Abstract
Lignans are widespread polyphenolic secondary plant metabolites possessing high biological activity. One of the most promising industrial-scale sources of such compounds is coniferous knotwood, containing a large number of polyphenolic compounds. Their use in pharmaceutical and other industries is limited by the difficulty in obtaining high-purity preparations from plant material and the requirement of advanced separation techniques. In this study, supercritical fluid chromatography on polar stationary phases was proposed for the efficient separation and identification of spruce, pine, fir, and larch knotwood extractives. Among the six tested sorbents, the best results were shown by silica with grafted diol and 2-ethylpyridine groups under conditions of gradient elution with a carbon dioxide–methanol mobile phase, which ensured the efficient retention and separation of analytes due to donor–acceptor interactions. Scaling up the method on a DIOL stationary phase provided a semi-preparative separation of extractives within 30 min to obtain 14 individual compounds with a purity of 90–99% and yields from 0.3 to 51% of the dry extract. These included eight lignans (nortrachelogenin, matairesinol, oxomatairesinol, α-conidendrin, 5-hydroxymatairesinol and its isomer, lariciresinol, and secoisolariciresinol), two oligolignans, three stilbenes (pinosylvin and its methyl ester, pterostilbene), and flavonoid taxifolin. The developed approach is distinguished with low operational costs, low consumption of organic solvents, environmental safety, and it is fully consistent with the principles of green chemistry.
1. Introduction
Lignans represent an extensive (more than one thousand compounds) group of polyphenolic plant secondary metabolites, the structure of which involves two phenylpropane units with a different degree of oxidation in the side-chain linked by a β–β′ carbon bond [1,2]. In addition to dimers, lignans also include the structures bearing three (sesquilignans), four (dilignans), and even more phenylpropane units. Due to their high biological activity, including estrogenic, antiviral, anticancer, antihypersensitive, and antioxidant properties [3,4,5], lignans attract increasing attention as promising precursors for the production of new generations of pharmaceuticals and dietary supplements. Chinese lemongrass (Schisandra chinensis), flax and sesame seeds, and some other plants containing lignans in amounts from 0.1 to 2% are commonly used as an industrial-scale source of these compounds. An important and still underestimated source of lignans is the compression wood of coniferous trees, located mainly in knots and root necks [6,7]. Active biosynthesis of lignans in this wood occurs in response to a mechanical stress and results in their extremely high content, reaching 20% [6,7]. The total number of lignans identified in coniferous knotwood reaches several tens, while their ratio varies widely depending on the tree species [8,9]. Their main representatives are secoisolariciresinol, 5-hydroxymatairesinol, nortrachelogenin, lariciresinol, todolactol, α-conidendrin, lignan A, and matairesinol (Figure 1). According to 2D NMR spectroscopy data [9], the first three compounds dominate (more than half of the total lignan content) in the compression wood of fir and larch, spruce, and pine, respectively.
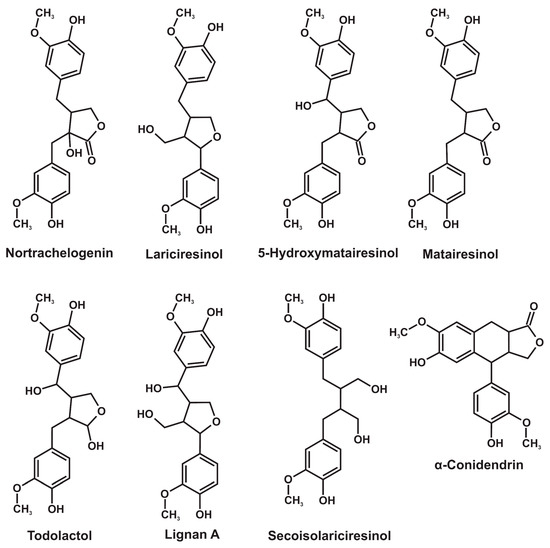
Figure 1.
Major representatives of coniferous knotwood lignans.
The use of knotwood as a feedstock for the production of lignans involves solving the problems of their isolation, separation from other extractives (steroids, flavonoids, low molecular weight fractions of lignin), as well as efficient separation to obtain pure individual compounds with the highest value. Methods for extracting lignans from plant materials are fairly well known and rely on maceration, ultrasonic extraction, supercritical fluid extraction (SFE) and pressurized liquid extraction techniques [10,11,12,13,14]. Ethanol, acetone, butanol and their mixtures with water or carbon dioxide (in the case of SFE) are used as extractants. At the same time, purification of obtained extracts and isolation of individual components is still a challenging task. The simplest approach to solving the problem is the co-precipitation of lignans from an ethanolic solution with potassium acetate [15,16], which makes it possible to obtain a 5-hydroxymatairesinol preparation from a spruce knot extract with a purity of about 90%. It is also known to precipitate lignans from alcohol solutions in the form of potassium or sodium salts upon the addition of concentrated solutions of KOH or NaOH, respectively [1]. Naturally, these methods are suitable for isolating a mixture of lignans, or a single component that is strongly dominant in the extract. To obtain a number of individual lignans, their chromatographic separation is required.
Numerous methods for the analytical and preparative chromatographic separation of lignans are described in the literature and summarized in well-known reviews [17,18]. Preparative thin layer chromatography [19,20] was used to obtain small amounts of analytes for further analysis, while larger-scale separations were most often performed by low- and medium-pressure normal-phase column chromatography on bare silica using dichloromethane/methanol, dichloromethane/acetone, chloroform/methanol/water, and ethyl acetate/n-hexane [21,22] as mobile phases. In addition, various authors also used reverse-phase HPLC, medium pressure chromatography on various non-polar stationary phases [17,23,24,25], and elution with methanol/water or acetonitrile/water systems. Given the rather high polarity of lignans, better retention and separation efficiency are achieved by using C8 or polar-functionalized sorbents instead of the common octadecyl stationary phase [17]. Another chromatographic method for the preparative separation of lignans is high-speed counter-current chromatography (HSCCC). Its advantage is that there is no need to use an expensive solid stationary phase and thus no problems occur with its contamination and poor separation reproducibility. HSCCC using two-phase solvent systems based on petroleum ether, ethyl acetate, alcohols, and water made it possible to isolate two new lignans from dandelion (Taraxacum mongolicum) [26] as well as to achieve simultaneous separation and purification of ten lignans extracted from water willow (Justicia procumbens) [27,28]. The drawback of all the mentioned chromatographic techniques is the significant consumption of organic solvents, most of which are toxic and flammable.
In our opinion, supercritical fluid chromatography (SFC) may be the most promising technique for separating lignans without the indicated disadvantages. Since the mobile phase in SFC consists of sub- or supercritical carbon dioxide, the eluting power of which is controlled by the addition of methanol (organic modifier), it is considered as a green separation technique also characterized by low operational costs [29,30]. Other important advantages of SFC are high separation speed and efficiency due to the low viscosity of the fluid compared to water and organic solvents, as well as selectivity due to the adsorption mechanism of analyte retention on polar stationary phases typical of SFC [30]. In the case of preparative separations, the use of SFC makes it possible to drastically reduce the complexity of isolating target compounds from the obtained fractions, since carbon dioxide spontaneously evaporates with decreasing pressure, providing a minimum volume of residual liquid (modifier and make-up solvent) in the receiving vessels. Despite the fact that, in recent years, SFC has been increasingly used for analytical and preparative separations of complex mixtures of plant metabolites [31,32,33,34], there are only a few examples of the use of this technique for lignans in the literature, and all of them are focused on the solution of purely analytical tasks. In this regard, two recent publications on SFC analysis of lignans in Chinese lemongrass extracts [13,35] as well as a work in which SFC was used in conjunction with reverse-phase and hydrophilic interaction HPLC for multi-stage separation of Fructus arctii lignans to obtain high-purity preparations [36] should be mentioned.
The present study is aimed at developing an approach to the rapid, environmentally friendly, and efficient separation of lignans from coniferous knotwood, as well as at obtaining on this basis pure preparations of a number of biologically active lignans isolated from the compression wood of spruce, fir, larch, and pine harvested in northern Europe on an industrial scale.
2. Materials and Methods
2.1. Chemicals and Materials
Hexane and acetone (chem. pure) were purchased from Komponent-Reaktiv (Moscow, Russia) and used in extraction procedures. Carbon dioxide (99.9%, BS Tekhnologiya, Arkhangelsk, Russia), HPLC gradient grade methanol and acetonitrile (Khimmed, Moscow, Russia), formic acid (ACS reagent, ≥96%, Sigma-Aldrich, Steinheim, Germany), and Type I ultra-pure Milli-Q water were used for the preparation of the mobile phase and sample solutions. Deuterated dimethyl sulfoxide (DMSO-d6, ≥99.8%, Merck, Darmstadt, Germany) was used as a sample solvent in NMR studies.
2.2. Plant Material and Extraction
The knotwood of four coniferous tree species typical for European North—pine (Pinus sylvestris), spruce (Picea abies), fir (Abies sibirica), and larch (Larix sibirica)—were chosen as the object of the study. The plant material were obtained from the trees with the age of 40–50, 60–70, 40–50, and 80–90 years, respectively, harvested in the Arkhangelsk region (Russia) for timber or pulp and paper industry. Parts of the tree trunk with the largest knots were sawn out at the harvesting site and delivered to the laboratory as quickly as possible (several days).
The inner part (located inside the tree trunk) of knots was drilled out and dried in a vacuum oven at 40 °C overnight. Then, the dry cuttings were additionally crushed on an ZM 200 centrifugal mill (Retsch, Haan, Germany) to attain a particle size of <1 mm. The prepared sawdust was carefully averaged and subjected to the two-stage Soxhlet extraction on a B-811 extractor (Buchi, Flawil, Switzerland). At the first stage (8 h), the sample (10 g) was de-resined with hexane and the non-polar compounds were removed (resin acids, lipids, terpenes). At the second stage, lignans and other phenolics were extracted with acetone for 8 h. The obtained acetone extract was concentrated on a rotary evaporator to a volume of 5–7 mL, diluted with deionized water to 100–150 mL, and thoroughly mixed. The resulting suspension was immediately frozen with liquid nitrogen and then dried in a FreeZone Triad (Labconco, Kansas City, MO, USA) freeze dryer. The obtained dry extracts, rich in lignans, were stored in glass vials at a temperature of 4 °C. The attained yields of extractives were 9.5, 16.1, 11.1, and 15.0% for oven-dried pine, fir, larch, and spruce knotwood samples, respectively.
2.3. Analytical SFC
The freeze-dried extracts were dissolved in methanol (0.25 mg mL–1) and centrifugated at 15,000 rpm immediately before SFC analysis. The chromatographic separation was carried out on a Nexera UC analytical SFC system (Shimazu, Kyoto, Japan), consisting of two LC-30AD HPLC pumps supplying organic modifier and make-up solvent, an LC-30ADSF carbon dioxide pump with a Peltier-cooled (4 °C) pump head, a CTO-20A column thermostat, an SIL-30AC autosampler, an SPD-M20A diode array UV-VIS spectrophotometric detector, and an SFC-30A back pressure regulator. The mobile phase was a mixture of carbon dioxide (A) and methanol (B, modifier) containing 0.1% formic acid. Neat methanol was used as a make-up solvent delivered through a T-tee to a capillary tubing after the back pressure regulator to avoid precipitation of analytes.
Six Aquity UPC2 (Waters, Milford, MA, USA) chromatographic columns (2.1 mm × 150 mm, particle size 1.7 µm) with different silica-based stationary phases were used: BEH (ethylene bridged hybrid silica), HSS C18 SB (grafted with octadecyl chains, non-endcapped), Torus DIOL (grafted ethylene glycol moieties), BEH 2-EP (grafted with 2-ethylpyridine groups), CSH Fluoro-Phenyl (grafted with propyl pentafluorophenyl groups), and HSS Cyano (grafted with cyanopropyl groups). The following chromatographic parameters were applied: mobile phase flow rate—1 mL min–1, make-up solvent flow rate—0.15 mL min–1, column temperature—40 °C, back pressure 150 bar, outlet valve temperature 50 °C, injection volume—2 µL. The separation was carried out in a gradient elution mode according to the following program: 0–3 min—10% B, 3–14 min—linear increase to 30% B, 14–15 min—30% B.
High-resolution mass spectrometry detection was carried out on a TripleTOF 5600+ (ABSciex, Concord, ON, Canada) quadrupole time-of-flight mass spectrometer equipped with a DuoSpray ion source working in negative ion atmospheric pressure chemical ionization mode (APCI–). The following ion source parameters were used: interface temperature (TEM)—500 °C, nebulizing gas pressure (GS2)—40 psi, curtain gas pressure (CUR)—25 psi, corona needle discharge voltage (ISVF) and declustering potential (DP)—4.5 kV and 80 V, respectively. Mass spectra were recorded in the m/z range of 150–1200. The mass scale was calibrated using sodium formate clusters to achieve the mass accuracy of 5 ppm in the entire m/z range. The obtained chromatograms were processed using the PeakView version 2.2 and FormulaFinder version 2.2 (ABSciex, Concord, ON, Canada) software.
2.4. Preparative SFC
Semi-preparative scale (tens of milligrams sample weight) separation was carried out using a Nexera UC Prep SFC preparative system (Shimadzu, Kyoto, Japan) consisting of an LC-20AP and an LC-40P SF preparative pump (maximum flow rate of 150 mL min–1) for the modifier and carbon dioxide supply, respectively, an LC-20AR make-up solvent pump, a CTO-40C column thermostat, an SIL-40C autosampler, an SFC-40P back pressure regulator, an FRC-40 fraction collector, and an SPD-40 UV-VIS spectrophotometric detector. Chromatographic separation was performed at 40 °C in a gradient elution mode on a Shim-pack US Diol II semi-preparative chromatographic column (Shimadzu, Kyoto, Japan), 250 × 10 mm, particle size 5 µm. A mixture of carbon dioxide (A) with methanol (B) containing 0.1% formic acid was used as an eluent with the following gradient program: 0–3 min—10% B, 3–25 min linear ramp to 35% B, 25–30 min—35% B. Neat methanol was used as a make-up solvent in a fraction collector. The mobile phase and make-up solvent flow rates were 20 and 2 mL min–1, respectively. The back pressure and outlet valve temperature were 130 bar and 50 °C, respectively, with injection volume of 500 μL. Detection was carried out at a wavelength of 280 nm.
All fractions were collected in 20 mL glass test tubes according to the fraction collector program established in a preliminary chromatographic run. Several (up to 30) consecutive chromatographic runs were used to separate 450–500 mg of the dry extract. After the separation, the fractions that contained the same component were combined, evaporated on a rotary evaporator to a volume of 0.5–1 mL, diluted with deionized water (20 mL), and thoroughly mixed. The suspensions were immediately frozen with liquid nitrogen and then freeze dried.
2.5. Analytical Methods
One-dimensional 1H and two-dimensional (2D) 1H-13C HSQC (heteronuclear single quantum correlation) and HMBC (heteronuclear multiple bond correlation) NMR spectra of the obtained fractions were recorded in DMSO-d6 (1–5 mg of a sample was dissolved in 550 µL of the solvent) at 298 K on an AVANCE III 600 spectrometer (Bruker, Ettlingen, Germany) with an operating frequency for protons of 600 MHz using sequences from the standard Bruker library. The Topspin 3.2 software (Bruker, Ettlingen, Germany) was used to register and process the experimental data. Cross-peak assignment to identify specific structures was performed by combining data from the HSQC and HMBC spectra using the ACD/Structure Elucidator expert system software version 2019 (ACD/Labs, Toronto, ON, Canada) including the NMR spectra database. The maximum permissible deviation for protons did not exceed 1 ppm, and for carbon atoms it did not exceed 5 ppm.
The purity assessment by HPLC with UV-VIS spectrophotometric detection was carried out on a Nexera LC-30 system (Shimazu, Kyoto, Japan), consisting of two LC-30AD chromatographic pumps, a CTO-20A column thermostat, a SIL-30AC autosampler, and an SPD-M20A diode array detector. A 0.5 mg sample was dissolved in 1 mL of a mixture of acetonitrile with water (50/50) and injected (2 μL) to the HPLC system. The mobile phase was a mixture of water (A) and acetonitrile (B), both containing 0.1% formic acid. Chromatographic separation was carried out in a reversed phase mode on a Nucleodur PFP column (Macherey-Nagel, Duren, Germany), 150 × 2.1 mm, particle size 1.8 μm, with a propyl pentafluorophenyl stationary phase. The mobile phase flow rate was 0.25 mL min–1, the column oven temperature was 40 °C. The following gradient elution program was used: 0–2 min—15% B, 2–25 min—a linear increase to 100% B, 25–30 min—100% B. UV-VIS spectra acquisition was carried out in the wavelength range of 200–500 nm with a frequency of 10 Hz and a spectral slit width of 4 nm. The purity was calculated as a ratio of the peak areas of the target compound and all other eluting components detected at 280 nm.
3. Results and Discussion
3.1. Column Screening and SFC Separation Conditions
Due to the wide variety of sorbents with different chemistries used in SFC, the key issue in the development of the separation method is the proper selection of a stationary phase that provides acceptable retention and efficient separation of analytes. For this purpose, at the first stage of the study, six widely used SFC columns (Section 2.3) with the same geometry and different stationary phases (BEH silica, HSS C18 SB, Torus DIOL, BEH 2-EP, CSH Fluoro-Phenyl, HSS Cyano) were tested with the same gradient elution profile and chromatographic conditions. The initial content (10%) of the organic modifier, which makes it possible to avoid the precipitation of lignans poorly soluble in carbon dioxide, was established in preliminary experiments.
The obtained chromatograms on the example of the spruce knotwood extract (Figure 2) allowed for distinguishing two groups of stationary phases. The first one is characterized with the unacceptably low retention of analytes and represented by the three sorbents bearing low-polar moieties (octadecyl, pentafluorophenyl, and cyanopropyl) and capable of mainly non-polar and π-π interactions with analytes. It should be noted that, in the case of non-endcapped HSS C18 SB, the significant, and in some cases decisive, contribution to the retention of analytes with polar groups in SFC is provided by the residual silanols while octadecyl grafting plays an auxiliary role [31].
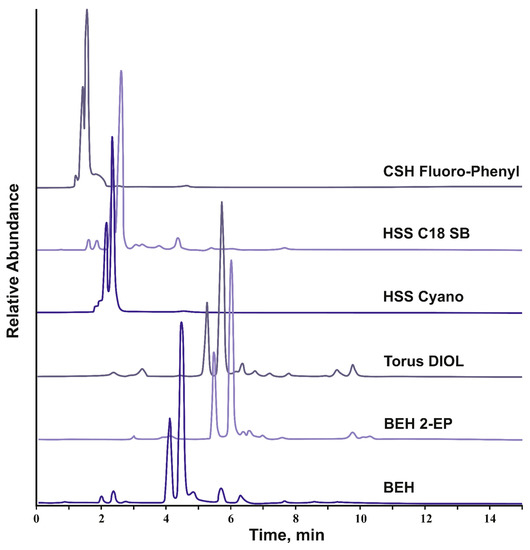
Figure 2.
SFC-APCI(-)-HRMS total ion current chromatograms of the spruce knotwood extract on CSH Fluoro-Phenyl, HSS C18 SB, HSS Cyano, Torus Diol, BEH 2-EP, and BEH silica columns under the same chromatographic conditions (linear ramp of methanol content from 10 to 30%, flow rate 1 mL min–1, column temperature —40 °C, back pressure 150 bar).
This is the reason for the better retention of analytes on octadecyl silica compared to the other two stationary phases of this group. It is quite natural that, in the second group of columns, represented by polar sorbents (BEH silica, Torus DIOL, and BEH 2-EP) and characterized by the highest retention of analytes, bare ethylene hybrid silica is closest to the octadecyl stationary phase, since the k values of analytes turned out to be significantly lower compared to DIOL and 2-EP. The latter two stationary phases have significant advantages in the retention and separation selectivity of the extractive substances and thus may be successfully used in analytical and preparative separations.
In general, the observed differences in analytes’ retention between the tested SFC columns are well explained involving the stationary phase classification based on the linear solvation energy relationship (LSER) concept and Abraham descriptors [37,38]. Thus, there is an apparent correlation between the retention factors of analytes (k’) and the value of the descriptor a in the Abraham’s equation. The latter characterizes the ability of the stationary phase to act as a hydrogen bond acceptor (basicity) when interacting with acidic analytes. According to the literature [37,39,40], by the value of this parameter, the tested sorbents can be arranged in the order CSH Fluoro-Phenyl < HSS C18 SB < HSS Cyano < BEH silica << BEH 2-EP < Torus DIOL, mostly coinciding with the retention of lignans. An exception is the HSS C18 SB/HSS Cyano and BEH 2-EP/Torus DIOL pairs, for which descriptor a values of 0.48/0.71 and 1.39/1.95, respectively, were reported. However, both sorbents, considered the most suitable for separating lignans, belong to the same SFC stationary phase group distinguishing with a high proton-acceptor ability [37]. This means that the predominant mechanism of lignan retention is specific donor–acceptor interactions with the stationary phase, in which phenolic hydroxyl groups of analytes act as proton donors. Naturally, this does not negate the possibility of implementing other types of interactions (π-π, dipole–dipole) whose contribution to the mixed retention mechanism turned out to be significantly smaller.
Of the two proposed stationary phases with similar analyte retention characteristics, a DIOL phase was chosen for further studies since, despite a slightly lower retention, it provided somewhat better chromatographic resolution of the peaks of some minor components. An important factor in this choice was also the lower cost and greater availability of sorbents with a grafted polyethylene glycol phase for HPLC/SFC separations supplied by various manufacturers.
To select separation conditions on a Torus DIOL column, the effects of back pressure, temperature, and gradient profile on the selectivity of separation of the knotwood extractives separation were estimated. The influence of the two former parameters, when varied within the reasonable range (100–200 bar and 30–50 °C) limited by the SFC system characteristics, as well as the need to maintain sufficient dissolving power of the mobile phase and chemical stability of the sorbent, turned out to be insignificant when compared with the mobile phase composition. This agrees well with the recent results of Ovchinnikov et al. [39], demonstrating the secondary role of pressure and temperature in optimizing the SFC of separations on polar stationary phases. The selection of the gradient of methanol in carbon dioxide was based on the need to ensure complete separation of the main analytes in the shortest time. Considering the indicated criteria, the following SFC parameters were used in further analytical separations of the knotwood extractives: linear ramp of methanol content from 10 to 30%, run time 15 min, flow rate 1 mL min–1, column temperature 40 °C, and back pressure 150 bar. The latter three values are typical for SFC separations.
3.2. Analytical Separation of Knotwood Extractives and Peak Assignment
An SFC-APCI(-)-HRMS analysis of the obtained extracts of spruce, fir, pine, and larch knotwood under the proposed chromatographic conditions showed the presence of 23 major components (Table 1), presumably identified by their exact masses and tandem mass-spectra using literature data [8,9,17,41]. Nineteen of them are represented by lignans (including four sesquilignans and one dilignan). The remaining four compounds of non-lignan nature were identified as stilbenes pinosylvin, its monomethyl ether, and pterostilbene (detected in pine knotwood), as well as the flavonoid taxifolin, which is present in large quantities in larch knotwood extract.

Table 1.
Results of SFC-APCI(-)-HRMS analysis of coniferous knotwood extracts.
Of the four studied tree species, spruce had the highest content and variety of major components (16 lignans) of the knotwood extract. It is characterized by the predominance of 5-hydroxymatairesinol (~75% of the total peak area) and its isomer (9%). The nortrachelogenin peak dominates in the chromatogram of the pine knotwood extract (>80% of the total peak area), while the remaining components are mainly represented by the already mentioned pinosylvin, as well as olivil and matairesinol. The extracts of fir and larch are distinguished with the predominance of secoisolariciresinol. However, they also contain 5-hydroxymatairesinol and its isomer (fir) and nortrachelogenin (larch) in comparable amounts. It should be noted that three sesquilignans with elemental compositions C30H36O10, C30H36O9, and C30H38O10, as well as dilignan C40H50O12 are also among the main components of the fir knotwood extract. Their reliable mass spectrometric identification without isolating pure preparations is difficult.
In general, the results presented in Table 1 are in a good agreement with the previously obtained data from two-dimensional NMR spectroscopy and high-performance liquid chromatography–high-resolution mass spectrometry [9]. At the same time, a distinctive feature of SFC is a higher separation speed and increased selectivity in the separation of compounds with close polarities. The latter factor is achieved in SFC due to the implementation of the adsorption retention mechanism based on specific donor–acceptor interactions of analytes with the stationary phase. An illustrative example of this advantage of SFC is the baseline separation of the two main components of the spruce knotwood extract—5-hydroxymatairesinol and its isomer—co-eluting under reversed-phase HPLC conditions [9].
3.3. Method Scaling and Semi-Preparative Fractionation of Extracts
In separation scaling to a semi-preparative level, a Shim-pack US Diol II chromatographic column (250 × 10 mm) with a polyethylene glycol-grafted stationary phase similar to that in analytical SFC column was used. Some loss of separation efficiency due to the larger particle size of the sorbent was partially compensated by an increase in the length of the column. Preliminary tests showed that, in general, the transition from one diol-type stationary phase to another did not lead to changes in the retention order of the components or substantial deterioration in chromatographic resolution at comparable sample loadings. An exception is the observed inversion of the elution order of two analytes—nortrachelogenin and hydroxymatairesinol isomer.
The key issue in the chromatographic method scaling is in maintaining an acceptable resolution of the target peaks under conditions of extremely high analyte content in the injected sample (concentration and volume overload), which is typical for preparative separations [42]. The maximum allowed amount of an injected sample in SFC is also limited by the solubility of its components in the non-polar supercritical fluid. Preliminary experiments showed that the maximum concentration of the knotwood extractives in the injected sample should not exceed 50 mg mL–1 for pine and 20 mg mL–1 for other coniferous tree species. Otherwise, a sharp increase in the system pressure and a malfunction of the injection valve would occur due to the precipitation of the most polar compounds when the sample solution is mixed with a mobile phase based on non-polar carbon dioxide. Even when working with the indicated limiting contents of extractive substances, periodic short-term flushing of the SFC system with a mixture of methanol and water (50/50) is required to effectively clean the chromatographic tract.
The use of the established maximum sample concentrations and an injection volume of 0.5 mL (typical for the used column geometry) provides a sample loading of 10 to 25 mg on column, which is at least four orders of magnitude higher compared to analytical separation. Concentration overloading leads to a significant broadening of the chromatographic peaks and a loss of chromatographic resolution, which drastically reduces the number of extract components that can be isolated with a sufficient degree of purity. To overcome this problem, the gradient elution profile was modified by reducing the gradient rate, which led to an increase in the separation time to 30 min (Section 2.4). This allowed for the baseline separation of the most components, although a few compounds were not completely separated. The attained separation efficiency made it possible to obtain a total of 33 fractions dissolved in methanol, including five pine, eight spruce, nine fir, and eleven larch knotwood major extractives (Figure 3).
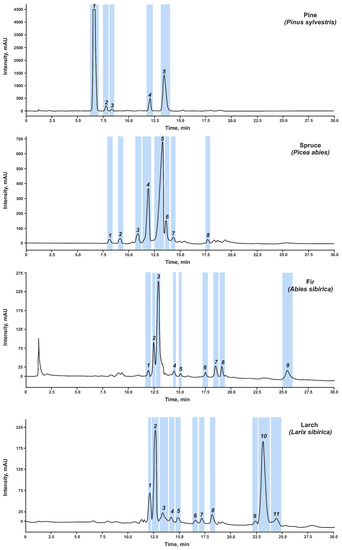
Figure 3.
Semi-preparative scale SFC-UV (280 nm) chromatograms of coniferous knotwood extracts and fraction collection zones (highlighted in blue and numbered).
It is worth noting that, despite the increased duration of the chromatographic run, the consumption of solvents in this case is only 140 g of methanol and 225 g of CO2, which ensures more than 100 consecutive separations using a common 40 L cylinder of food-grade carbon dioxide. Methanol can be easily recycled through distillation of eluent wastes and evaporation of fractions since they do not contain water or other organic solvents which are required for reversed or normal-phase HPLC, as well as counter-current chromatography. This allows for the consideration of the proposed method for separating knotwood extractives as consistent with the principles of green chemistry and significantly superior in environmental friendliness to other chromatographic approaches.
3.4. Chemical Composition the Obtained Fractions
All the obtained 33 fractions were accumulated in quantities from units to hundreds of milligrams during repeated semi-preparative separations of each extract and then were tested for their purity by reversed-phase HPLC with spectrophotometric detection. The choice of this technique was due to the “orthogonality” of separations in the implementation of reversed-phase and adsorption mechanisms, which makes it possible to separate compounds co-eluting under SFC conditions. The obtained results showed that 16 predominantly minor fractions (Supplementary Table S1) did not meet our purity requirements and contained from 30 to 86% of the main substance due to partial overlapping of neighboring chromatographic peaks and co-eluting of components. Their further purification can be carried out using additional SFC separations after appropriate optimization of chromatographic conditions. Seventeen other fractions had a main substance content of more than 90% (Table 2), and ten were distinguished by a purity in the range of 95–99%, which is quite sufficient even for their use as analytical standards.
Table 2.
High-purity (≥90%) preparative SFC fractions of coniferous knotwood extractives.
The isolated compounds were identified by high-resolution mass spectrometry (determination of elemental composition) and 1D (1H) and 2D (1H-13C) NMR spectroscopy (Supplementary Table S2, Figures S1–S17), which made it possible to fully confirm the correct assignment of peaks in the SFC-MS analysis (Table 1). As can be seen from Table 2, all lignans dominated in extracts (nortrachelogenin, 5-hydroxymatairesinol, and secoisolariciresinol) were successfully isolated as high-purity preparations with the yields close to their contents estimated earlier by 2D NMR [9]. Thus, for example, one of the most important biologically active lignans, 5-hydroxymatairesinol, can be obtained as a result of single SFC separation in amounts exceeding 50% of the spruce knotwood extract with a purity of 99%. Obtaining pure preparations of minor lignans (hydroxymatairesinol isomer, matairesinol, lariciresinol, α-conidendrin, and oxomatairesinol) is no less important since these compounds are either commercially unavailable or extremely expensive due to the difficulty of obtaining them synthetically. In this regard, special attention should be paid to the possibility of one-step isolation of two secoisolariciresinol-derived oligolignans from fir and larch knotwood—sesquilignan C30H38O10 and dilignan C40H50O12. According to NMR spectra they were identified as guaiacyl glycerol ether of secoisolariciresinol and secoisolariciresinol dimer with aryl-aryl (5-5) bonding (Figure 4). The former compound was identified earlier in the wood of Moroccan fir (Abies Marocana) and was named sesquimarocanol B [43,44], while the latter one was first isolated in the present study. It should be noted that the similar dimeric lignan formed by unsymmetrical 5-5 coupling of 5-hydroximatairesinol was described earlier as a component of spruce knotwood extract [45].
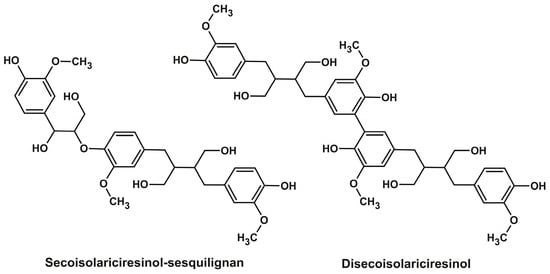
Figure 4.
Structural formulas of the oligolignans isolated from fir and larch knotwood extracts.
The last, but not least, group of isolated compounds is represented by non-lignan extractives, namely, flavonoid taxifolin and three stilbene derivatives—pterostilbene, pinosylvin and its methyl ether. The latter two compounds are of the greatest interest since they were isolated as preparations with a high purity (99%) and a total yield of about 50% by weight of the pine knotwood extract. Considering the antimicrobial, anti-inflammatory, antioxidant, anti-fungi, and even anti-cancer activities of pinosylvin, this compound can be claimed by the pharmaceutical industry [46]. Given the lack of effective technologies for its isolation and synthesis, the proposed SFC approach can be considered as very promising and economically justified for use on an industrial scale along with the production of lignans.
4. Conclusions
Supercritical fluid chromatography on bonded DIOL and 2-ethyl pyridine stationary phases provides fast and highly selective separation of lignans due to specific donor–acceptor interactions of analytes with the active sites of the sorbent. The use of this technique for the analysis of extractive substances of spruce, pine, fir, and larch knotwood in the gradient elution mode in combination with spectrophotometric and mass spectrometric detection made it possible to detect and tentatively identify 23 major components, including 19 lignans. The method scaling with additional tuning of the gradient elution profile allowed for a semi-preparative separation of the main components of the extracts with a chromatographic run duration of 30 min and a sample loading of up to 25 mg on a 10 mm (i.d.) column. The analysis of the obtained fractions showed the possibility of single-stage isolation of eight lignans with a purity of >90%, while, in the case of the dominant components (nortrachelogenin, 5-hydroxymatairesinol, and secoisolariciresinol), it is 98–99%. Along with lignans, in the case of larch and pine knotwood extracts, SFC makes it possible to obtain preparations of the flavonoid taxifolin and stilbenes, including pinosylvin and its methyl ether with a high yield and a purity of 99%. The proposed approach is distinguished with simplicity, low consumption of organic solvents, and environmental friendliness, which is consistent with the principles of green chemistry. Its further scaling will make it possible to develop industrial technologies for processing wood wastes to obtain a number of valuable polyphenolic compounds that have high potential to be used in the pharmaceutical industry and other applications.
Supplementary Materials
The following supporting information can be downloaded at: https://www.mdpi.com/article/10.3390/separations10080449/s1, Table S1: Low-purity preparative SFC fractions of coniferous knotwood extractives; Table S2: 1H NMR spectra of the isolated pure compounds; Figure S1: 1H-13C HSQC NMR spectrum of Fraction 1 isolated from pine (Pinus Sylvestris) extract; Figure S2: 1H-13C HSQC NMR spectrum of Fraction 2 isolated from pine (Pinus Sylvestris) extract; Figure S3: 1H-13C HSQC NMR spectrum of Fraction 4 isolated from pine (Pinus Sylvestris) extract; Figure S4: 1H-13C HSQC NMR spectrum of Fraction 5 isolated from pine (Pinus Sylvestris) extract; Figure S5: 1H-13C HSQC NMR spectrum of Fraction 1 isolated from spruce (Picea abies) extract; Figure S6: 1H-13C HSQC NMR spectrum of Fraction 2 isolated from spruce (Picea abies) extract; Figure S7: 1H-13C HSQC NMR spectrum of Fraction 3 isolated from spruce (Picea abies) extract; Figure S8: 1H-13C HSQC NMR spectrum of Fraction 4 isolated from spruce (Picea abies) extract; Figure S9: 1H-13C HSQC NMR spectrum of Fraction 5 isolated from spruce (Picea abies) extract; Figure S10: 1H-13C HSQC NMR spectrum of Fraction 1 isolated from fir (Abies sibirica) extract; Figure S11: 1H-13C HSQC NMR spectrum of Fraction 2 isolated from fir (Abies sibirica) extract; Figure S12: 1H-13C HSQC NMR spectrum of Fraction 3 isolated from fir (Abies sibirica) extract; Figure S13: 1H-13C HSQC NMR spectrum of Fraction 7 isolated from fir (Abies sibirica) extract; Figure S14: 1H-13C HSQC NMR spectrum of Fraction 9 isolated from fir (Abies sibirica) extract; Figure S15: 1H-13C HSQC NMR spectrum of Fraction 2 isolated from larch (Larix sibirica) extract; Figure S16: 1H-13C HSQC NMR spectrum of Fraction 8 isolated from larch (Larix sibirica) extract; Figure S17: 1H-13C HSQC NMR spectrum of Fraction 10 isolated from larch (Larix sibirica) extract.
Author Contributions
Conceptualization, D.S.K. and N.V.U.; methodology, D.S.K. and N.V.U.; validation, N.S.G. and D.V.O.; investigation, A.A.O., A.V.F., N.V.U., and D.V.O.; resources, N.V.U.; data curation, N.S.G. and A.V.F.; writing—original draft preparation, A.A.O. and N.V.U.; writing—review and editing, D.S.K.; visualization, N.V.U. and N.S.G.; supervision, N.V.U. and D.S.K.; project administration, N.V.U.; funding acquisition, N.V.U. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Russian Science Foundation and the Government of the Arkhangelsk Region (grant No. 22-13-20071).
Data Availability Statement
The data presented in this study are available in the article and Supplementary Materials.
Acknowledgments
This study was performed using an instrumentation of the Core Facility Center “Arktika” of the Northern (Arctic) Federal University.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Calvo-Flores, F.G.; Dobado, J.A.; Isac-García, J.; Martin-Martinez, F.J. Lignin and Lignans as Renewable Raw Materials: Chemistry, Technology and Applications; John Wiley & Sons, Ltd.: Chichester, West Sussex, UK, 2015; 512p. [Google Scholar] [CrossRef]
- Teponno, R.B.; Kusari, S.; Spiteller, M. Recent advances in research on lignans and neolignans. Nat. Prod. Rep. 2016, 33, 1044–1092. [Google Scholar] [CrossRef]
- Zálešák, F.; Bon, D.J.-Y.D.; Pospíšil, J. Lignans and Neolignans: Plant secondary metabolites as a reservoir of biologically active substances. Pharmacol. Res. 2019, 146, 104284. [Google Scholar] [CrossRef]
- Sun, W.; Shahrajabian, M.H. Therapeutic Potential of Phenolic Compounds in Medicinal Plants—Natural Health Products for Human Health. Molecules 2023, 28, 1845. [Google Scholar] [CrossRef]
- Wang, L.-X.; Wang, H.-L.; Huang, J.; Chu, T.-Z.; Peng, C.; Zhang, H.; Chen, H.-L.; Xiong, Y.-A.; Tan, Y.-Z. Review of lignans from 2019 to 2021: Newly reported compounds, diverse activities, structure-activity relationships and clinical applications. Phytochemistry 2022, 202, 113326. [Google Scholar] [CrossRef]
- Willför, S.; Hemming, J.; Reunanen, M.; Eckerman, C.; Holmbom, B. Lignans and Lipophilic Extractives in Norway Spruce Knots and Stemwood. Holzforschung 2003, 57, 27–36. [Google Scholar] [CrossRef]
- Willför, S.; Nisula, L.; Hemming, J.; Reunanen, M.; Holmbom, B. Bioactive phenolic substances in industrially important tree species. Part 1: Knots and stemwood of different spruce species. Holzforschung 2004, 58, 335–344. [Google Scholar] [CrossRef]
- Kumar, R.; Tsvetkov, D.E.; Varshney, V.K.; Nifantiev, N.E. Chemical constituents from temperate and subtropical trees with reference to knotwood. Ind. Crops Prod. 2020, 145, 112077. [Google Scholar] [CrossRef]
- Ul’yanovskii, N.V.; Onuchina, A.A.; Faleva, A.V.; Gorbova, N.S.; Kosyakov, D.S. Comprehensive Characterization of Chemical Composition and Antioxidant Activity of Lignan-Rich Coniferous Knotwood Extractives. Antioxidants 2022, 11, 2338. [Google Scholar] [CrossRef] [PubMed]
- Patyra, A.; Kołtun-Jasion, M.; Jakubiak, O.; Kiss, A.K. Extraction Techniques and Analytical Methods for Isolation and Characterization of Lignans. Plants 2022, 11, 2323. [Google Scholar] [CrossRef] [PubMed]
- Willför, S.M.; Ahotupa, M.O.; Hemming, J.E.; Reunanen, M.H.T.; Eklund, P.C.; Sjöholm, R.E.; Eckerman, C.S.E.; Pohjamo, S.P.; Holmbom, B.R. Antioxidant Activity of Knotwood Extractives and Phenolic Compounds of Selected Tree Species. J. Agric. Food Chem. 2003, 51, 7600–7606. [Google Scholar] [CrossRef] [PubMed]
- Razgonova, M.; Zakharenko, A.; Pikula, K.; Kim, E.; Chernyshev, V.; Ercisli, S.; Cravotto, G.; Golokhvast, K. Rapid Mass Spectrometric Study of a Supercritical CO2-extract from Woody Liana Schisandra chinensis by HPLC-SPD-ESI-MS/MS. Molecules 2020, 25, 2689. [Google Scholar] [CrossRef]
- Dai, Z.; Xin, H.; Fu, Q.; Hao, H.; Li, Q.; Liu, Q.; Jin, Y. Exploration and optimization of conditions for quantitative analysis of lignans in Schisandra chinensis by an online supercritical fluid extraction with supercritical fluid chromatography system. J. Sep. Sci. 2019, 42, 2444–2454. [Google Scholar] [CrossRef]
- Lee, S.; Ban, H.S.; Kim, Y.P.; Kim, B.K.; Cho, S.H.; Ohuchi, K.; Shin, K.H. Lignans from Acanthopanax chiisanensis having an inhibitory activity on prostaglandin E2 production. Phytother. Res. 2005, 19, 103–106. [Google Scholar] [CrossRef] [PubMed]
- Freudenberg, K.; Knof, L. Die Lignane des Fichtenholzes. Chem. Ber. 1957, 90, 2857–2869. [Google Scholar] [CrossRef]
- Holmbom, B.; Eckerman, C.; Eklund, P.; Hemming, J.; Nisula, L.; Reunanen, M.; Sjöholm, R.; Sundberg, K.; Willför, S. Knots in trees–A new rich source of lignans. Phytochem. Rev. 2003, 2, 331–340. [Google Scholar] [CrossRef]
- Willfor, S.M.; Smeds, A.I.; Holmbom, B.R. Chromatographic analysis of lignans. J. Chromatogr. A 2006, 1112, 64–77. [Google Scholar] [CrossRef]
- Slanina, J.; Glatz, Z. Separation procedures applicable to lignan analysis. J. Chromatogr. B 2004, 812, 215–229. [Google Scholar] [CrossRef]
- Srivastava, V.; Singh, M.; Malasoni, R.; Shanker, K.; Verma, R.K.; Gupta, M.M.; Gupta, A.K.; Khanuja, S.P.S. Separation and quantification of lignans in Phyllanthus species by a simple chiral densitometric method. J. Sep. Sci. 2008, 31, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Owen, R.W.; Mier, W.; Giacosa, A.; Hull, W.E.; Spiegelhalder, B.; Bartsch, H. Identification of lignans as major components in the phenolic fraction of olive oil. Clin. Chem. 2000, 46, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Kawamura, F.; Kawai, S.; Ohashi, H. Sesquilignans and lignans from Tsuga heterophylla. Phytochemistry 1997, 44, 1351–1357. [Google Scholar] [CrossRef]
- Takaku, N.; Choi, D.-H.; Mikame, K.; Okunishi, T.; Suzuki, S.; Ohashi, H.; Umezawa, T.; Shimada, M. Lignans of Chamaecyparis obtusa. J. Wood Sci. 2001, 47, 476–482. [Google Scholar] [CrossRef]
- Wang, P.; Liu, Y.; Chen, T.; Xu, W.; You, J.; Liu, Y.; Li, Y. One-step separation and purification of three lignans and one flavonol from Sinopodophyllum emodi by medium-pressure liquid chromatography and high-speed counter-current chromatography. Phytochem. Anal. 2013, 24, 603–607. [Google Scholar] [CrossRef]
- Zhu, J.; Li, W.; Xiao, W.; Ding, Y.; Huang, W.; Tu, P.; Wang, Y. Separation of lignans from Schisandra chinensis by two-dimensional reversed-phase liquid chromatography. Chin. J. Chromatogr. 2018, 36, 464–473. [Google Scholar] [CrossRef]
- Westcott, N.D.; Muir, A.D. Process for Extracting Lignans from Flaxseed. U.S. Patent US5705618A, 6 January 1998. [Google Scholar]
- Shi, S.; Zhang, Y.; Huang, K.; Liu, S.; Zhao, Y. Application of preparative high-speed counter-current chromatography for separation and purification of lignans from Taraxacum mongolicum. Food Chem. 2008, 108, 402–406. [Google Scholar] [CrossRef]
- Zhou, P.; Luo, Q.; Ding, L.; Fang, F.; Yuan, Y.; Chen, J.; Zhang, J.; Jin, H.; He, S. Preparative Isolation and Purification of Lignans from Justicia procumbens Using High-Speed Counter-Current Chromatography in Stepwise Elution Mode. Molecules 2015, 20, 7048–7058. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Dong, H.; Wang, T.; Zhao, R.; Mu, Y.; Geng, Y.; Zheng, Z.; Wang, X. A Strategy for Preparative Separation of 10 Lignans from Justicia procumbens L. by High-Speed Counter-Current Chromatography. Molecules 2017, 22, 2024. [Google Scholar] [CrossRef]
- Chester, T.L.; Pinkston, J.D. Supercritical Fluid and Unified Chromatography. Anal. Chem. 2004, 76, 4606–4613. [Google Scholar] [CrossRef]
- Farrell, W.P. Practical Approaches to Column Selection for Supercritical Fluid Chromatography. In Supercritical Fluid Chromatography; Poole, C.F., Ed.; Elsevier: Amsterdam, The Netherlands, 2017; pp. 57–101. [Google Scholar] [CrossRef]
- Falev, D.I.; Ovchinnikov, D.V.; Voronov, I.S.; Faleva, A.V.; Ul’yanovskii, N.V.; Kosyakov, D.S. Supercritical Fluid Chromatography—Tandem Mass Spectrometry for Rapid Quantification of Pentacyclic Triterpenoids in Plant Extracts. Pharmaceuticals 2022, 15, 629. [Google Scholar] [CrossRef]
- Zhang, Q.W.; Lin, L.G.; Ye, W.C. Techniques for extraction and isolation of natural products: A comprehensive review. Chin. Med. 2018, 13, 20. [Google Scholar] [CrossRef]
- Chen, M.; Wen, S.-S.; Wang, R.; Ren, Q.-X.; Guo, C.-W.; Li, P.; Gao, W. Advanced Development of Supercritical Fluid Chromatography in Herbal Medicine Analysis. Molecules 2022, 27, 4159. [Google Scholar] [CrossRef]
- Kingston, J.; Leek, H.; Buica, A.; Öhlén, K.; Proctor, K.; Raubo, J.; Sanders, M.; Thunberg, L. Application of preparative SFC in the pharmaceutical industry. In Practical Application of Supercritical Fluid Chromatography for Pharmaceutical Research and Development; Hicks., M., Ferguson, P., Eds.; Academic Press: London, UK, 2022; pp. 133–165. [Google Scholar] [CrossRef]
- Onay, S.; Hofer, S.; Ganzera, M. Rapid analysis of nine lignans in Schisandra chinensis by supercritical fluid chromatography using diode array and mass spectrometric detection. J. Pharm. Biomed. Anal. 2020, 185, 113254. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Xin, H.; Wang, F.; Cai, J.; Liu, Y.; Fu, Q.; Jin, Y.; Liang, X. Purification of lignans from Fructus Arctii using off-line two-dimensional supercritical fluid chromatography/reversed-phase liquid chromatography. J. Sep. Sci. 2017, 40, 3231–3238. [Google Scholar] [CrossRef]
- West, C.; Lemasson, E.; Bertin, S.; Hennig, P.; Lesellier, E. An improved classification of stationary phases for ultra-high performance supercritical fluid chromatography. J. Chromatogr. A 2016, 1440, 212–228. [Google Scholar] [CrossRef]
- Lesellier, E.; West, C. The many faces of packed column supercritical fluid chromatography--a critical review. J. Chromatogr. A 2015, 1382, 2–46. [Google Scholar] [CrossRef] [PubMed]
- Ovchinnikov, D.V.; Pokrovskiy, O.I.; Kosyakov, D.S.; Bogolitsyn, K.G.; Ul’yanovskii, N.V.; Falev, D.I. Evaluation of temperature and pressure effects on retention in supercritical fluid chromatography on polar stationary phases. J. Chromatogr. A 2020, 1610, 460600. [Google Scholar] [CrossRef]
- Ovchinnikov, D.V.; Ul’yanovskii, N.V.; Kosyakov, D.S.; Pokrovskiy, O.I. Some aspects of additives effects on retention in supercritical fluid chromatography studied by linear free energy relationships method. J. Chromatogr. A 2022, 1665, 462820. [Google Scholar] [CrossRef]
- Tsvetkov, D.E.; Dmitrenok, A.S.; Tsvetkov, Y.E.; Men´shov, V.M.; Nifantiev, N.E. Polyphenolic components of knotwood extracts from Abies sibirica. Russ. Chem. Bull. 2021, 70, 1356–1362. [Google Scholar] [CrossRef]
- Vajda, P.; Kamarei, F.; Felinger, A.; Guiochon, G. Comparison of volume and concentration overloadings in preparative enantio-separations by supercritical fluid chromatography. J. Chromatogr. A 2014, 1341, 57–64. [Google Scholar] [CrossRef]
- Barrero, A.F.; Haïdour, A.; Dorado, M.M.; Cuerva, J.M. Two sesquilignans from the wood of Abies marocana. Phytochemistry 1996, 41, 605–609. [Google Scholar] [CrossRef]
- Yashunsky, D.V.; Men´shov, V.M.; Tsvetkov, D.E.; Bel´ko, A.A.; Vasiyarov, G.G.; Titova, E.V.; Pimenov, A.V.; Onuchin, A.A.; Dokichev, V.A.; Tomilov, Y.V.; et al. Analysis of content of (–)-secoisolariciresinol and related polyphenols in different morphological parts and anatomical structures of larch wood from Siberia. Russ. Chem. Bull. 2014, 63, 2571–2576. [Google Scholar] [CrossRef]
- Willför, S.M.; Reunanen, M.; Eklund, P.C.; Eklund, P.; Sjöholm, R.; Kronberg, L.; Fardim, P.; Pietarinen, S.; Holmbom, B. Oligolignans in Norway spruce and Scots pine knots and Norway spruce stemwood. Holzforschung 2004, 58, 345–354. [Google Scholar] [CrossRef]
- Bakrim, S.; Machate, H.; Benali, T.; Sahib, N.; Jaouadi, I.; Omari, N.E.; Aboulaghras, S.; Bangar, S.P.; Lorenzo, J.M.; Zengin, G.; et al. Natural Sources and Pharmacological Properties of Pinosylvin. Plants 2022, 11, 1541. [Google Scholar] [CrossRef] [PubMed]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).