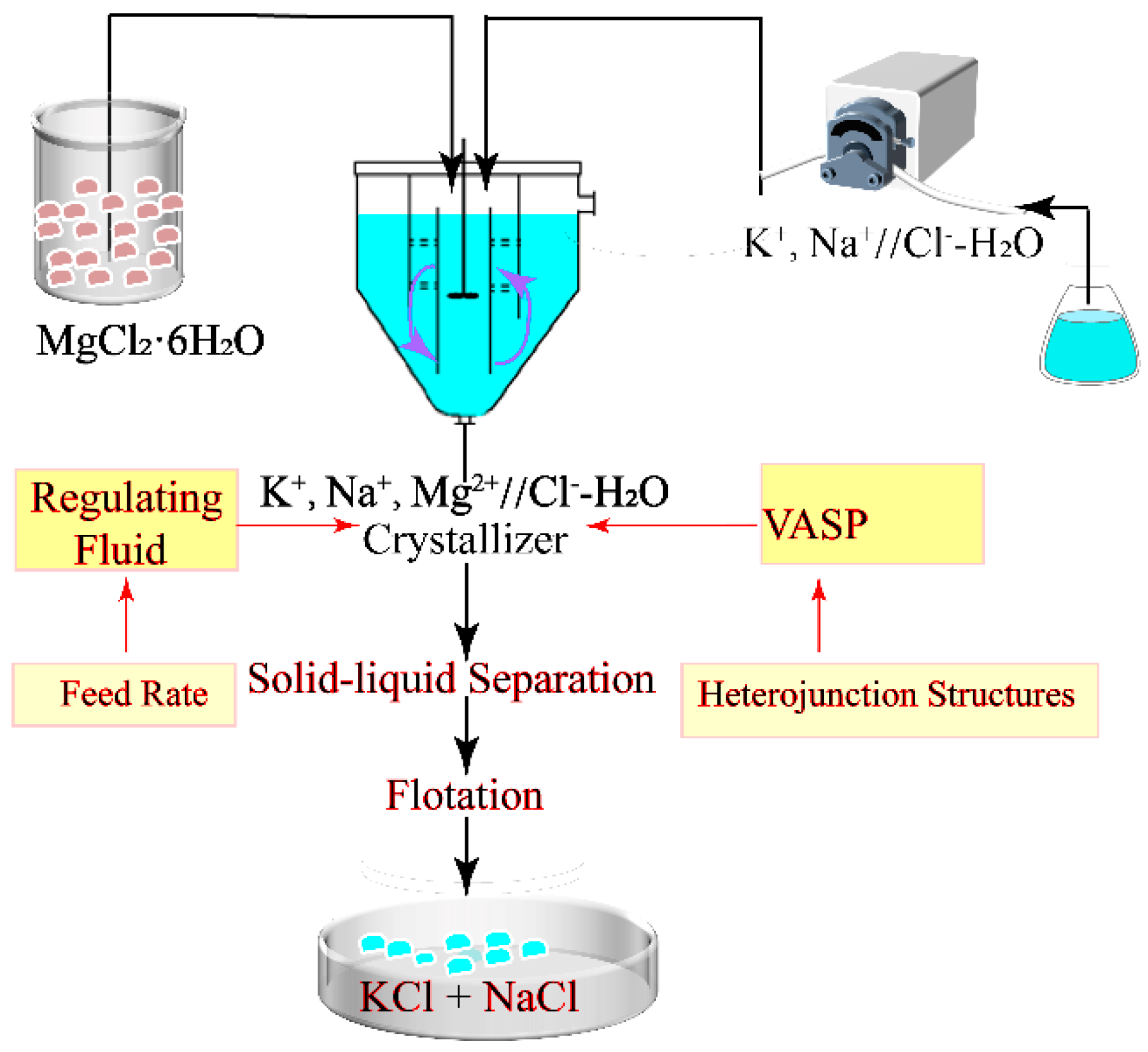
3.2.1. VASP Calculation Model and Its Method
- (1)
Methodology
The calculations are implemented using VASP code [
28]. The calculation of the correlation potentials is conducted using the GGA-PBE functional [
29]. A Weak van der Waals interaction is considered using the DFT-D3 functional [
30]. The cut-off energy for the plane-wave is 400 eV. The Gamma point in the Brillouin-zone is chosen for the integration. The total energies of the systems converge to 10
−5 eV in the iteration solution of the Kohn−Sham equation. The optimized force per atom is 0.05 eV/Å.
- (2)
Model Construction
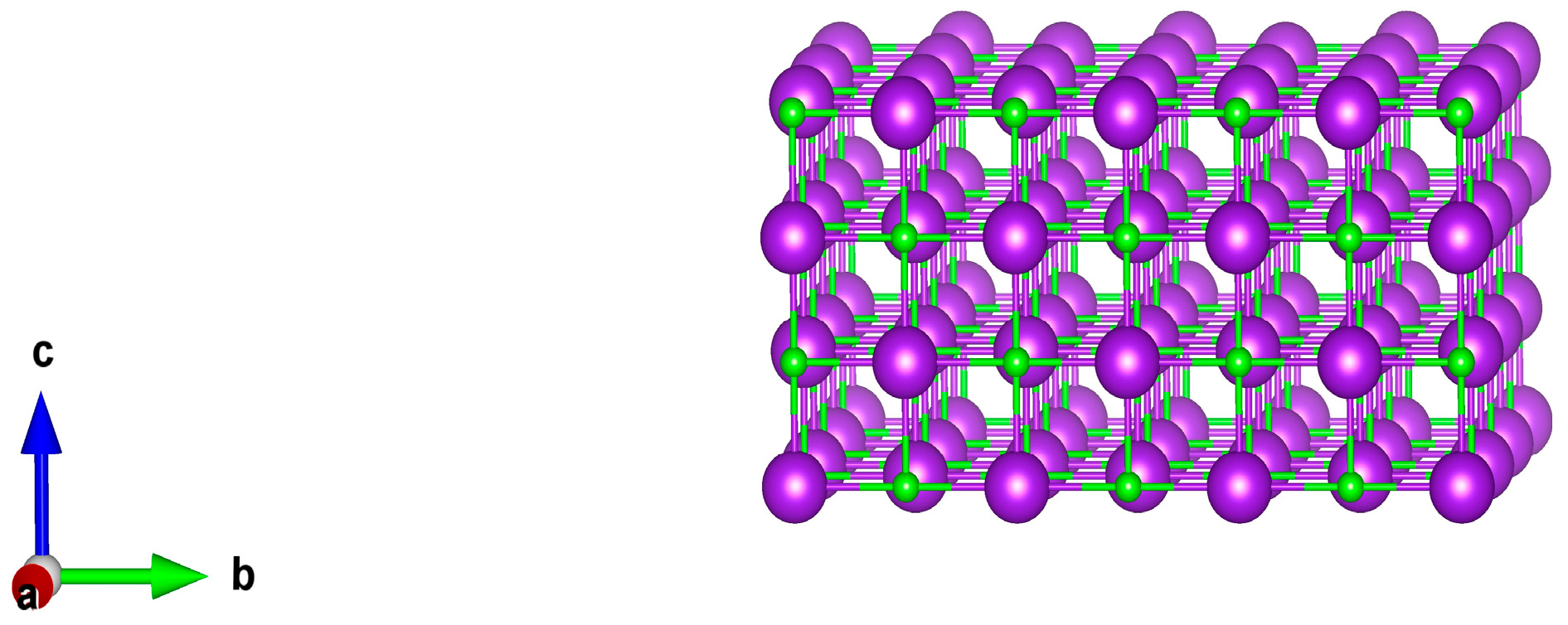
Firstly, the optimized crystal structure of KCl is shown in
Table 8. After optimization, the lattice parameters are similar to the experimental values and the model is reliable. Comparative results are shown in
Figure 5.
- (3)
Adsorption energy calculation
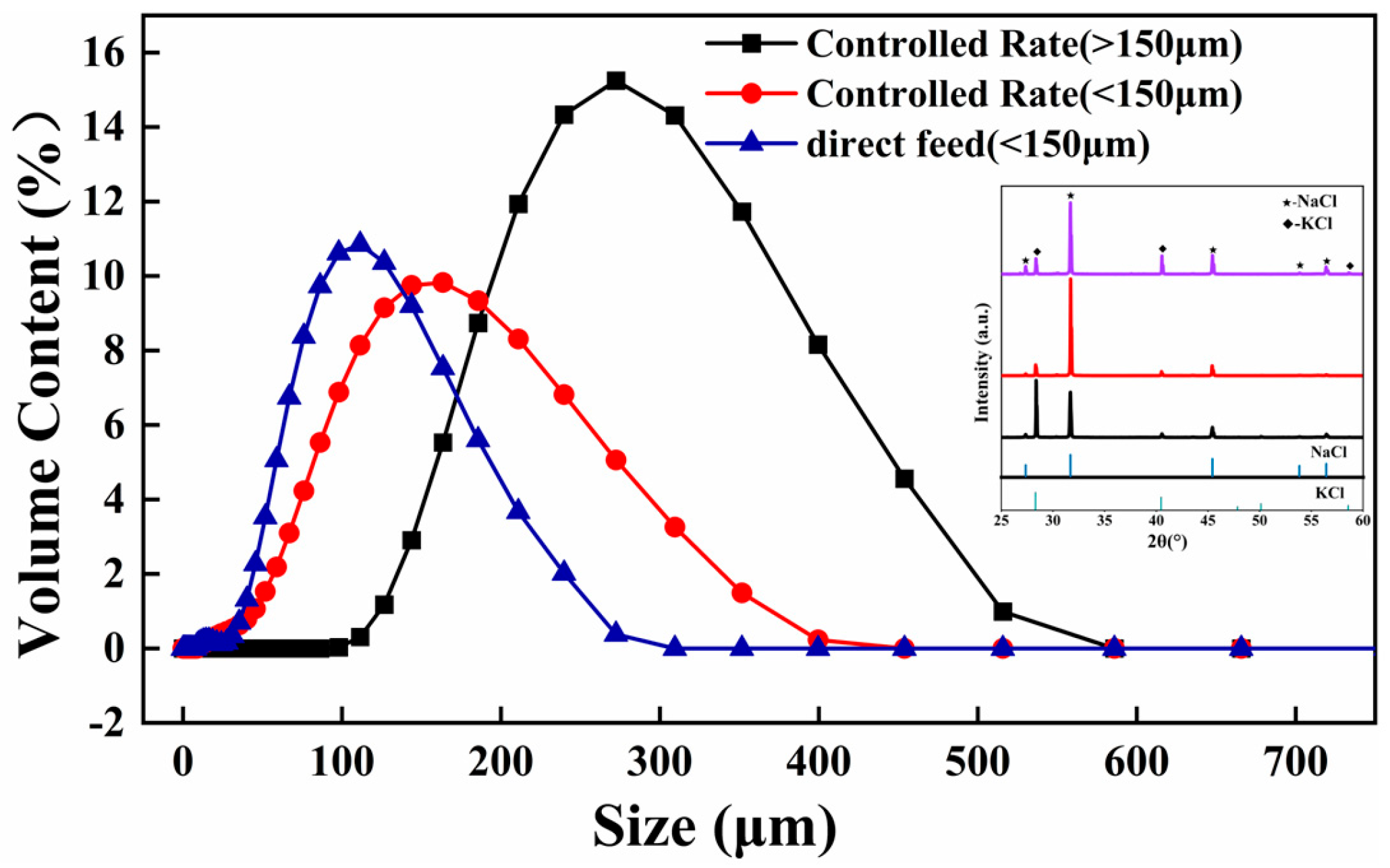
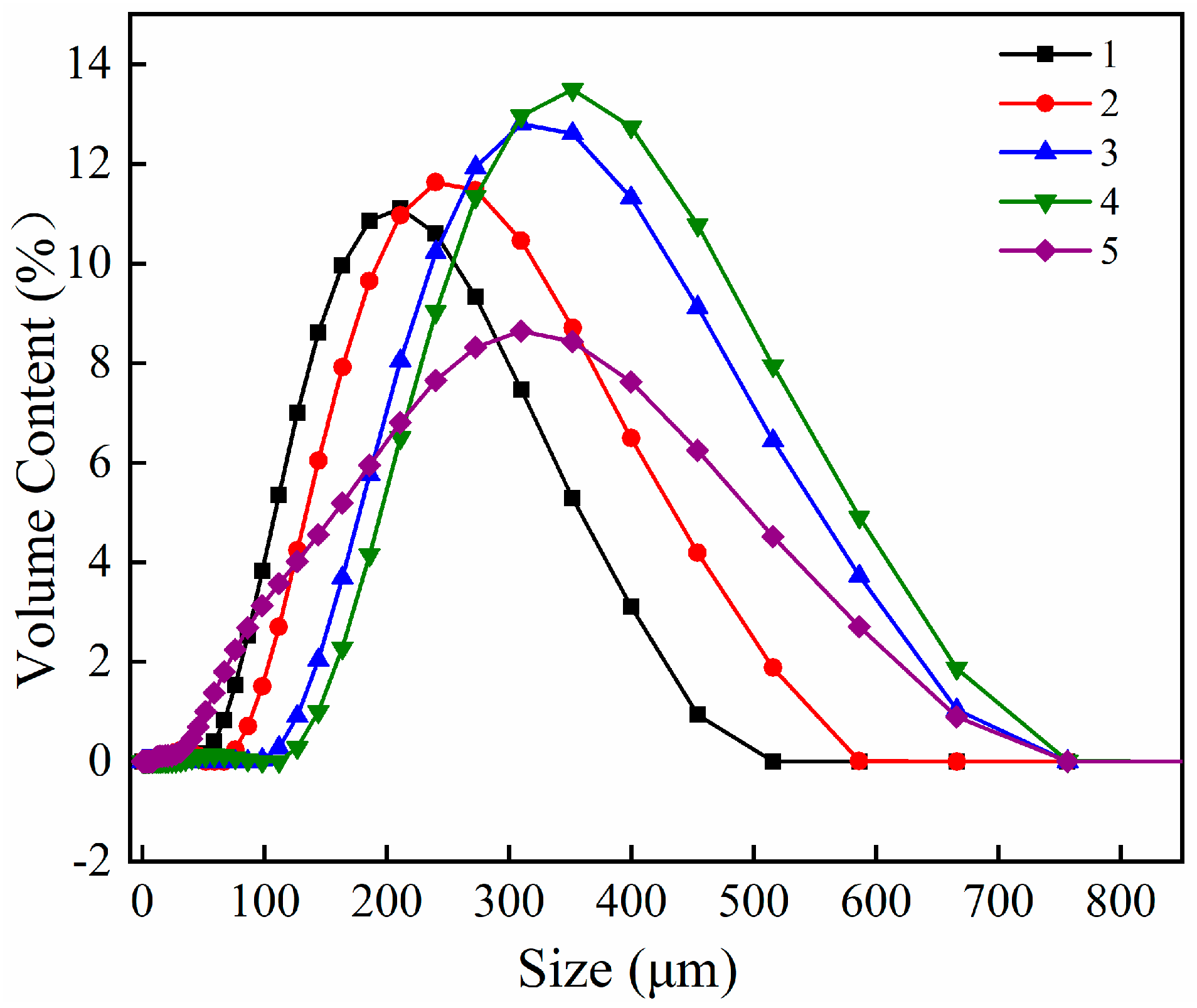
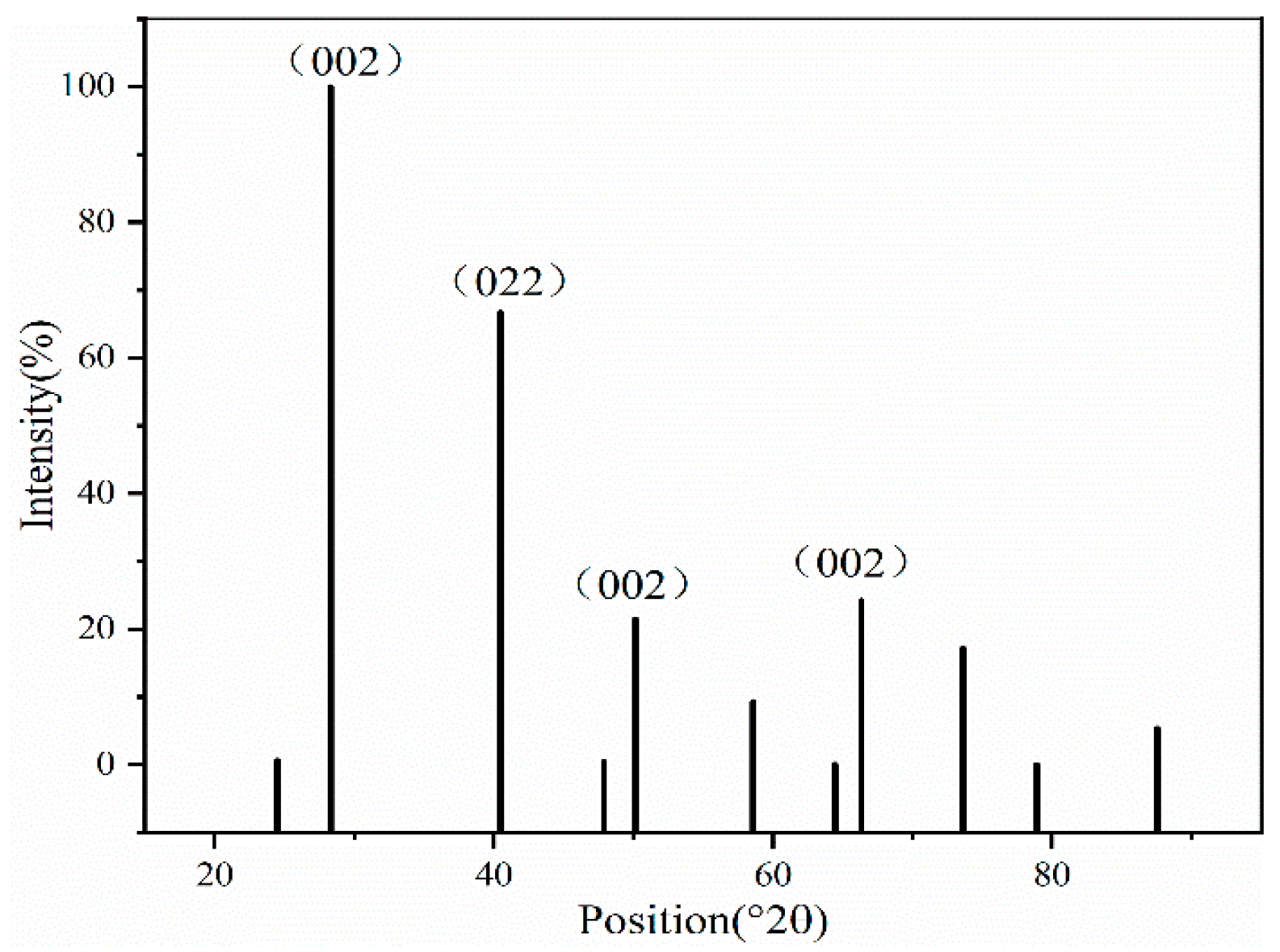
The XRD patterns of the KCl crystal during brine evaporation are shown in
Figure 6 and
Figure 7. The adsorption behavior of Na
+ on the KCl (002) surface of each crystal of KCl is calculated and analyzed using the first-nature principle to examine the adsorption intensity of Na
+ on the crystal surfaces. The crystal face model is constructed from the supercell structure of the crystal, fixing the atoms at the bottom of the surface of each of its faces, and adding a 15 Å thick vacuum layer in the Z-direction of the crystal face to prevent the neighboring layers from interacting with each other and affecting the results. Refs. [
31,
32] is defined as shown in Equation (1):
Eads is the adsorption energy;
Esurface+absorbate refers to total energy of absorbate on the crystal surface.
Esurface—is the crystal surface energy;
Eabsorbate is the energy of the adsorbed substance. According to this definition, negative values indicate exothermic and favorable adsorption processes, positive values indicate adsorptive and unfavorable adsorption processes [
33].
- (4)
Defect formation energy calculation
Defect formation can be used to assess the stability of the material defects. Formation energy is used to determine the stability of the doped system [
34].
The formation energy of the Na atom replacing the K atom and Na atom in KCl crystal and NaCl crystals (2) [
35,
36]:
where
E(
D) is the total energy of system containing defect
D;
E(
pure) is the total energy of the system with intact cells;
μK, μNa denote the chemical potentials of K atom and Na atom, respectively.
The formation energy of Na atoms in the interstitial sites of KCl crystals and NaCl crystals (3) [
35,
36]:
where
E(
D) is the total energy of the system with defect
D in charge state
q;
E(
pure) is the total energy of the system with intact cell;
EVBM is the valence band top of the defect-free system;
EF is the Fermi energy level; and
μNa denotes the K atomic chemical potential. The chemical potential of the Na atom in an Na-rich environment is (4):
The chemical potential of the Na atom in an Na-poor environment is (5):
where
E represents the energy of various bulk materials. Na-rich and Na-poor environments are those with the highest and lowest Na atomic chemical potentials, respectively.
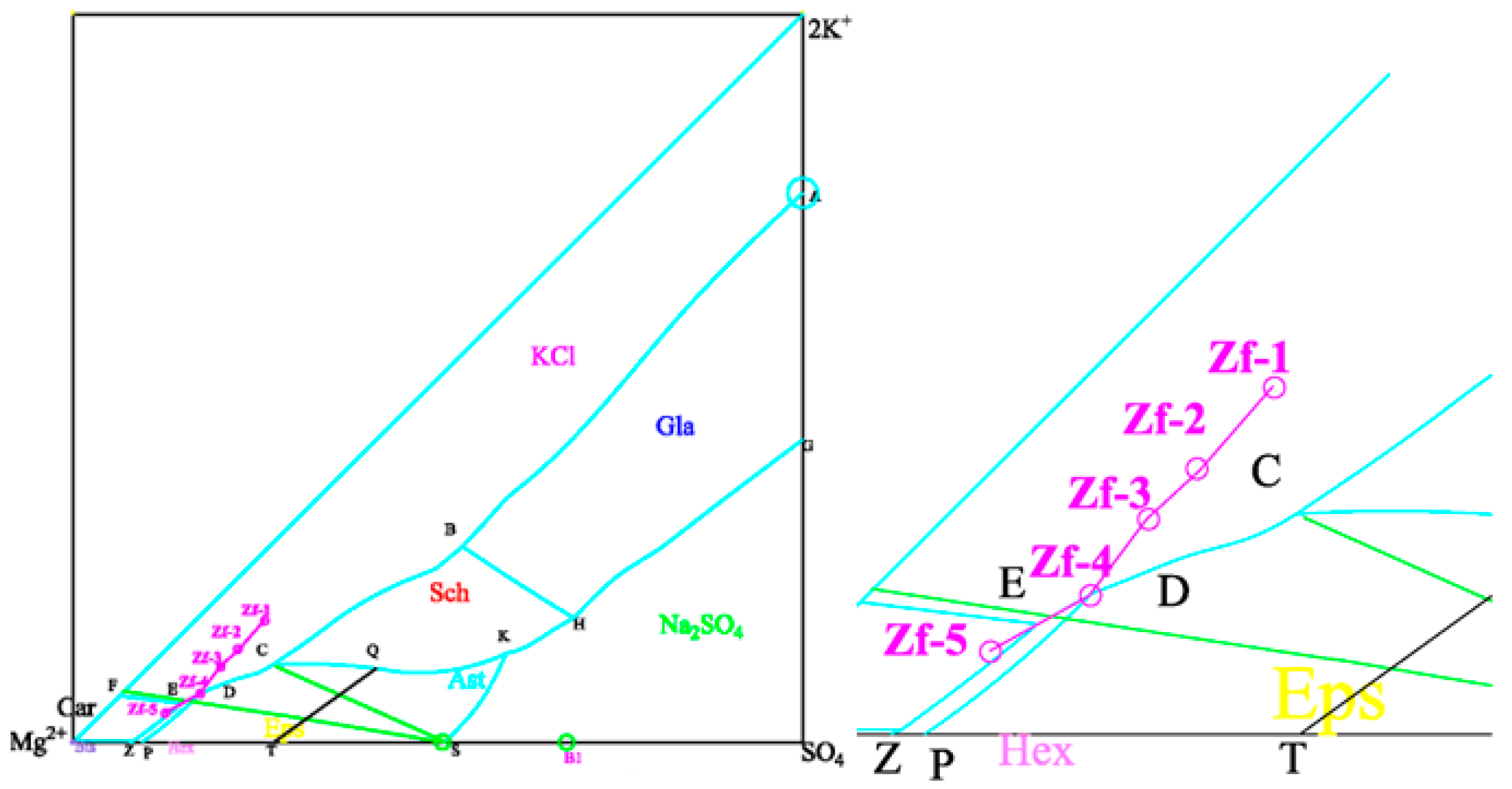
3.2.2. Study of Na Adsorption Behavior on the Surface of the Crystal Cell
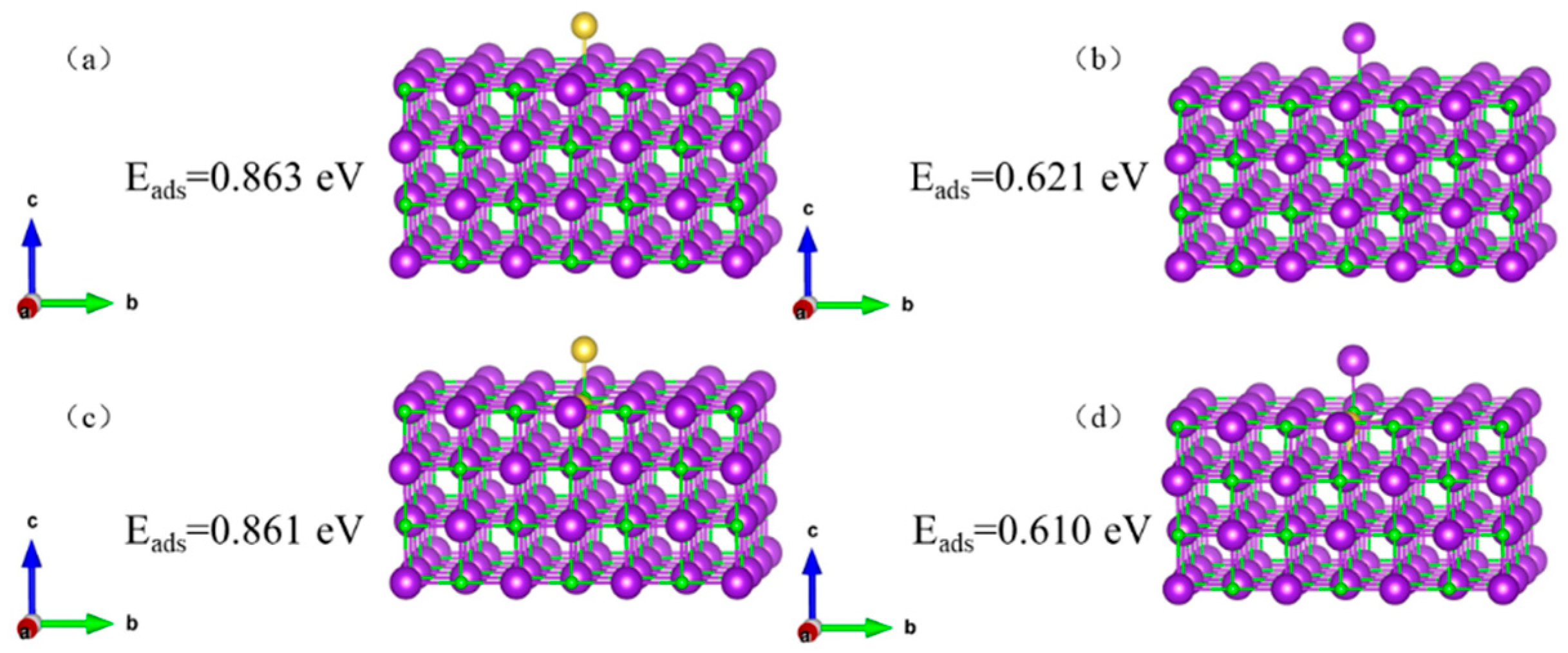
Adsorption structure and adsorption energy of K/Na on the KCl (002) surface and KCl (002) surface with K vacancy (Na replaces K) are calculated respectively, as shown in
Figure 8.
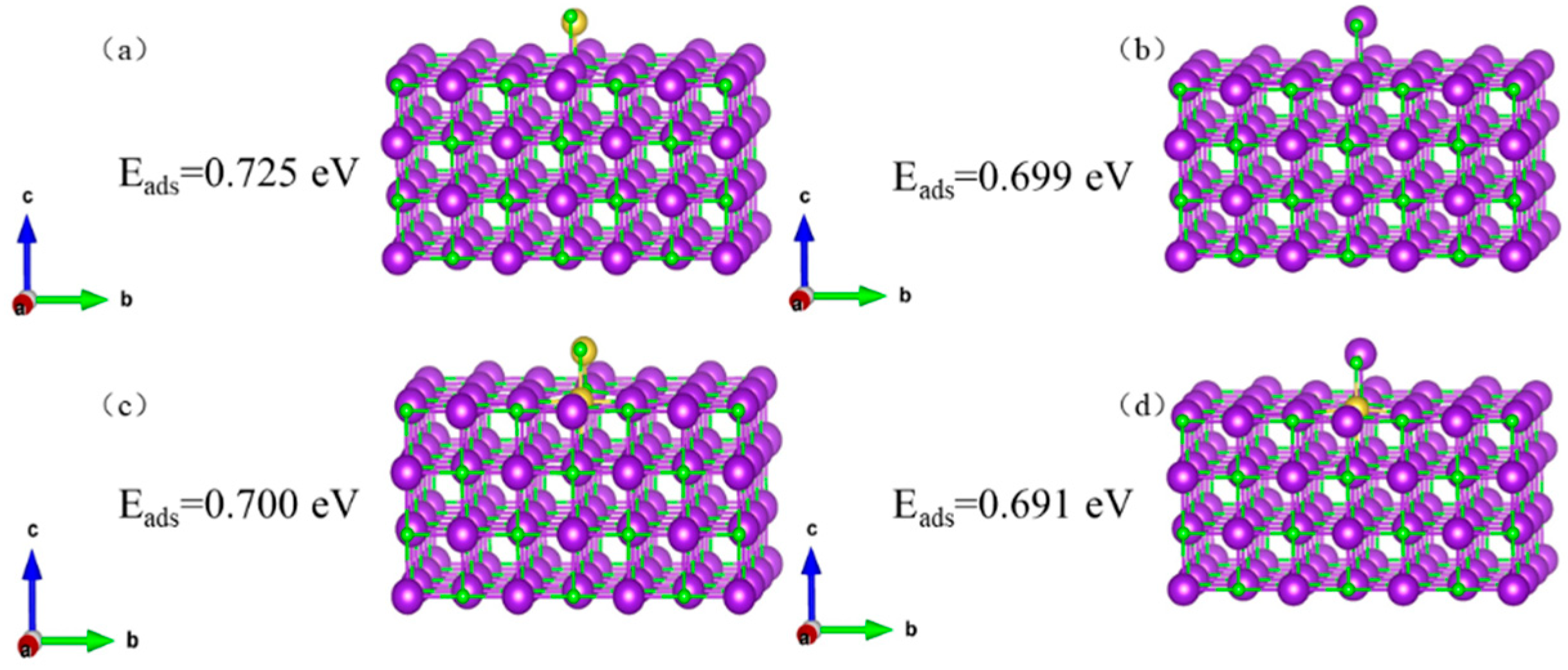
Adsorption structures and energies were calculated for KCl/NaCl on the KCl (002) surface and the KCl (002) surface with K replaced by Na, as shown in
Figure 9.
The adsorption behavior of the KCl crystal surface was studied, and the adsorption structures and adsorption energies of K
2Cl
2 and Na
2Cl
2 dimers on KCl (002) and KCl (002) surfaces were calculated, respectively, after K was replaced by Na, as shown in
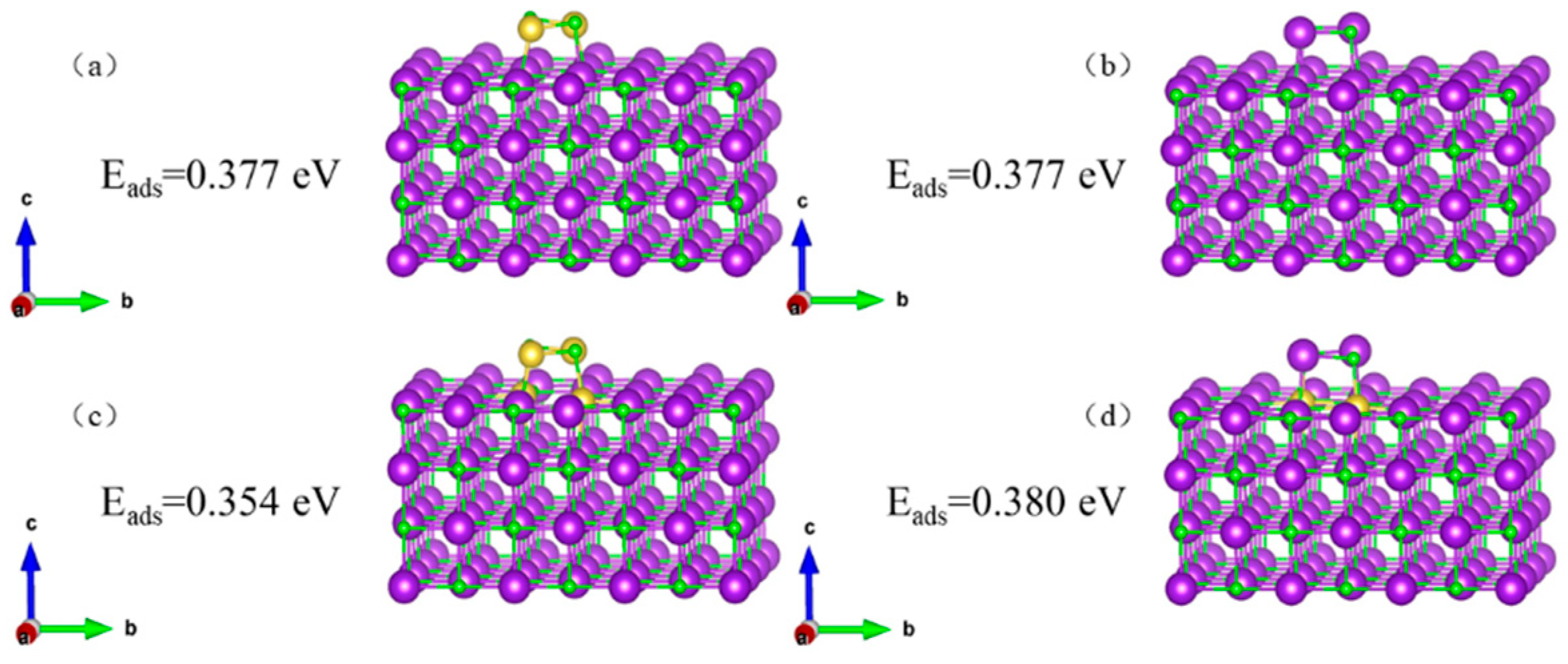
Figure 10.
Figure 8 shows that the KCl crystal has a stronger ability to adsorb K than to adsorb Na (0.863 eV > 0.621 eV), and the defective KCl crystal has a stronger ability to adsorb K than to adsorb Na (0.861 eV > 0.610 eV).
Figure 9 indicates that the KCl crystal has a slightly stronger ability to adsorb KCl than to adsorb NaCl (0.725 eV > 0.699 eV), and the defective KCl crystal has a slightly stronger ability to adsorb KCl than to adsorb NaCl (0.700 eV > 0.691 eV).
Figure 10 shows that the KCl crystal has an equal ability to adsorb K
2Cl
2 and Na
2Cl
2 (0.377 eV), while the defective KCl crystal has a weaker ability to adsorb 2 KCl compared to 2 NaCl (0.354 eV < 0.380 eV).
The ability of the KCl crystal to adsorb Na (0.863 eV), NaCl (0.725 eV), and 2 NaCl (0.377 eV) gradually increases, while the ability of the defective KCl crystal to adsorb Na (0.861 eV), NaCl (0.700 eV), and 2 NaCl (0.354 eV) also gradually increases.
It is more difficult for the non-defective KCl surface to adsorb Na atoms than K atoms (with a higher Na adsorption energy of 0.242 eV), and the adsorption energy for NaCl molecules is 0.026 eV higher than that for KCl, indicating that if single molecules or dimers of NaCl form in the solution, it favors the nucleation of NaCl.
Furthermore, the adsorption energy of NaCl on the defective KCl crystal surface is 0.009 eV higher than that of KCl, and the adsorption energy of Na2Cl2 dimer is 0.026 eV lower than that of KCl dimer. This indicates that the surface substitution of K by Na favors the nucleation of NaCl, and the growth of NaCl crystals is relatively easier (with a smaller difference in adsorption energy).
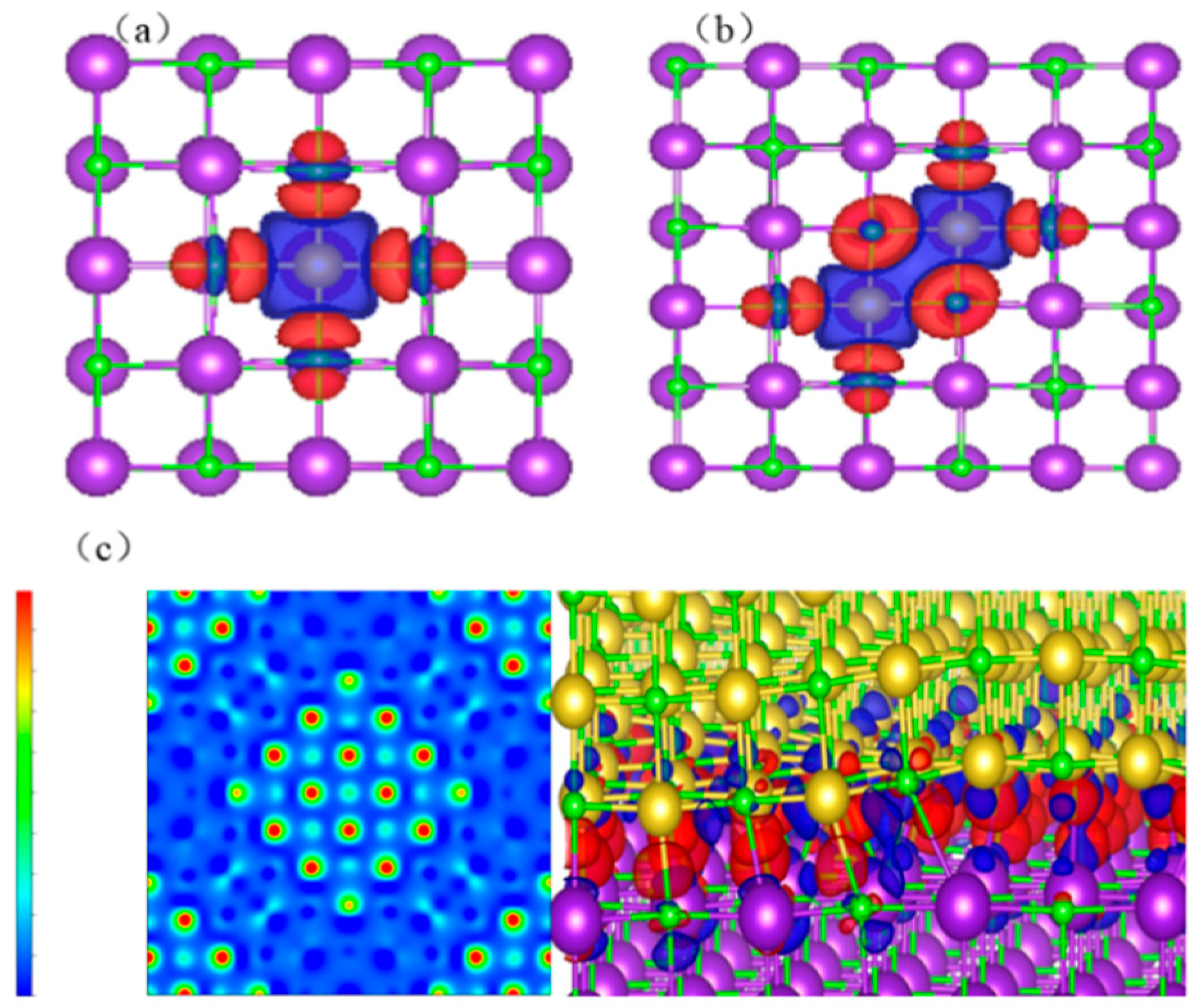
The differential charge density on the surfaces of KCl crystals, KCl (002) surfaces with K replaced by Na, and the heterojunction of KCl and NaCl crystals are shown in
Figure 11.
Through the aforementioned studies, it was observed that NaCl and KCl readily formed heterojunction structures in the coexisting system. The formation energies for heterojunctions and defective KCl crystals were both 0.49 eV, and the transfer of electrons between Na and K was nearly identical. However, from
Figure 6, it can be observed that the charge density distribution gradually increased for KCl crystals, defective KCl crystals, and the heterojunction structure of KCl and NaCl, confirming the hypothesis that NaCl and KCl form heterojunctions in the coexisting system.
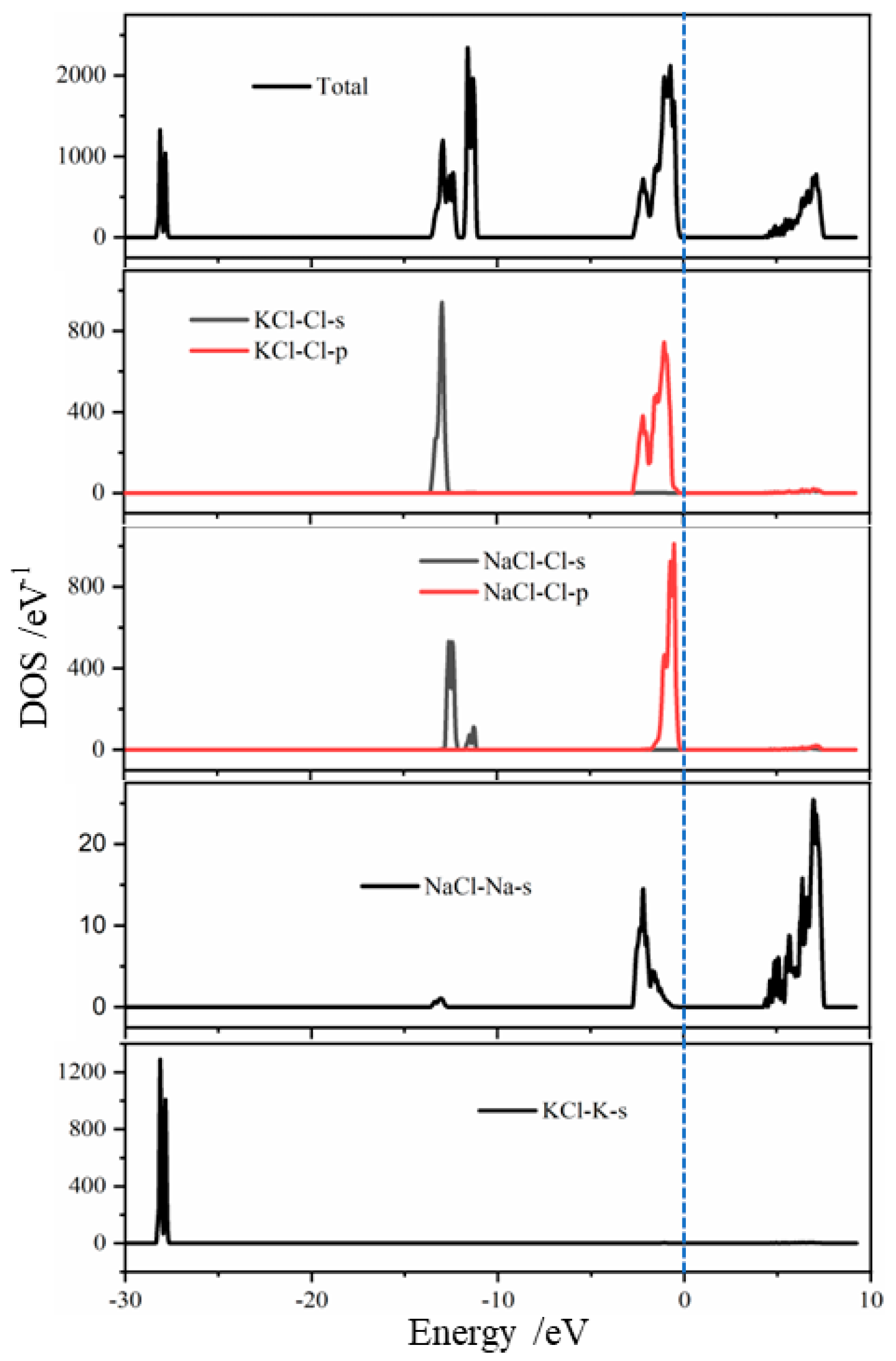
Density of states is defined as the number of electronic states that can occur within a unit energy interval. By analyzing the system’s density of states, we can gain further insights into each other’s mechanisms between ions and surface ions [
37]. In order to investigate the electronic structure changes of NaCl and KCl in greater depth, the density of states before and after adsorption on various crystal surfaces was calculated, using the energy zero point as the Fermi level. The results are shown in
Figure 12. There was a negative shift in the energy of K-s orbitals within the crystal, and the position of the Fermi level (5–10 eV) due to Na-s orbitals enhanced the overall density of states. This further confirmed the ease with which NaCl and KCl formed heterojunctions in the coexisting system.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}