Sci. 2004, 42, 402–409. [Google Scholar] [CrossRef]
- Bicchi, C.; Cordero, C.; Liberto, E.; Rubiolo, P.; Sgorbini, B. Automated headspace solid-phase dynamic extraction to analyse the volatile fraction of food matrices. J. Chromatogr. A 2004, 1024, 217–226. [Google Scholar] [CrossRef]
- Risticevic, S.; Vuckovic, D.; Lord, H.L.; Pawliszyn, J. 2.21—Solid-Phase Microextraction. In Comprehensive Sampling and Sample Preparation; Academic Press: Cambridge, MA, USA, 2012; pp. 419–460. [Google Scholar] [CrossRef]
- Lord, H.L.; Pfannkoch, E.A. Sample Preparation Automation for GC Injection; Elsevier: Amsterdam, Netherland, 2012. [Google Scholar] [CrossRef]
- Ross, C.F. 2.02—Headspace Analysis; Elsevier: Amsterdam, Netherland, 2012. [Google Scholar] [CrossRef]
- Bicchi, C.; Cordero, C.; Liberto, E.; Sgorbini, B.; Rubiolo, P. Headspace sampling of the volatile fraction of vegetable matrices. J. Chromatogr. A 2008, 1184, 220–233. [Google Scholar] [CrossRef]
- Cordero, C.; Schmarr, H.G.; Reichenbach, S.E.; Bicchi, C. Current Developments in Analyzing Food Volatiles by Multidimensional Gas Chromatographic Techniques. J. Agric. Food Chem. 2018, 66, 2226–2236. [Google Scholar] [CrossRef]
- Cordero, C.; Kiefl, J.; Schieberle, P.; Reichenbach, S.E.; Bicchi, C. Comprehensive two-dimensional gas chromatography and food sensory properties: Potential and challenges. Anal. Bioanal. Chem. 2015, 407, 169–191. [Google Scholar] [CrossRef]
- Oliver-Pozo, C.; Trypidis, D.; Aparicio, R.; Garciá-González, D.L.; Aparicio-Ruiz, R. Implementing Dynamic Headspace with SPME Sampling of Virgin Olive Oil Volatiles: Optimization, Quality Analytical Study, and Performance Testing. J. Agric. Food Chem. 2019, 67, 2086–2097. [Google Scholar] [CrossRef]
- Chin, S.T.; Eyres, G.T.; Marriott, P.J. Cumulative solid phase microextraction sampling for gas chromatography-olfactometry of Shiraz wine. J. Chromatogr. A 2012, 1255, 221–227. [Google Scholar] [CrossRef]
- Bicchi, C.; Cordero, C.; Liberto, E.; Sgorbini, B.; David, F.; Sandra, P.; Rubiolo, P. Influence of polydimethylsiloxane outer coating and packing material on analyte recovery in dual-phase headspace sorptive extraction. J. Chromatogr. A 2007, 1164, 33–39. [Google Scholar] [CrossRef]
- Bicchi, C.; Cordero, C.; Liberto, E.; Rubiolo, P.; Sgorbini, B.; David, F.; Sandra, P. Dual-phase twisters: A new approach to headspace sorptive extraction and stir bar sorptive extraction. J. Chromatogr. A 2005, 1094, 9–16. [Google Scholar] [CrossRef]
- Giddings, J.C. Sample dimensionality: A predictor of order-disorder in component peak distribution in multidimensional separation. J. Chromatogr. A 1995, 703, 3–15. [Google Scholar] [CrossRef]
- NIST/EPA/NIH Mass Spectral Library with Search Program Data Version: NIST v17, (n.d.); U.S. Department of Commerce: New York, NY, USA, 2008.
- Adams, R.P. Identification of Essential Oil Components by Gas Chromatography—Mass Spectroscopy; Allured Publishing: New York, NY, USA, 1995. [Google Scholar]
- Feussner, I.; Wasternack, C. The Lipoxygenase Pathway. Annu. Rev. Plant Biol. 2002, 53, 275–297. [Google Scholar] [CrossRef]
- Berlitz, H.D.; Grosch, W.; Schieberle, P. Food Chemistry; Springer: New York, NY, USA, 2009. [Google Scholar]
- Angerosa, F.; Camera, L.; D’Alessandro, N.; Mellerio, G. Characterization of Seven New Hydrocarbon Compounds Present in the Aroma of Virgin Olive Oils. J. Agric. Food Chem. 1998, 46, 648–653. [Google Scholar] [CrossRef]
- Reichenbach, S.E.; Tian, X.; Cordero, C.; Tao, Q. Features for non-targeted cross-sample analysis with comprehensive two-dimensional chromatography. J. Chromatogr. A 2012, 1226, 140–148. [Google Scholar] [CrossRef]
- Reichenbach, S.E.; Carr, P.W.; Stoll, D.R.; Tao, Q. Smart Templates for peak pattern matching with comprehensive two-dimensional liquid chromatography. J. Chromatogr. A 2009, 1216, 3458–3466. [Google Scholar] [CrossRef]
- GC ImageTM. GC Image GCxGC Edition Users’ Guide. 2017. Available online: https://www.gcimage.com/gcxgc/usersguide/index.html (accessed on 12 July 2019).
- Cordero, C.; Liberto, E.; Bicchi, C.; Rubiolo, P.; Reichenbach, S.E.; Tian, X.; Tao, Q. Targeted and non-targeted approaches for complex natural sample profiling by GCxGC-qMS. J. Chromatogr. Sci. 2010, 48, 251–261. [Google Scholar] [CrossRef]
- Reichenbach, S.E.; Tian, X.; Tao, Q.; Stoll, D.R.; Carr, P.W. Comprehensive feature analysis for sample classification with comprehensive two-dimensional LC. J. Sep. Sci. 2010, 33, 1365–1374. [Google Scholar] [CrossRef]
- Reichenbach, S.E.; Tian, X.; Boateng, A.A.; Mullen, C.A.; Cordero, C.; Tao, Q. Reliable peak selection for multisample analysis with comprehensive two-dimensional chromatography. Anal. Chem. 2013, 85, 4974–4981. [Google Scholar] [CrossRef]
- Rempe, D.W.; Reichenbach, S.E.; Tao, Q.; Cordero, C.; Rathbun, W.E.; Zini, C.A. Effectiveness of Global, Low-Degree Polynomial Transformations for GCxGC Data Alignment. Anal. Chem. 2016, 88, 10028–10035. [Google Scholar] [CrossRef]
- Sgorbini, B.; Cagliero, C.; Liberto, E.; Rubiolo, P.; Bicchi, C.; Cordero, C.E.I. Strategies for Accurate Quantitation of Volatiles from Foods and Plant-Origin Materials: A Challenging Task. J. Agric. Food Chem. 2019, 67, 1619–1630. [Google Scholar] [CrossRef]
- Cordero, C.; Guglielmetti, A.; Sgorbini, B.; Bicchi, C.; Allegrucci, E.; Gobino, G.; Baroux, L.; Merle, P. Odorants quantitation in high-quality cocoa by multiple headspace solid phase micro-extraction: Adoption of FID-predicted response factors to extend method capabilities and information potential. Anal. Chim. Acta. 2019, 1052, 190–201. [Google Scholar] [CrossRef]
- Kolb, B.; Ettre, L.S. Static Headspace-Gas Chromatography: Theory and Practice, Wiley-VCH, New York. Available online: https://books.google.com/books?hl=en&lr=&id=nGPmpb4VvEgC&oi=fnd&pg=PR5&ots=6SZHNIygx6&sig=lcvdWEsXW-3H8BzCWuKuK2PnjiI#v=onepage&q&f=false (accessed on 12 July 2019).
- Cordero, C.; Liberto, E.; Bicchi, C.; Rubiolo, P.; Schieberle, P.; Reichenbach, S.E.; Tao, Q. Profiling food volatiles by comprehensive two-dimensional gas chromatography coupled with mass spectrometry: Advanced fingerprinting approaches for comparative analysis of the volatile fraction of roasted hazelnuts (Corylus avellana L.) from different ori. J. Chromatogr. A 2010, 1217, 5848–5858. [Google Scholar] [CrossRef]
- Brevard, H.; Cantergiani, E.; Cachet, T.; Chaintreau, A.; Demyttenaere, J.; French, L.; Gassenmeier, K.; Joulain, D.; Koenig, T.; Leijs, H.; et al. Guidelines for the quantitative gas chromatography of volatile flavouring substances, from the Working Group on Methods of Analysis of the International Organization of the Flavor Industry. Flavour Fragr. J. 2011, 26, 297–299. [Google Scholar] [CrossRef]
- Vichi, S.; Pizzale, L.; Conte, L.S.; Buxaderas, S.; Lopez-Tamames, E. Solid phase microextraction in the analysis of virgin olive oil volatile fraction: Characterization of virgin oils from two distinct geographical areas of northern Italy. J. Agric. Food Chem. 2003, 51, 6564–6571. [Google Scholar] [CrossRef]
- Cavalli, J.F.; Fernandez, X.; Lizzani-Cuvelier, L.; Loiseau, A.M. Comparison of Static Headspace, Headspace Solid Phase Microextraction, Headspace Sorptive Extraction, and Direct Thermal Desorption Techniques on Chemical Composition of French Olive Oils. J. Agric. Food Chem. 2003, 51, 7709–7716. [Google Scholar] [CrossRef]
- Morales, M.T.; Luna, G.; Aparicio, R. Comparative study of virgin olive oil sensory defects. Food Chem. 2005, 91, 293–301. [Google Scholar] [CrossRef]
- Nigri, S.; Oumeddour, R.; Fernandez, X. Analysis of some Algerian virgin olive oils by headspace solid phase micro-extraction coupled to gas chromatography/mass spectrometry. Riv. Ital. Delle Sostanze Grasse 2012, 89, 54–61. [Google Scholar] [CrossRef]
- Costa, R.; Albergamo, A.; Bua, G.D.; Saija, E.; Dugo, G. Determination of flavor constituents in particular types of flour and derived pasta by heart-cutting multidimensional gas chromatography coupled with mass spectrometry and multiple headspace solid-phase microextraction. LWT 2017, 86, 99–107. [Google Scholar] [CrossRef]
- Cagliero, C.; Bicchi, C.; Cordero, C.; Rubiolo, P.; Sgorbini, B.; Liberto, E. Fast headspace-enantioselective GC-mass spectrometric-multivariate statistical method for routine authentication of flavoured fruit foods. Food Chem. 2012, 132, 1071–1079. [Google Scholar] [CrossRef]
- Nicolotti, L.; Cordero, C.; Cagliero, C.; Liberto, E.; Sgorbini, B.; Rubiolo, P.; Bicchi, C. Quantitative fingerprinting by headspace-Two-dimensional comprehensive gas chromatography-mass spectrometry of solid matrices: Some challenging aspects of the exhaustive assessment of food volatiles. Anal. Chim. Acta. 2013, 798, 115–125. [Google Scholar] [CrossRef]
- Sgorbini, B.; Bicchi, C.; Cagliero, C.; Cordero, C.; Liberto, E.; Rubiolo, P. Herbs and spices: Characterization and quantitation of biologically-active markers for routine quality control by multiple headspace solid-phase microextraction combined with separative or non-separative analysi. J. Chromatogr. A 2015, 1376, 9–17. [Google Scholar] [CrossRef]
- Griglione, A.; Liberto, E.; Cordero, C.; Bressanello, D.; Cagliero, C.; Rubiolo, P.; Bicchi, C.; Sgorbini, B. High-quality Italian rice cultivars: Chemical indices of ageing and aroma quality. Food Chem. 2015, 172, 305–313. [Google Scholar] [CrossRef]
- Kremser, A.; Jochmann, M.A.; Schmidt, T.C. PAL SPME Arrow—Evaluation of a novel solid-phase microextraction device for freely dissolved PAHs in water. Anal. Bioanal. Chem. 2016, 408, 943–952. [Google Scholar] [CrossRef]
- Helin, A.; Rönkkö, T.; Parshintsev, J.; Hartonen, K.; Schilling, B.; Läubli, T.; Riekkola, M.L. Solid phase microextraction Arrow for the sampling of volatile amines in wastewater and atmosphere. J. Chromatogr. A 2015, 1426, 56–63. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Acronym | Sampling Approach | Sample Weight/Volume | Temperature and Time | Other |
|---|---|---|---|---|
| STME-TRIF | HS-SPME—DVB/CAR/PDMS | 1.500 g oil Sampling vial: 20 mL | Temperature: 40 °C Sampling time: 60 min | Constant stirring Desorption time: 5 (min) S/SL injector: 250 °C Split ratio 1:10 |
| HSSE-TW1 | HSSE—Twister™ PDMS 1 cm | 1.500 g oil Sampling vial: 20 mL | Temperature: 40 °C Sampling time: 60 min | TDU conditions: from 30 °C to 27.0°C (5 min) at 60 °C/min; Flow mode: Splitless Transfer line: 270 °C. CIS-4 PTV injector temp: −50 °C Coolant: Liquid CO2; Injection temp program: From −50 °C to 270 °C (10 min) at 12 °C/s. Inlet operated in split mode: Split ratio 1:10. |
| HSSE-TW2 | HSSE—Twister™ PDMS 2 cm | |||
| HSSE-PDMS/CPB | HSSE—Twister™ PDMS—Carbopack B™ | |||
| HSSE-PDMS/EG | HSSE—Twister™ PDMS—Ethylene glycol EG | |||
| MMSE-ODS | MMSE ODS | |||
| MMSE-ODS/GC | MMSE ODS—Graphite carbon | |||
| DHS-TENAX | D-HS TENAX TA™ | 1.500 g oil Sampling vial: 20 mL | Incubation: 40 °C Sampling: room temperature Carrier: nitrogen Sampling flow: 10 mL/min Sampling time: 20 min | |
| DHS-PDMS | D-HS PDMS (foam) |
| Compound | 1tR (min) | 2tR (s) | IT | Attributes | Odor Quality | OT (mg/kg) | Ref |
|---|---|---|---|---|---|---|---|
| Heptane | 4.34 | 1.09 | 750 | Alkane | |||
| Octane | 5.59 | 1.89 | 800 | F/V/R | Alkane | 0.94 | [2] |
| 1-Octene | 6.09 | 1.68 | 820 | M | - | 0.08 | |
| Ethyl acetate | 6.75 | 1.35 | 850 | F/V | Pineapple | 0.94 | [2] |
| Butanal | 7.00 | 1.04 | 857 | F/M | Pungent, green | 0.018 | |
| Ethanol | 7.67 | 1.14 | 883 | V | Alcohol | 30 | [2] |
| Pentanal | 7.75 | 1.35 | 892 | - | |||
| Nonane | 7.82 | 2.34 | 895 | Alkane | |||
| 3,4-Diethyl-1,5-hexadiene (RS+SR) | 8.50 | 2.36 | 917 | - | |||
| 3,4-Diethyl-1,5-hexadiene (meso) | 8.66 | 2.40 | 923 | - | |||
| 3-Methylbutanal | 8.75 | 2.61 | 927 | F/V | Malty | 0.0054 | [3] |
| 3-Pentanone | 8.84 | 1.47 | 930 | V | Ether | 70 | [2] |
| (Z)-3-Ethyl-1,5-octadiene | 9.92 | 2.61 | 973 | - | |||
| 1-Penten-3-one | 10.17 | 1.47 | 983 | M | Mustard | 0.00073 | [3] |
| (E)-3-Ethyl-1,5-octadiene | 10.42 | 2.61 | 993 | - | |||
| Ethyl butanoate | 10.59 | 1.77 | 1000 | F | Sweet, fruity | 0.03 | [2] |
| (E)-2-Butenal | 10.75 | 1.38 | 1010 | Green, fruit | |||
| Butyl acetate | 12.00 | 1.73 | 1046 | F | Green, fruity, pungent, sweet | 0.3 | [2] |
| Hexanal | 12.25 | 1.77 | 1054 | F/Mo/V/R | Green apple, grassy | 0.08 | [2] |
| (E,Z)-3,7-Decadiene | 12.25 | 2.74 | 1054 | - | |||
| (E,E)-3,7-Decadiene | 12.58 | 2.74 | 1065 | - | |||
| (Z)-Pent-2-enal | 14.00 | 1.51 | 1108 | Mo | Strawberry, fruit, tomato, green, pleasant | ||
| (E)-Pent-2-enal | 14.08 | 1.52 | 1110 | V | Green, apple, tomato, pungent | 0.3 | [2] |
| Ethyl benzene | 14.25 | 1.78 | 1115 | Fr | Strong | ||
| 1-Penten-3-ol | 14.70 | 0.20 | 1129 | ||||
| 1-Butanol | 15.42 | 1.26 | 1142 | V/M | Winey | 0.15 | [3] |
| 2-Heptanone | 16.42 | 1.89 | 1161 | V | Sweet, fruity | 0.3 | |
| Heptanal | 16.50 | 1.89 | 1169 | R | Oily, fatty, woody | 0.5 | |
| Limonene | 16.91 | 2.15 | 1181 | Citrus, mint | |||
| 1-Pentanol | 17.17 | 1.45 | 1190 | F/M/V | Fruity | 3 | [2] |
| (Z)-2-Hexenal | 17.54 | 1.61 | 1198 | Fr | Green leaves, cut grass | 0.003 | [4] |
| (E)-2-Hexenal | 18.00 | 1.64 | 1208 | Mo/V/F/R | Bitter almond, green | 0.42 | [3] |
| 3-Methylbutan-1-ol | 18.35 | 0.94 | 1215 | F/M/Mo | Whiskey, malt, burnt | 0.1 | [2] |
| Ethyl hexanoate | 18.36 | 1.94 | 1216 | F | Apple peel, fruit | ||
| 1-Hexanol | 19.00 | 1.71 | 1231 | Fr | Fruity, banana, soft | 0.4 | [2] |
| Styrene | 19.25 | 1.35 | 1237 | Balsamic, gasoline | |||
| Hexyl acetate | 20.33 | 2.02 | 1263 | Fr | Green, fruity, sweet | 1.04 | [4] |
| 2-Octanone | 20.83 | 2.06 | 1274 | V | Mold, green | 0.51 | |
| Octanal | 21.08 | 2.02 | 1280 | Mo/R | Fatty, sharp | 0.32 | [2] |
| 1-Octen-3-one | 21.58 | 1.89 | 1292 | Mo | Mushroom, mold | 0.01 | |
| (Z)-2-Penten-1-ol | 21.75 | 1.22 | 1296 | Butter, pungent | |||
| (E)-4,8-Dimethyl-1,3,7-nonatriene | 21.92 | 2.36 | 1300 | - | |||
| (Z)-3-Hexen-1-ol acetate | 22.08 | 1.68 | 1304 | Green, banana | |||
| (E)-2-Penten-1-ol | 22.09 | 1.02 | 1304 | Butter, pungent | 1.04 | [4] | |
| 1-Heptanol | 22.31 | 1.89 | 1309 | Herb | |||
| (Z)-2-Heptenal | 22.58 | 1.77 | 1315 | R | Oxidized, tallowy | 0.042 | [2] |
| (E)-2-Heptenal | 22.67 | 1.81 | 1317 | Mo/R | - | 0.005 | [2] |
| Ethyl pentanoate | 23.08 | 2.27 | 1327 | M | - | 0.0015 | |
| 6-Methylhept-5-en-2-one | 23.17 | 1.85 | 1329 | Mo/F/R | Pungent, green | 1 | [2] |
| (Z)-3-Hexen-1-ol | 24.08 | 1.30 | 1350 | F/R/V | Green | 1.5 | [2] |
| (E)-3-Hexen-1-ol | 24.92 | 1.35 | 1369 | V/F | Green | 6 | [4] |
| 1-Octanol | 25.50 | 1.96 | 1383 | Mo | Moss, nut, mushroom | 0.1 | |
| Nonanal | 25.75 | 2.19 | 1388 | R | Fatty, waxy, pungent | 0.15 | [2] |
| (E,Z)-2,4-Hexadienal | 25.76 | 1.46 | 1388 | Green | |||
| (E)-2-Hexen-1-ol | 25.92 | 1.26 | 1392 | V | Green grass, leaves | 5 | [2] |
| (E,E)-2,4-Hexadienal | 26.00 | 1.48 | 1395 | - | |||
| (Z)-2-Octenal | 27.15 | 1.86 | 1420 | Green leaf, walnut | |||
| (E)-2-Octenal | 27.25 | 1.89 | 1424 | R | Green, nut, fat | 0.004 | [2] |
| Ethyl octanoate | 27.50 | 2.36 | 1429 | V | Fruit, fat | 10 | |
| 1-Octen-3-ol | 27.83 | 1.47 | 1437 | Mo | Mold, earthy | 0.05 | |
| Acetic acid | 28.50 | 0.97 | 1453 | F/V/R | Sour, vinegary | 0.5 | [2] |
| (Z,E)-2,4-Heptadienal | 28.58 | 1.60 | 1455 | R/Mo/F | Fatty, rancid | 0.36 | [2] |
| (E,Z)-2,4-Heptadienal | 28.66 | 3.24 | 1457 | R/Mo/F | Fatty, rancid | 10 | [2] |
| (E,E)-2,4-Heptadienal | 28.75 | 1.73 | 1459 | R/Mo/F | Fatty, rancid | 4 | [2] |
| 1-Nonanol | 30.02 | 2.02 | 1487 | Fresh, clean, floreal | 0.28 | [3] | |
| Copaene | 30.16 | 2.99 | 1492 | Wood, spice | |||
| Decanal | 30.25 | 2.23 | 1494 | R | Penetrating, sweet, waxy | 0.65 | [2] |
| (E)-Octa-3,5-dien-2-one | 30.91 | 1.73 | 1507 | V/Mo | Geranium-like | 0.0005 | [4] |
| (Z)-2-Nonenal | 31.43 | 1.94 | 1521 | R | Green, fatty | 0.0045 | [3] |
| (E)-2-Nonenal | 31.75 | 1.98 | 1530 | R | Paper-like, fatty | 0.9 | [3] |
| Propanoic acid | 32.24 | 0.78 | 1541 | Pungent, acidic | |||
| (E)-6-Methylhepta-3,5-dien-2-one | 33.83 | 1.64 | 1568 | V/Mo | - | 0.38 | [2] |
| Undecanal | 34.49 | 2.02 | 1597 | Waxy, aldehydic, soapy | |||
| Methyl benzoate | 34.91 | 1.43 | 1608 | Phenolic, prune, lettuce | |||
| Butanoic acid | 35.75 | 1.01 | 1630 | - | |||
| (E,E)-2,4-Nonadienal | 35.82 | 1.63 | 1632 | R | Watermelon | 2.5 | [2] |
| Ethyl decanoate | 35.91 | 2.48 | 1634 | V | Grape | 10 | [2] |
| (E)-2-Decenal | 36.08 | 2.02 | 1638 | R | Painty, fishy, fatty | 0.01 | [2] |
| 1-Decanol | 36.20 | 2.06 | 1642 | Fatty, waxy, floral, orange | |||
| Methyl butanoate | 37.41 | 1.05 | 1672 | F | Ether, fruit, sweet | 0.06 | |
| γ-Hexalactone | 37.91 | 1.56 | 1684 | Herbal, coconut, sweet | |||
| Dodecanal | 38.65 | 2.12 | 1704 | Soapy, waxy, citrus | |||
| 3,4-Dimethyl-2,5-Furandione | 39.16 | 1.35 | 1717 | - | |||
| α-Farnesene | 40.15 | 2.23 | 1744 | Wood, sweet | |||
| Pentanoic acid | 40.16 | 1.05 | 1751 | Sweet, acidic, sharp | |||
| (E,Z)-2,4-Decadienal | 40.58 | 1.73 | 1756 | R | Deep-fried | 0.01 | [2] |
| Phenylethyl alcohol | 40.80 | 0.26 | 1763 | ||||
| γ-Heptalactone | 41.66 | 1.60 | 1783 | Sweet, coconut, nutty | |||
| 1-Undecanol | 41.93 | 2.10 | 1792 | Fresh, waxy, rose, soapy | |||
| (E,E)-2,4-Decadienal | 42.25 | 1.64 | 1800 | R | Deep-fried | 0.18 | [2] |
| Tridecanal | 42.57 | 2.24 | 1809 | R | Flower, sweet, must, clean | ||
| Geranylacetone | 43.90 | 1.85 | 1846 | Magnolia, green | |||
| Butyl benzoate | 43.99 | 1.64 | 1848 | Balsamic, mild, fruity | |||
| Hexanoic acid | 44.16 | 1.01 | 1853 | Sweet, sour, fatty | 0.7 | [3] | |
| γ-Octalactone | 45.66 | 1.77 | 1891 | Sweet, coconut, creamy | |||
| Tetradecanal | 46.31 | 2.36 | 1914 | R | Fatty, lactonic, coconut, woody | ||
| 1-Dodecanol | 47.57 | 2.14 | 1951 | Soapy, waxy, clean | |||
| Heptanoic acid | 47.99 | 1.01 | 1963 | Waxy, cheesy, fruity | 0.1 | [2] | |
| γ-Nonalactone | 49.49 | 1.85 | 2007 | Fatty, coconut | |||
| Pentadecanal | 49.89 | 2.48 | 2020 | R | Fresh, waxy | ||
| Octanoic acid | 51.58 | 1.01 | 2072 | Rancid, soapy, cheesy | 3 | [2] | |
| γ-Decalactone | 53.16 | 1.98 | 2109 | Fruity, fresh, peach | |||
| Hexadecanal | 53.31 | 2.58 | 2126 | R | Cardboard | ||
| 1-Tridecanol | 54.23 | 2.17 | 2155 | Musty | |||
| Nonanoic acid | 54.91 | 1.05 | 2176 | Fatty, waxy, cheesy | |||
| Methyl palmitate | 55.90 | 2.44 | 2208 | Oily, waxy, fatty, orris | |||
| Ethyl palmitate | 57.06 | 2.61 | 2247 | Waxy, fruity, creamy | |||
| Decanoic acid | 58.24 | 1.05 | 2286 | Soapy, waxy, fruity | |||
| Palmitic acid | 59.56 | 2.69 | 2332 | Waxy, creamy, fatty, soapy | |||
| Heptadecanal | 59.72 | 2.74 | 2338 | R | - | ||
| 1-Tetradecanol | 60.40 | 2.21 | 2361 | Coconut | |||
| Butyl palmitate | 62.23 | 3.11 | 2438 |
| Sampling Amount 0.100 g | Sampling Amount 1.500 g | MHS-SPME-TRIF (0.100 g) | |||||
|---|---|---|---|---|---|---|---|
| Norm. Volume | % Volume | Norm. Volume | % Volume | % Error | β | Decay Function | |
| 1-Octanol | 1.10 | 9.42 × 10−3 | 0.11 | 6.75 × 10−4 | −89.75 | 0.67 | y = −0.40x + 14.1 |
| (E)-2-Decenal | 0.92 | 7.82 × 10−3 | 0.35 | 2.07 × 10−3 | −62.06 | 0.59 | y = −0.54x + 14.7 |
| Heptanal | 1.36 | 1.16 × 10−2 | 0.77 | 4.62 × 10−3 | −43.25 | 0.47 | y = −0.76x + 14.6 |
| 2-Octanone | 0.20 | 1.71 × 10−3 | 0.15 | 8.95 × 10−4 | −25.33 | 0.76 | y = −0.28x + 12.5 |
| 2-Nonanone | 0.17 | 1.46 × 10−3 | 0.15 | 8.91 × 10−4 | −12.96 | 0.62 | y = −0.48x + 12.7 |
| 1-Butanol | 3.38 | 2.88 × 10−2 | 2.97 | 1.77 × 10−2 | −12.05 | 0.37 | y = −1.00x + 16.1 |
| (E)-2-Pentenal | 25.42 | 2.17 × 10−1 | 22.45 | 1.34 × 10−1 | −11.65 | 0.28 | y = −1.28x + 17.5 |
| (Z)-2-Penten-1-ol | 9.06 | 7.73 × 10−2 | 8.81 | 5.26 × 10−2 | −2.74 | 0.39 | y = −0.94x + 17.1 |
| Hexanal | 366.32 | 3.13 × 100 | 360.14 | 2.15 × 100 | −1.69 | 0.48 | y = −0.74x + 20.6 |
| (E)-2-Hexenal | 0.15 | 1.24 × 10−3 | 0.15 | 8.68 × 10−4 | 0.21 | 0.67 | y = −0.74x + 23.6 |
| Ethyl acetate | 0.61 | 5.23 × 10−3 | 0.62 | 3.68 × 10−3 | 0.67 | 0.63 | y = −0.46x + 13.4 |
| 2-Pentanol | 0.19 | 1.61 × 10−3 | 0.19 | 1.15 × 10−3 | 2.84 | 0.69 | y = −0.37x + 12.7 |
| 1-Hexanol | 304.90 | 2.60 × 100 | 384.66 | 2.30 × 100 | 26.16 | 0.75 | y = −0.28x + 19.8 |
| Toluene | 1.14 | 9.72 × 10−3 | 1.46 | 8.71 × 10−3 | 28.23 | 0.44 | y = −0.81x + 14.6 |
| (Z)-3-Hexen-1-ol | 71.87 | 6.13 × 10−1 | 92.98 | 5.55 × 10−1 | 29.37 | 0.74 | y = −0.30x + 18.5 |
| (E)-2-Penten-1-ol | 220.87 | 1.88 × 100 | 301.14 | 1.80 × 100 | 36.34 | 0.28 | y = −0.82x + 20.3 |
| 1-Penten-3-ol | 191.90 | 1.64 × 100 | 267.17 | 1.59 × 100 | 39.23 | 0.44 | y = −0.81x + 20.0 |
| (E)-2-Hexen-1-ol | 171.70 | 1.46 × 100 | 247.77 | 1.48 × 100 | 44.30 | 0.73 | y = −0.32x + 19.3 |
| Decane | 0.34 | 2.90 × 10−3 | 0.49 | 2.93 × 10−3 | 44.54 | 0.73 | y = −0.32x + 13.0 |
| (E)-2-Octenal | 0.41 | 3.48 × 10−3 | 0.59 | 3.52 × 10−3 | 44.89 | 0.44 | y = −0.40x + 13.1 |
| Acetic acid | 15.99 | 1.36 × 10−1 | 37.64 | 2.25 × 10−1 | 135.36 | 0.45 | y = −0.81x + 17.9 |
| (E,E)-2,4-Hexadienal | 19.17 | 1.64 × 10−1 | 50.81 | 3.03 × 10−1 | 165.05 | 0.48 | y = −0.73x + 17.9 |
| 1-Pentanol | 3.54 | 3.02 × 10−2 | 9.89 | 5.90 × 10−2 | 179.08 | 0.51 | y = −0.68x + 15.8 |
| 2-Methylfuran | 0.25 | 2.17 × 10−3 | 1.11 | 6.64 × 10−3 | 338.20 | 0.57 | y = −0.56x + 13.1 |
| Nonanal | 0.69 | 5.88 × 10−3 | 7.88 | 4.70 × 10−2 | 1043.06 | 0.58 | y = −0.55x + 15.6 |
| Cyclohexane | 15.81 | 1.35 × 10−1 | 273.40 | 1.63 × 100 | 1629.52 | 0.34 | y = −1.07x + 18.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stilo, F.; Cordero, C.; Sgorbini, B.; Bicchi, C.; Liberto, E. Highly Informative Fingerprinting of Extra-Virgin Olive Oil Volatiles: The Role of High Concentration-Capacity Sampling in Combination with Comprehensive Two-Dimensional Gas Chromatography. Separations 2019, 6, 34. https://doi.org/10.3390/separations6030034
Stilo F, Cordero C, Sgorbini B, Bicchi C, Liberto E. Highly Informative Fingerprinting of Extra-Virgin Olive Oil Volatiles: The Role of High Concentration-Capacity Sampling in Combination with Comprehensive Two-Dimensional Gas Chromatography. Separations. 2019; 6(3):34. https://doi.org/10.3390/separations6030034
Chicago/Turabian StyleStilo, Federico, Chiara Cordero, Barbara Sgorbini, Carlo Bicchi, and Erica Liberto. 2019. "Highly Informative Fingerprinting of Extra-Virgin Olive Oil Volatiles: The Role of High Concentration-Capacity Sampling in Combination with Comprehensive Two-Dimensional Gas Chromatography" Separations 6, no. 3: 34. https://doi.org/10.3390/separations6030034
APA StyleStilo, F., Cordero, C., Sgorbini, B., Bicchi, C., & Liberto, E. (2019). Highly Informative Fingerprinting of Extra-Virgin Olive Oil Volatiles: The Role of High Concentration-Capacity Sampling in Combination with Comprehensive Two-Dimensional Gas Chromatography. Separations, 6(3), 34. https://doi.org/10.3390/separations6030034