Hydrophilic Monomethyl Auristatin E Derivatives as Novel Candidates for the Design of Antibody-Drug Conjugates

 , and
, and 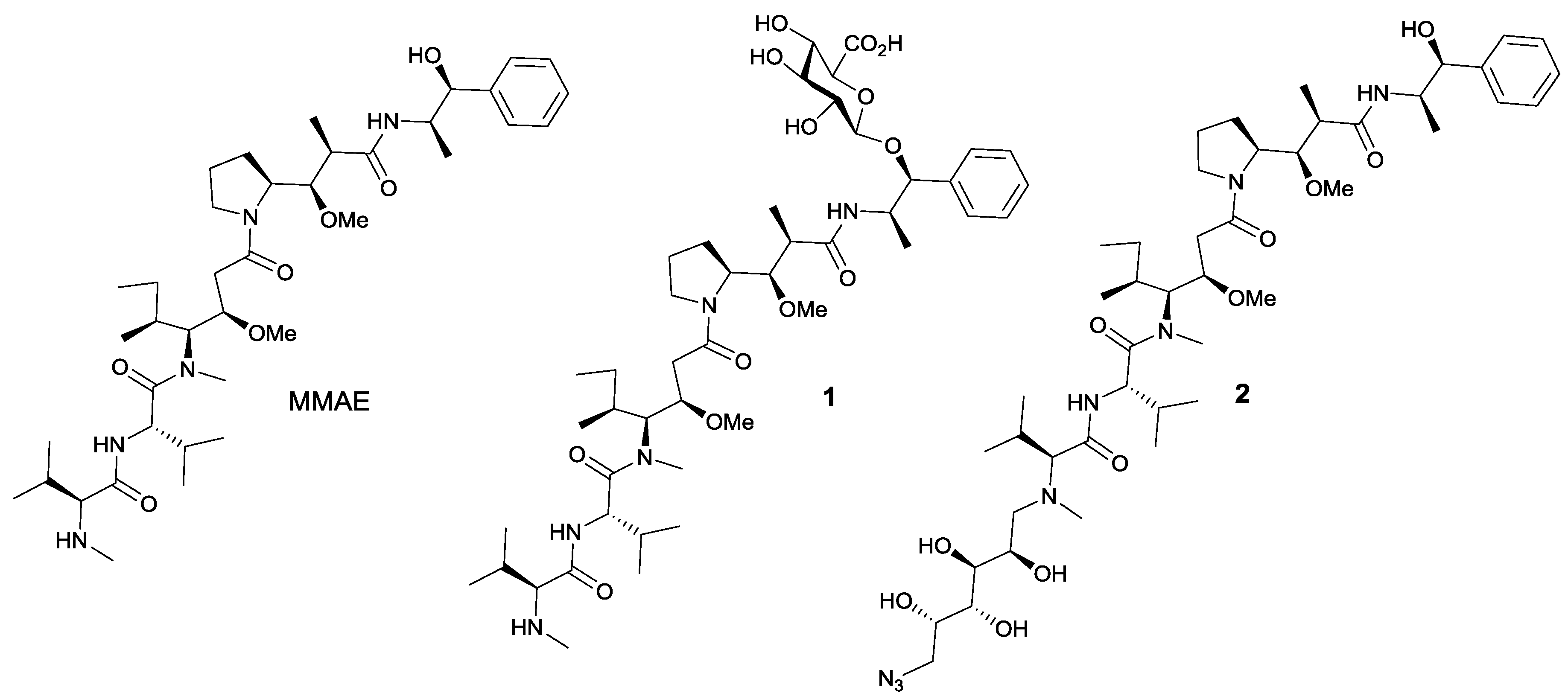{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Capillary Electrophoresis
2.3. Calculations of Retention Factors and Distribution Constants
2.4. Determination of the Critical Micelle Concentration
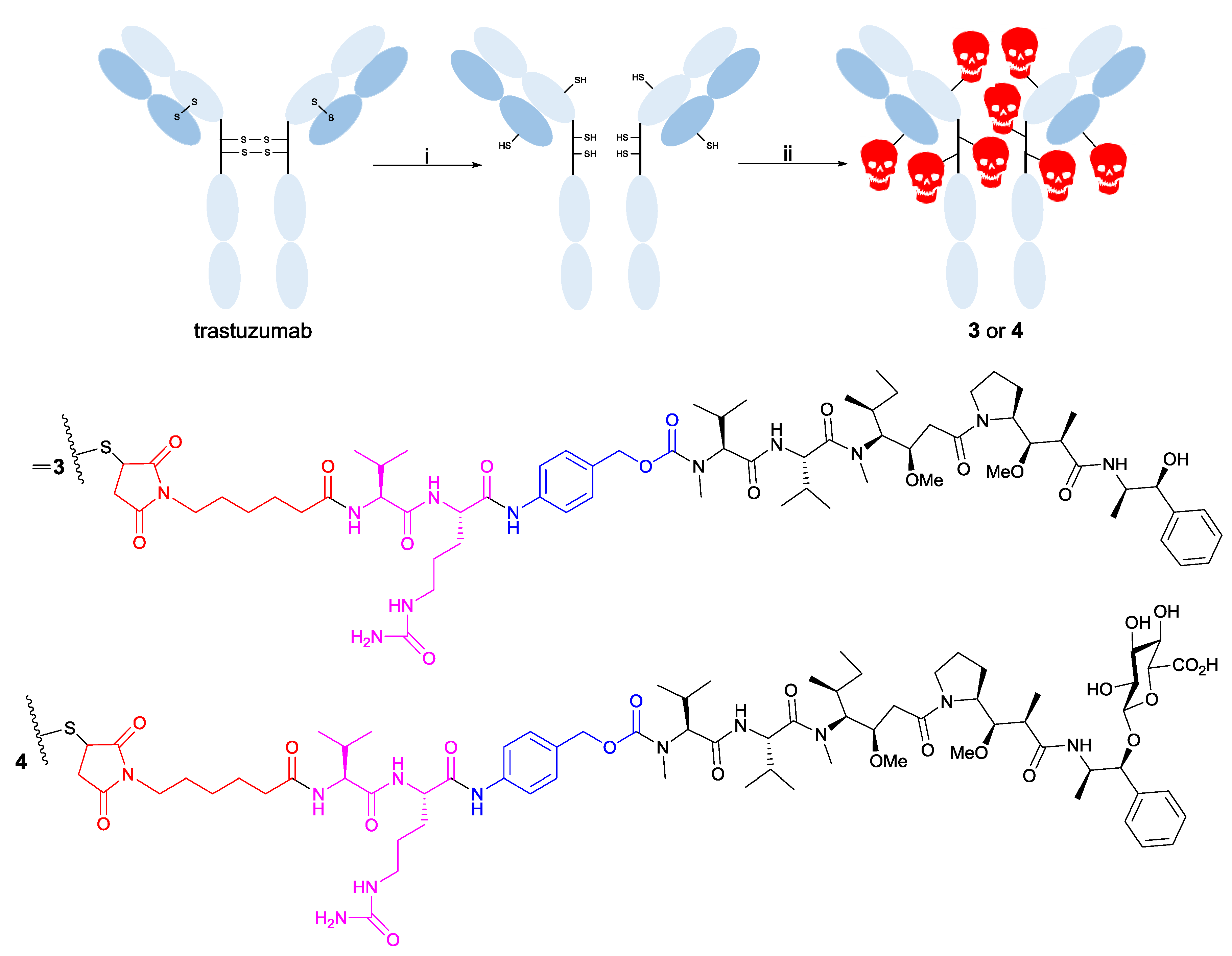
2.5. Preparation of Drug-Linker Compounds and Antibody-Drug Conjugates
2.6. Cytotoxicity Assay
3. Results and Discussion
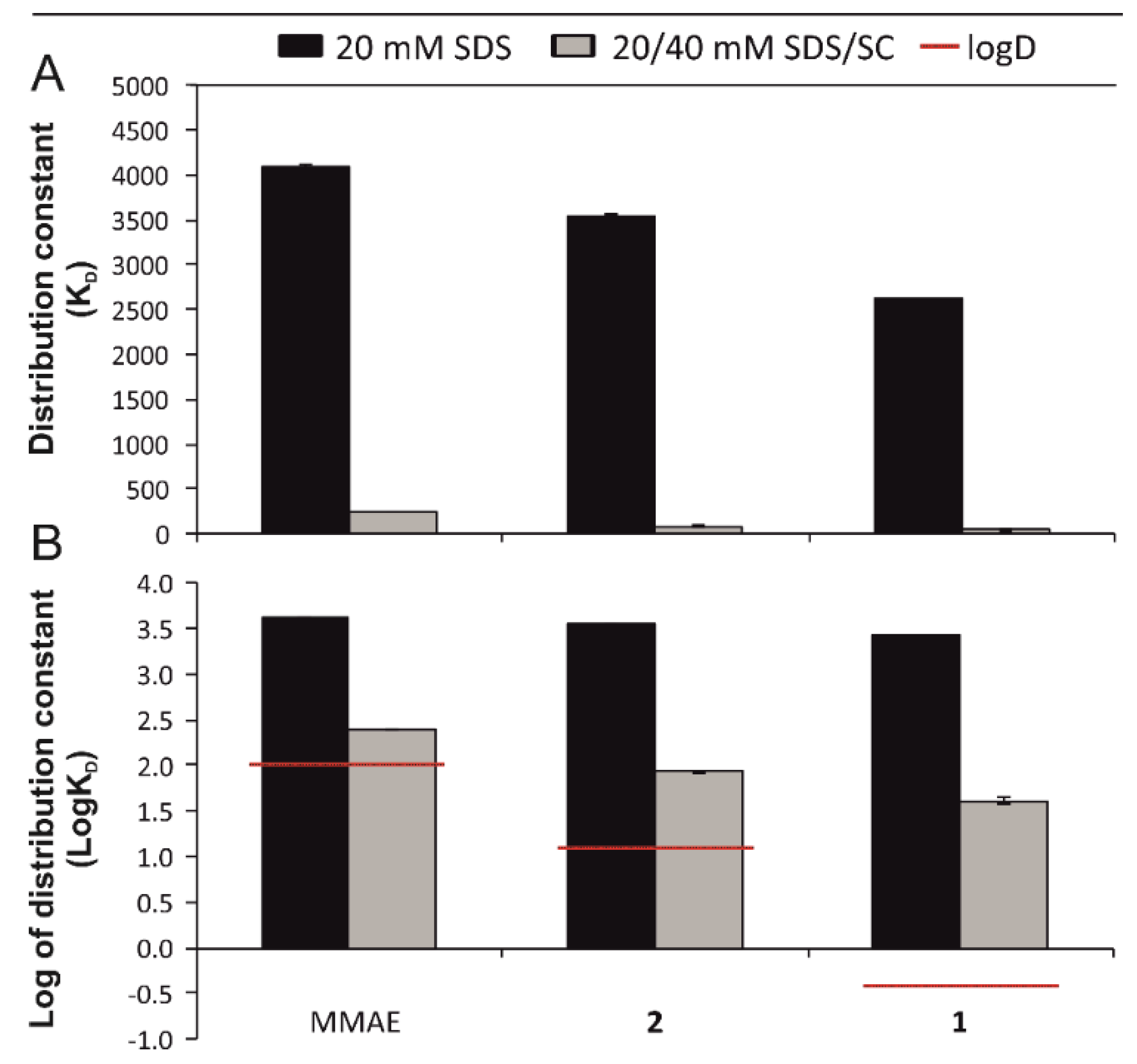
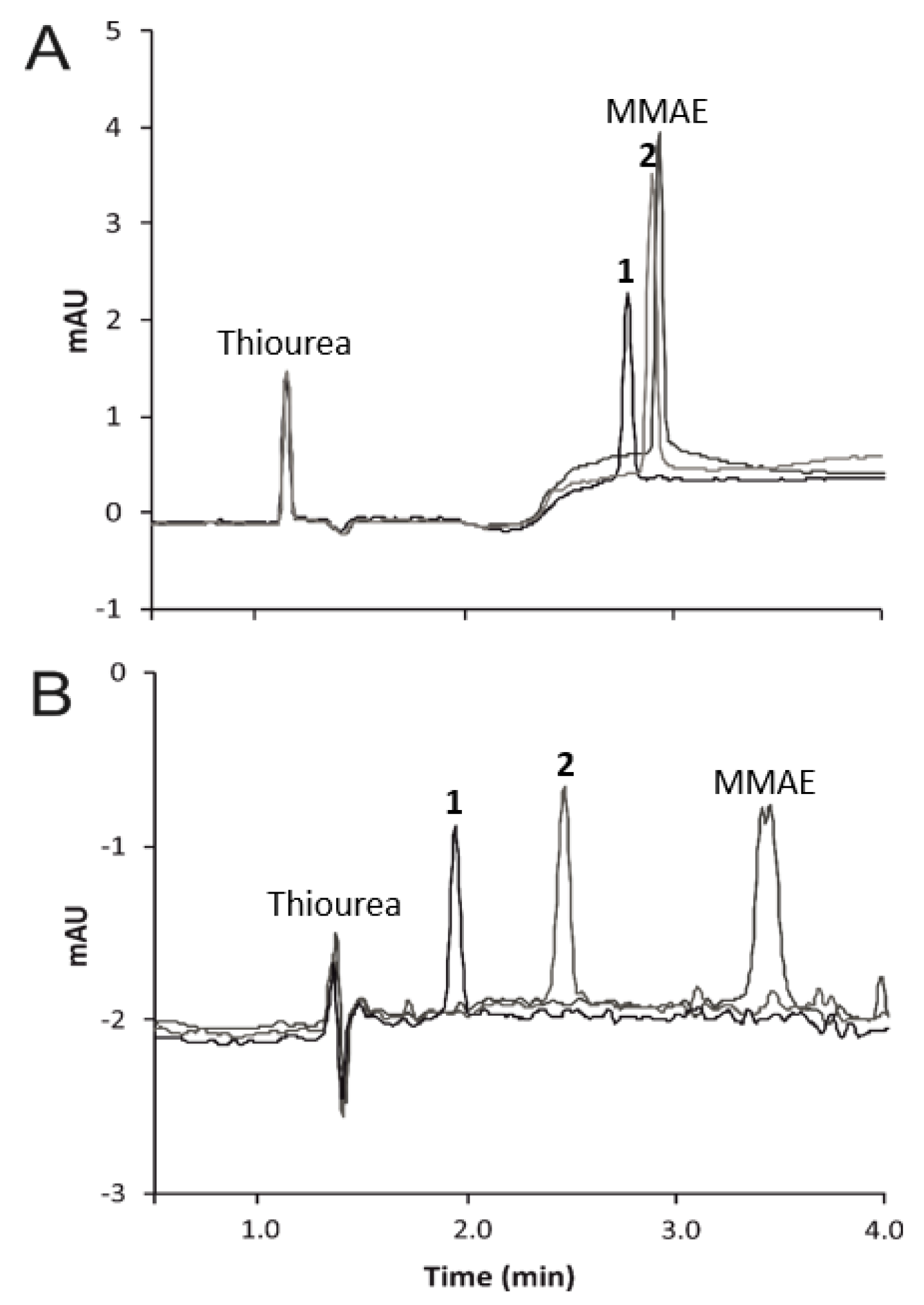
3.1. Micellar Electrokinetic Chromatography
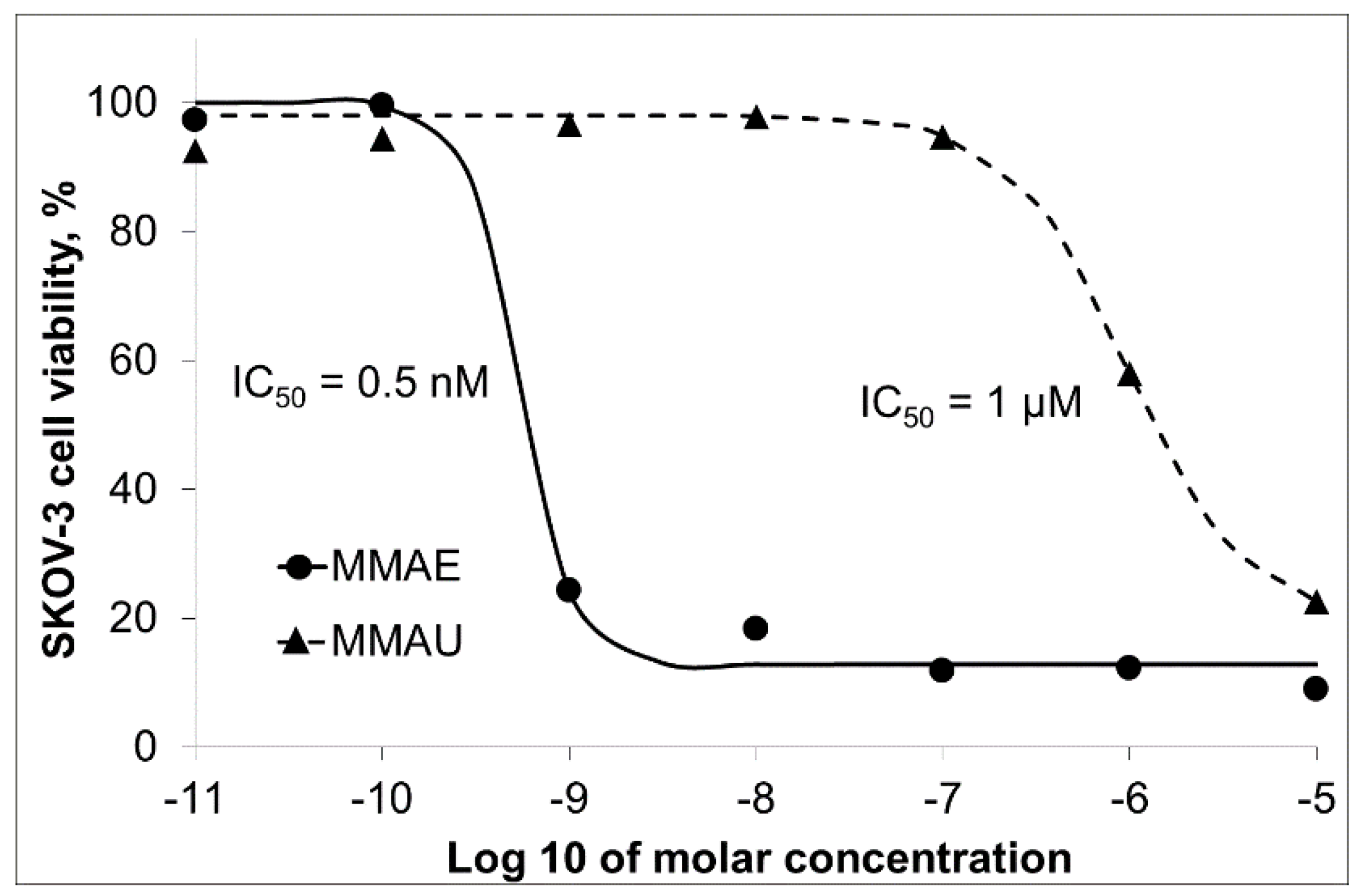
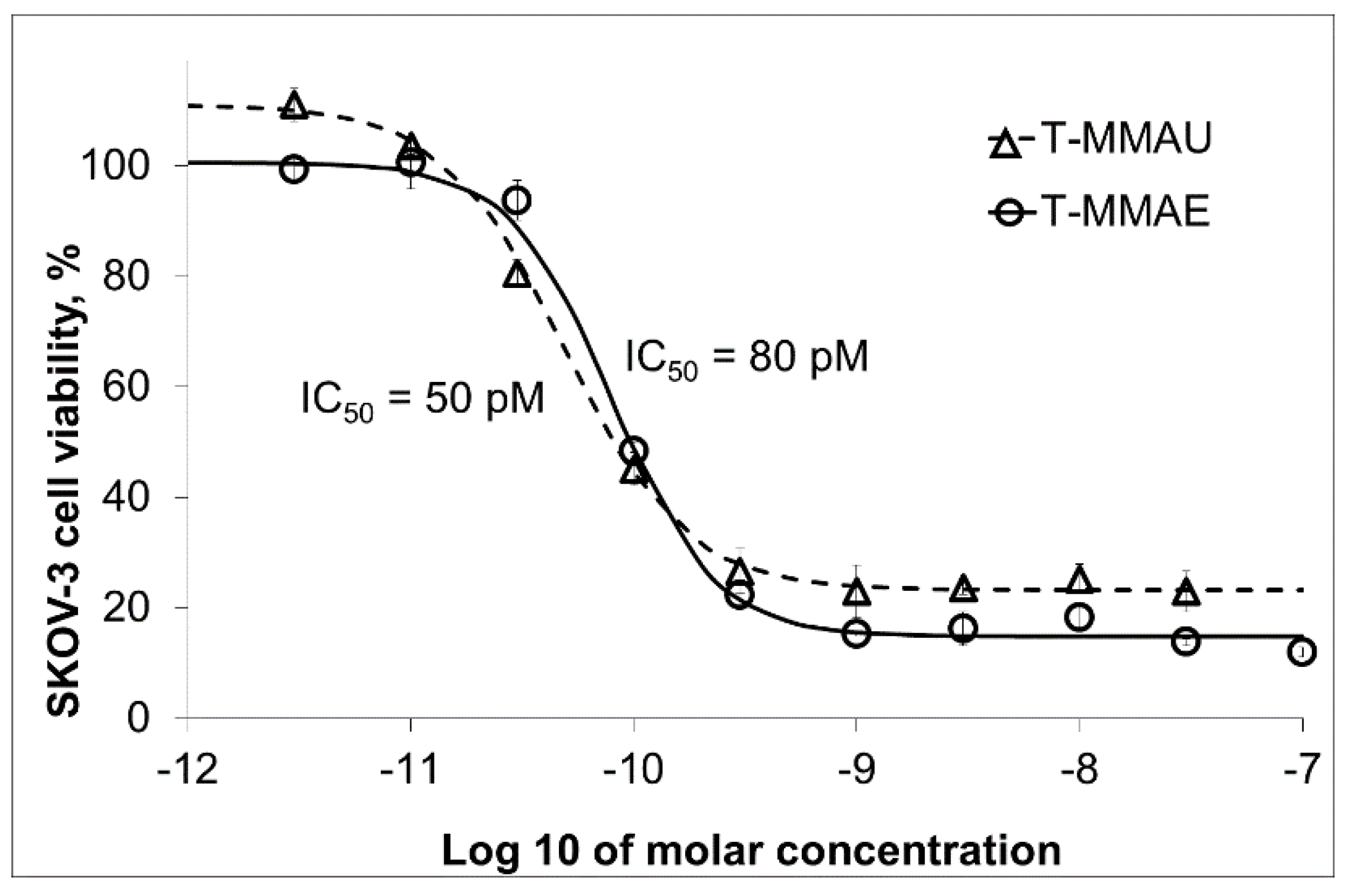
3.2. Trastuzumab Conjugates and Cytotoxicity Assays
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Schrama, D.; Reisfeld, R.A.; Becker, J.C. Antibody targeted drugs as cancer therapeutics. Nat. Rev. Drug Discov. 2006, 5, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Alley, S.C.; Okeley, N.M.; Senter, P.D. Antibody–drug conjugates: targeted drug delivery for cancer. Curr. Opin. Chem. Biol. 2010, 14, 529–537. [Google Scholar] [CrossRef] [PubMed]
- Younes, A.; Bartlett, N.L.; Leonard, J.P.; Kennedy, D.A.; Lynch, C.M.; Sievers, E.L.; Forero-Torres, A. Brentuximab vedotin (SGN-35) for relapsed CD30-positive lymphomas. N. Eng. J. Med. 2010, 363, 1812–1821. [Google Scholar] [CrossRef] [PubMed]
- Verma, S.; Miles, D.; Gianni, L.; Krop, I.E.; Welslau, M.; Baselga, J.; Pegram, M.; Oh, D.-Y.; Diéras, V.; Guardino, E. Trastuzumab emtansine for HER2-positive advanced breast cancer. N. Eng. J. Med. 2012, 367, 1783–1791. [Google Scholar] [CrossRef] [PubMed]
- Rowe, J.M.; Löwenberg, B. Gemtuzumab ozogamicin in acute myeloid leukemia: a remarkable saga about an active drug. Blood 2013, 121, 4838–4841. [Google Scholar] [CrossRef] [PubMed]
- Rytting, M.; Triche, L.; Thomas, D.; O’brien, S.; Kantarjian, H. Initial experience with CMC-544 (inotuzumab ozogamicin) in pediatric patients with relapsed B-cell acute lymphoblastic leukemia. Pediatr. Blood Cancer 2014, 61, 369–372. [Google Scholar] [CrossRef] [PubMed]
- Lyon, R.P.; Bovee, T.D.; Doronina, S.O.; Burke, P.J.; Hunter, J.H.; Neff-LaFord, H.D.; Jonas, M.; Anderson, M.E.; Setter, J.R.; Senter, P.D. Reducing hydrophobicity of homogeneous antibody-drug conjugates improves pharmacokinetics and therapeutic index. Nat. Biotechnol. 2015, 33, 733–735. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Wilhelm, S.D.; Audette, C.; Jones, G.; Leece, B.A.; Lazar, A.C.; Goldmacher, V.S.; Singh, R.; Kovtun, Y.; Widdison, W.C. Synthesis and evaluation of hydrophilic linkers for antibody–maytansinoid conjugates. J. Med. Chem. 2011, 54, 3606–3623. [Google Scholar] [CrossRef] [PubMed]
- Ekholm, F.S.; Pynnönen, H.; Vilkman, A.; Pitkänen, V.; Helin, J.; Saarinen, J.; Satomaa, T. Introducing glycolinkers for the functionalization of cytotoxic drugs and applications in antibody–drug conjugation chemistry. ChemMedChem 2016, 11, 2501–2505. [Google Scholar] [CrossRef]
- Johansson, M.P.; Maaheimo, H.; Ekholm, F.S. New insight on the structural features of the cytotoxic auristatins MMAE and MMAF revealed by combined NMR spectroscopy and quantum chemical modelling. Sci. Rep. 2017, 7, 15920. [Google Scholar] [CrossRef] [PubMed]
- Rostami, S.; Qazi, I.; Sikorski, R. The clinical landscape of antibody-drug conjugates. ADC Rev. 2014. [Google Scholar] [CrossRef]
- Barok, M.; Joensuu, H.; Isola, J. Trastuzumab emtansine: mechanisms of action and drug resistance. Breast Cancer Res. 2014, 16, 209. [Google Scholar] [CrossRef] [PubMed]
- Hamblett, K.J.; Senter, P.D.; Chace, D.F.; Sun, M.M.; Lenox, J.; Cerveny, C.G.; Kissler, K.M.; Bernhardt, S.X.; Kopcha, A.K.; Zabinski, R.F. Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin. Cancer Res. 2004, 10, 7063–7070. [Google Scholar] [CrossRef] [PubMed]
- Bushey, M.M.; Jorgenson, J.W. Separation of dansylated methylamine and dansylated methyl-d3-amine by micellar electrokinetic capillary chromatography with methanol-modified mobile phase. Anal. Chem. 1989, 61, 491–493. [Google Scholar] [CrossRef]
- Laine, J.; Lokajová, J.; Parshintsev, J.; Holopainen, J.M.; Wiedmer, S.K. Interaction of a commercial lipid dispersion and local anesthetics in human plasma: Implications for drug trapping by “lipid-sinks”. Anal. Bioanal. Chem. 2010, 396, 2599–2607. [Google Scholar] [CrossRef] [PubMed]
- Ahlstrom, D.M.; Hoyos, Y.M.; Arslan, H.; Akbay, C. Binary mixed micelles of chiral sodium undecenyl leucinate and achiral sodium undecenyl sulfate: I. Characterization and application as pseudostationary phases in micellar electrokinetic chromatography. J. Chromatogr. A 2010, 1217, 375–385. [Google Scholar] [CrossRef] [PubMed]
- González-Gaitano, G.; Compostizo, A.; Sánchez-Martín, L.; Tardajos, G. Speed of sound, density, and molecular modeling studies on the inclusion complex between sodium cholate and β-cyclodextrin. Langmuir 1997, 13, 2235–2241. [Google Scholar] [CrossRef]
- Satomaa, T.; Pynnönen, H.; Vilkman, A.; Kotiranta, T.; Pitkänen, V.; Heiskanen, A.; Herpers, B.; Price, L.S.; Helin, J.; Saarinen, J. Hydrophilic Auristatin Glycoside Payload Enables Improved Antibody-Drug Conjugate Efficacy and Biocompatibility. Antibodies 2018, 7, 15. [Google Scholar] [CrossRef]
- Terabe, S.; Otsuka, K.; Ichikawa, K.; Tsuchiya, A.; Ando, T. Electrokinetic separations with micellar solutions and open-tubular capillaries. Anal. Chem. 1984, 56, 111–113. [Google Scholar] [CrossRef]
- Nishi, H.; Terabe, S. Micellar electrokinetic chromatography perspectives in drug analysis. J. Chromatogr. A 1996, 735, 3–27. [Google Scholar]
- Silva, M. Micellar electrokinetic chromatography: A review of methodological and instrumental innovations focusing on practical aspects. Electrophoresis 2013, 34, 141–158. [Google Scholar] [CrossRef] [PubMed]
- Deeb, S.E.; Dawwas, H.A.; Gust, R. Recent methodological and instrumental development in MEKC. Electrophoresis 2013, 34, 1295–1303. [Google Scholar] [CrossRef] [PubMed]
- Ji, A.J.; Nunez, M.F.; Machacek, D.; Ferguson, J.E.; Iossi, M.F.; Kao, P.C.; Landers, J.P. Separation of urinary estrogens by micellar electrokinetic chromatography. J. Chromatogr. B 1995, 669, 15–26. [Google Scholar] [CrossRef]
- Jumppanen, J.H.; Wiedmer, S.K.; Siren, H.; Riekkola, M.L.; Haario, H. Optimized Separation of 7 Corticosteroids by Micellar Electrokinetic Chromatography. Electrophoresis 1994, 15, 1267–1272. [Google Scholar] [CrossRef] [PubMed]
- Wiedmer, S.K.; Jumppanen, J.H.; Haario, H.; Riekkola, M.L. Optimization of selectivity and resolution in micellar electrokinetic capillary chromatography with a mixed micellar system of sodium dodecyl sulfate and sodium cholate. Electrophoresis 1996, 17, 1931–1937. [Google Scholar] [CrossRef] [PubMed]
- Cole, R.O.; Sepaniak, M.J.; Hinze, W.L.; Gorse, J.; Oldiges, K. Bile salt surfactants in micellar electrokinetic capillary chromatography: application to hydrophobic molecule separations. J. Chromatogr. A 1991, 557, 113–123. [Google Scholar] [CrossRef]
- Yang, S.; Bumgarner, J.G.; Kruk, L.F.; Khaledi, M.G. Quantitative structure-activity relationships studies with micellar electrokinetic chromatography influence of surfactant type and mixed micelles on estimation of hydrophobicity and bioavailability. J. Chromatogr. A 1996, 721, 323–335. [Google Scholar] [CrossRef]
- Doronina, S.O.; Mendelsohn, B.A.; Bovee, T.D.; Cerveny, C.G.; Alley, S.C.; Meyer, D.L.; Oflazoglu, E.; Toki, B.E.; Sanderson, R.J.; Zabinski, R.F. Enhanced activity of monomethylauristatin F through monoclonal antibody delivery: effects of linker technology on efficacy and toxicity. Bioconjug. Chem. 2006, 17, 114–124. [Google Scholar] [CrossRef]
- Lambert, J.M.; Chari, R.V. Ado-trastuzumab Emtansine (T-DM1): an antibody–drug conjugate (ADC) for HER2-positive breast cancer. J. Med. Chem. 2014, 57, 6949–6964. [Google Scholar] [CrossRef]
- Alley, S.C.; Benjamin, D.R.; Jeffrey, S.C.; Okeley, N.M.; Meyer, D.L.; Sanderson, R.J.; Senter, P.D. Contribution of linker stability to the activities of anticancer immunoconjugates. Bioconjug. Chem. 2008, 19, 759–765. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The discovery and development of brentuximab vedotin for use in relapsed Hodgkin lymphoma and systemic anaplastic large cell lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Waight, A.B.; Bargsten, K.; Doronina, S.; Steinmetz, M.O.; Sussman, D.; Prota, A.E. Structural basis of microtubule destabilization by potent auristatin anti-mitotics. PLoS ONE 2016, 11, e0160890. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Benz, F.W.; Wu, Y.; Wang, Q.; Chen, Y.; Chen, X.; Li, H.; Zhang, Y.; Zhang, R.; Yang, J. Structural Insights into the Pharmacophore of Vinca Domain Inhibitors of Microtubules. Mol. Pharmacol. 2016, 89, 233–242. [Google Scholar] [CrossRef] [PubMed]
- Phillips, G.D.L.; Li, G.; Dugger, D.L.; Crocker, L.M.; Parsons, K.L.; Mai, E.; Blättler, W.A.; Lambert, J.M.; Chari, R.V.; Lutz, R.J. Targeting HER2-positive breast cancer with trastuzumab-DM1, an antibody–cytotoxic drug conjugate. Cancer Res. 2008, 68, 9280–9290. [Google Scholar] [CrossRef] [PubMed]
- Zhao, R.Y.; Erickson, H.K.; Leece, B.A.; Reid, E.E.; Goldmacher, V.S.; Lambert, J.M.; Chari, R.V. Synthesis and biological evaluation of antibody conjugates of phosphate prodrugs of cytotoxic DNA alkylators for the targeted treatment of cancer. J. Med. Chem. 2012, 55, 766–782. [Google Scholar] [CrossRef] [PubMed]






© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ekholm, F.S.; Ruokonen, S.-K.; Redón, M.; Pitkänen, V.; Vilkman, A.; Saarinen, J.; Helin, J.; Satomaa, T.; Wiedmer, S.K. Hydrophilic Monomethyl Auristatin E Derivatives as Novel Candidates for the Design of Antibody-Drug Conjugates. Separations 2019, 6, 1. https://doi.org/10.3390/separations6010001
Ekholm FS, Ruokonen S-K, Redón M, Pitkänen V, Vilkman A, Saarinen J, Helin J, Satomaa T, Wiedmer SK. Hydrophilic Monomethyl Auristatin E Derivatives as Novel Candidates for the Design of Antibody-Drug Conjugates. Separations. 2019; 6(1):1. https://doi.org/10.3390/separations6010001
Chicago/Turabian StyleEkholm, Filip S., Suvi-Katriina Ruokonen, Marina Redón, Virve Pitkänen, Anja Vilkman, Juhani Saarinen, Jari Helin, Tero Satomaa, and Susanne K. Wiedmer. 2019. "Hydrophilic Monomethyl Auristatin E Derivatives as Novel Candidates for the Design of Antibody-Drug Conjugates" Separations 6, no. 1: 1. https://doi.org/10.3390/separations6010001
APA StyleEkholm, F. S., Ruokonen, S.-K., Redón, M., Pitkänen, V., Vilkman, A., Saarinen, J., Helin, J., Satomaa, T., & Wiedmer, S. K. (2019). Hydrophilic Monomethyl Auristatin E Derivatives as Novel Candidates for the Design of Antibody-Drug Conjugates. Separations, 6(1), 1. https://doi.org/10.3390/separations6010001