Abstract
The subcellular localization of long non-coding RNA (lncRNA) provides important insights and opportunities for an in-depth understanding of cell biology, revealing disease mechanisms, drug development, and innovation in the biomedical field. Although several computational methods have been proposed to identify the subcellular localization of lncRNA, it is difficult to accurately predict the subcellular localization of lncRNA effectively with these methods. In this study, a new deep-learning predictor called PreSubLncR has been proposed for accurately predicting the subcellular localization of lncRNA. This predictor firstly used the word embedding model word2vec to encode the RNA sequences, and then combined multi-scale one-dimensional convolutional neural networks with attention and bidirectional long short-term memory networks to capture the different characteristics of various RNA sequences. This study used multiple RNA subcellular localization datasets for experimental validation, and the results showed that our method has higher accuracy and robustness compared with other state-of-the-art methods. It is expected to provide more in-depth insights into cell function research.
1. Introduction
Long non-coding RNAs (lncRNAs) are a type of RNA molecule that typically exceeds 200 nucleotides in length [1,2,3,4,5]. Unlike protein-encoded RNA (mRNA), lncRNA does not encode proteins but rather performs various functions in cells [6,7], including gene expression regulation, chromatin conformation adjustment, signal transduction, and cell cycle regulation [3,8,9,10,11,12,13]. Numerous studies have shown that lncRNA is involved in cellular mechanisms ranging from gene expression to protein translation and maintaining protein stability, playing an important role in development, homeostasis, and maintaining cell fate. The abnormal expression of lncRNA is closely related to various diseases, including cancer and cardiovascular diseases [14,15,16], and can provide new biomarkers and drug targets [17,18,19]. Therefore, in recent years, there has been increasing research on the function of lncRNA in the field of biology. The diversity of the subcellular localization of lncRNAs indicates that they perform diverse functions within cells [20,21,22,23,24]. The function of lncRNA is often closely related to its subcellular localization within cells. The nucleus and cytoplasm are the most common subcellular locations, but there are also lncRNAs localized in specific subcellular organelles, such as ribosomes or exosomes [25,26,27,28]. Different subcellular localization may reflect the different functions of lncRNA in different cellular processes, from gene regulation to cellular signaling. Therefore, studying the subcellular localization of lncRNA has become a crucial step in deeply understanding its function and regulatory mechanisms [29,30].
At present, some predictors for calculating the subcellular localization of lncRNA have been proposed. The first predictor is the four-mer feature and advanced feature extracted by LncLocator using a stacked autoencoder [25], which is fed into two classifiers: support vector machine and random forest. Then, different ensemble strategies are used to combine the results of different classifiers to obtain the final prediction result. During the training process, a supervised oversampling algorithm is used to balance the proportion of different classes. Su et al. proposed that iLoc-lncRNA uses eight-mer features to encode lncRNA sequences. Considering that the eight-mer feature dimension is too large, iLoc-lncRNA adopts a feature selection method based on binomial distribution to select the optimal feature. Then, the optimal features are input into the support vector machine (SVM) to obtain the prediction results [26]. Zeng et al. developed DeepLncLoc [29], a deep learning-based lncRNA subcellular localization predictor. It encodes lncRNA sequences using a combination of subsequence embeddings. Firstly, a sequence is divided into several consecutive subsequences, and the patterns of each subsequence are extracted using an average pooling layer [30]. Finally, these patterns are combined to obtain a complete representation of the lncRNA sequence. Then, textCNN is used to learn advanced features and perform prediction tasks. Li et al. developed GraphLncLoc [26], a graph neural network-based model that uses only lncRNA sequences to predict the subcellular localization of lncRNA. GraphLncLoc converts lncRNA into a de Bruijn graph [31], where the nodes are four-mer and the direction of the edges is determined by order. Then, GraphLncLoc uses pre-trained four-mer word2vec embedding vectors as node features and assigns weights to edges. GraphLncLoc uses graph convolutional networks (GCNs) to learn potential representations and extract advanced features from de Bruijn graphs [31].
While these existing predictors have made significant progress in different aspects, there are still some potential challenges and limitations. Some of the existing predictors employ the k-mer method to extract features from lncRNA sequences. The k-mer method does not take into account the sequential information of the sequences, which can be crucial for understanding the biological function of lncRNA sequences. While some other predictors leverage TextCNN or graph convolutional networks for extracting advanced features and model construction, they fall short in capturing the multi-scale characteristics of sequences. By considering features at various scales, multi-scale features provide a more comprehensive representation of the sequence. This approach captures both local and global patterns, which can be crucial for understanding complex biological sequences. To overcome these limitations [32,33,34,35,36,37], we developed a new deep learning-based subcellular localization predictor of lncRNAs, named PreSubLncR. We firstly use the k-mer and word2vec model to encode the lncRNA sequence. We secondly employ multi-scale one-dimensional convolutional neural networks with attention to extract multi-scale local discriminative features, and then concatenate these local features. We then extract long-range-dependent information from these combined multi-scale local features by using the bidirectional LSTM model and finally utilize the fully connected network to make predictions. The PreSubLncR predictor has two advantages: (1) Multi-scale convolution can capture feature information at different scales, and the combination of an attention mechanism can further improve the model’s ability to extract important features, which is helpful to improve the prediction accuracy of the model. (2) Bidirectional LSTM can effectively capture long-term dependencies in sequences, which is helpful for better modeling the characteristics of lncRNA sequences. Through the bidirectional information transfer of bidirectional LSTM, the characteristics of the sequence can be understood more comprehensively.
2. Materials and Methods
2.1. Datasets
Creating a robust benchmark dataset is essential for developing a reliable machine learning model. For this study, we utilize the same dataset as GraphLncLoc [26], sourced from the RNALocate v1.0 database [38]. This database encompasses 2383 entries for lncRNA subcellular localization. Given that the majority of lncRNAs are annotated with a single localization in the database, the lncRNAs that are located in single subcellular localization are selected in the work. To minimize redundancy, the CD-HIT-EST tool [39] is employed to eliminate duplicate sequences with a similarity threshold of 80%. The resulting dataset spans seven distinct subcellular localizations. Categories with fewer than ten lncRNAs are excluded, leading to the removal of those in the two subcellular localizations. Considering the ambiguous annotation of the cytoplasm and the cytosol localization, our analysis is concentrated solely on the cytoplasmic compartment, and then, those lncRNAs located in the cytosol are excluded. Consequently, a benchmark dataset is compiled which is composed of 769 lncRNAs across four subcellular localizations: cytoplasm, nucleus, ribosome, and exosome. Formula (1) divides the data into four different subcellular localizations.
Table 1 shows the distribution of the benchmark dataset, where S1 represents 328 lncRNAs from the cytoplasm, S2 represents 325 lncRNAs from the nucleus, S3 represents 88 lncRNAs from the ribosome, and S4 represents 28 lncRNAs from the exosome.

Table 1.
Distribution of benchmark data sets.
2.2. Network Framework of PreSubLncR
In this study, we proposed a new deep-learning prediction method for lncRNA subcellular localization. The predictor in this study, called PreSubLncR, uses a multi-layer deep neural network structure that combines one-dimensional convolutional neural networks (1D-CNNs), attention mechanisms, and bidirectional long short-term memory networks (BiLSTM). Figure 1 shows the entire network framework of PreSubLncR, which consists of three modules of Feature Encoding, Model Construction. and Prediction. The Feature Encoding module is designed to efficiently characterize lncRNA sequences. This module combines two different feature encoding methods, k-mer and word2vec, to represent the sequence as the feature vector. The Model Construction module is a combination of the 1D-CNN, attention, and BiLSTM models. The 1D-CNNs use three convolutional kernels of sizes 1, 3, and 5, so that each convolutional kernel considers embedding 1, 3, and 5 adjacent words to identify sequence features at different scales. By calculating the attention weights, the attention layer is applied to weight the output of the CNN layer. The use of an attention layer can further improve the model’s ability to extract important features. Then, the combined features are fed into the BiLSTM layer for further extraction of temporal–sequential features. The Prediction module adopts a fully connected network with two hidden layers, which map the features extracted from the BiLSTM layer to the final lncRNA subcellular localization label. The final output layer consists of four nodes corresponding to the cytoplasm, nucleus, ribosome, and exosome. The values of these nodes represent the predicted probability distribution, that is, the probability that a long non-coding RNA is likely to exist in each subcellular location. Suppose our neural network makes a prediction for a long non-coding RNA and the output is [0.6, 0.1, 0.2, 0.1]. This means that the model believes that the RNA has a 60% probability of being in the cytoplasm, a 10% probability of being in the nucleus, a 20% probability of being in the ribosome, and a 10% probability of being in the exosome. That is, the model prefers to classify the RNA as being located in the cytoplasm. It is worth noting that the output layer uses the softmax function to convert the raw output into a probability value. The range of the output probability value is [0–1], and the sum of the output probability value is 1.
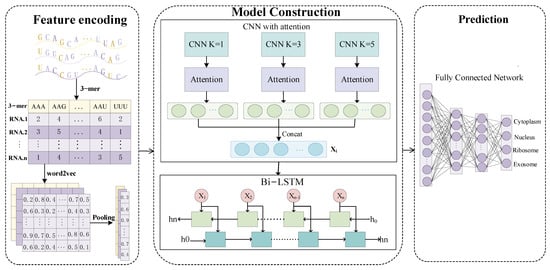
Figure 1.
Overview of PreSubLncR. PreSubLncR consists of three modules of Feature Encoding, Model Construction, and Prediction. The Feature Encoding module combines two different feature encoding methods, k-mer and word2vec, to represent the sequence as the feature vector. The Model Construction module is a combination of the 1D-CNN, attention and BiLSTM models. The 1D-CNNs extract sequence features at different scales using different kernels. The attention layer is applied to weight the output of the CNN layer. The combined features are fed into the BiLSTM layer for the further extraction of temporal–sequential features. The Prediction module adopts a fully connected network with two hidden layers to make the final prediction.
2.3. Feature Encoding
This study uses the word embedding technology word2vec to encode lncRNA sequences, which is an important technique in natural language processing (NLP). Word2vec is a technique that maps discrete vocabulary to continuous vector spaces. The word2vec model maps vocabulary with similar contexts to similar vector spaces by learning the distributed representation of vocabulary in a corpus. RNA sequences are strings composed of four bases (adenine A, guanine G, cytosine C, and uracil U). To convert RNA sequences into vocabulary sequences, we chose k-mer [33] as the basic vocabulary unit. K-mer refers to a continuous subsequence with a length of k, and by selecting the appropriate parameter k value, an appropriate segmentation method can be found. Using k-mer as a vocabulary to capture local features in lncRNA sequences, each lncRNA sequence is divided into segments of length k; that is, each lncRNA sequence is divided into k-mer subsequences, where k is the length of the predetermined subsequence k-mer. Assuming k = 4, ‘AUCGGCAUAG’ will be divided into [AUCG, UCGG, CGGC, GGCA, GCAU, CAUA, AUAG]. These k-mers will be used to build a vocabulary. The word2vec pre-training model is used to encode k-mer into a vector with dimension D. Each sequence has a length of L and is encoded with a length of . Since the length of each sequence is not fixed, average pooling is used to fix the dimension to D.
2.4. Multi-Scale One-Dimensional Convolutional Neural Network with Attention
Traditional convolutional neural networks (CNNs) are usually used to process image data, and their basic structure is two-dimensional [40,41]. However, text can be regarded as one-dimensional sequence data, so the idea of a one-dimensional CNN can be borrowed and applied to text data to extract text features, which is the basic concept of TextCNN. The attention mechanism has been widely used in the processing of sequential data, allowing models to assign different weights to inputs at different positions, thereby strengthening or suppressing the importance of different words or markers, to better capture key information in text. The 1D-CNN with an attention model combines the advantages of the CNN and the attention mechanism to process text data more comprehensively. Specifically, the 1D-CNN with attention first captures local features in input data through convolution operations. The convolutional kernel slides on the input data, enabling the recognition of sequence features at different scales, which helps the model understand the structure and patterns of the text. At the same time, the attention mechanism is used to enhance the output of CNN convolutional layers. It allows the model to focus on specific parts of the input sequence and focus more on information that is important for solving the task. Through convolutional layers, the model can capture features at different scales in the text, and through attention mechanisms, the model can assign different weights to features at different positions, allowing the model to consider both local and global information, thus better understanding the entire sequence. This enables the 1D-CNN with the attention model to have stronger expressive power and performance in text-processing tasks.
2.5. Bidirectional Long Short-Term Memory
To more effectively capture sequence dependencies in RNA sequences, this study introduced the bidirectional long short-term memory network (BiLSTM) as a key component. The number of hidden states for each BiLSTM unit is set to 6, and a bidirectional structure that includes two layers of BiLSTM is constructed. Bidirectional LSTM not only considers the contextual information before each moment but also considers the contextual information after each moment, providing more comprehensive sequence information for RNA subcellular localization tasks. By introducing BiLSTM, the model can better understand the relationships between different parts of RNA sequences, including temporal and contextual dependencies. This helps the model to more accurately capture features and patterns in RNA sequences, thereby improving the performance of RNA subcellular localization.
2.6. Performance Evaluation Metrics
To evaluate the classification performance of the PreSubLncR predictor, we chose to calculate four metrics: accuracy, precision, recall, and F1-score values, as shown in Equations (2)–(5):
where TP represents the number of true positives, FP represents the number of false positives, TN represents the number of true negatives, and FN represents the number of false negatives. True positives occur when the model correctly identifies an instance as positive. False positives are instances where the model incorrectly identifies an instance as positive when it is actually negative. True negatives are cases where the model correctly identifies an instance as negative. False negatives occur when the model incorrectly identifies an instance as negative when it is actually positive. These metrics provide insights into different aspects of the classifier’s performance. Accuracy measures the overall correctness of the classifier’s predictions, Precision quantifies the proportion of true positive predictions among all positive predictions made by the classifier, recall measures the proportion of actual positive instances that are correctly identified by the model, and F1-score is the harmonic mean of precision and recall, providing a single metric that balances the two. Considering these metrics together can help us evaluate the performance of the classifier more comprehensively and provide a direction for improving the classifier.
2.7. Hyperparameter Optimization
In the training process of PreSubLncR, AdamW is used as an optimizer to update the weight parameters of PreSubLncR with an initial learning rate of 0.01. ReLU is used as a transfer function. The reason why we chose the ReLU function as a transfer function is that it is simple and easy to calculate, and it can effectively alleviate the gradient disappearance problem and speed up the training of the model. To avoid overfitting, dropouts with rates of 0.2 and 0.3 were applied in the embedded and fully connected layers, respectively. Focal loss is used as a loss function to solve the class imbalance problem.
During the experimental process, 5-fold cross-validation was used, and hyperparameter tuning was performed in conjunction with F1-scores [42,43,44,45]. Multiple hyperparameters have a significant impact on model performance, including the k-value in k-mer, the dimension of pre-trained word2vec embedding vectors, the initial learning rate, and the packet loss rate. It should be noted that the sequence encoding method has a significant impact on the experimental results. Therefore, we adopted a grid search strategy to find the optimal combination of hyperparameters [46,47,48]. When selecting hyperparameters, we filtered from the following candidate values: a k value selected from {1, 2, 3, 4, 5, 6}, the dimension of pre-training vector selected from {32, 64, 128}, the dimension of the bidirectional LSTM hidden layer selected from {2, 4, 6, 8, 16, 32}, and the dimension of two hidden layers of the final fully connected network selected from {32, 64, 128, 256}. Through grid searching, the optimal hyperparameter combination was found, and it was ultimately determined that in the experiment, when the k-value was 3, the dimension of the pre-training vector was 64, the dimension of the bidirectional LSTM hidden layer was 6, and the dimensions of the two hidden layers of the final fully connected network were 128 and 64, the model performance reached its optimal state.
3. Results and Discussion
3.1. Comparison of Different k-mer Features
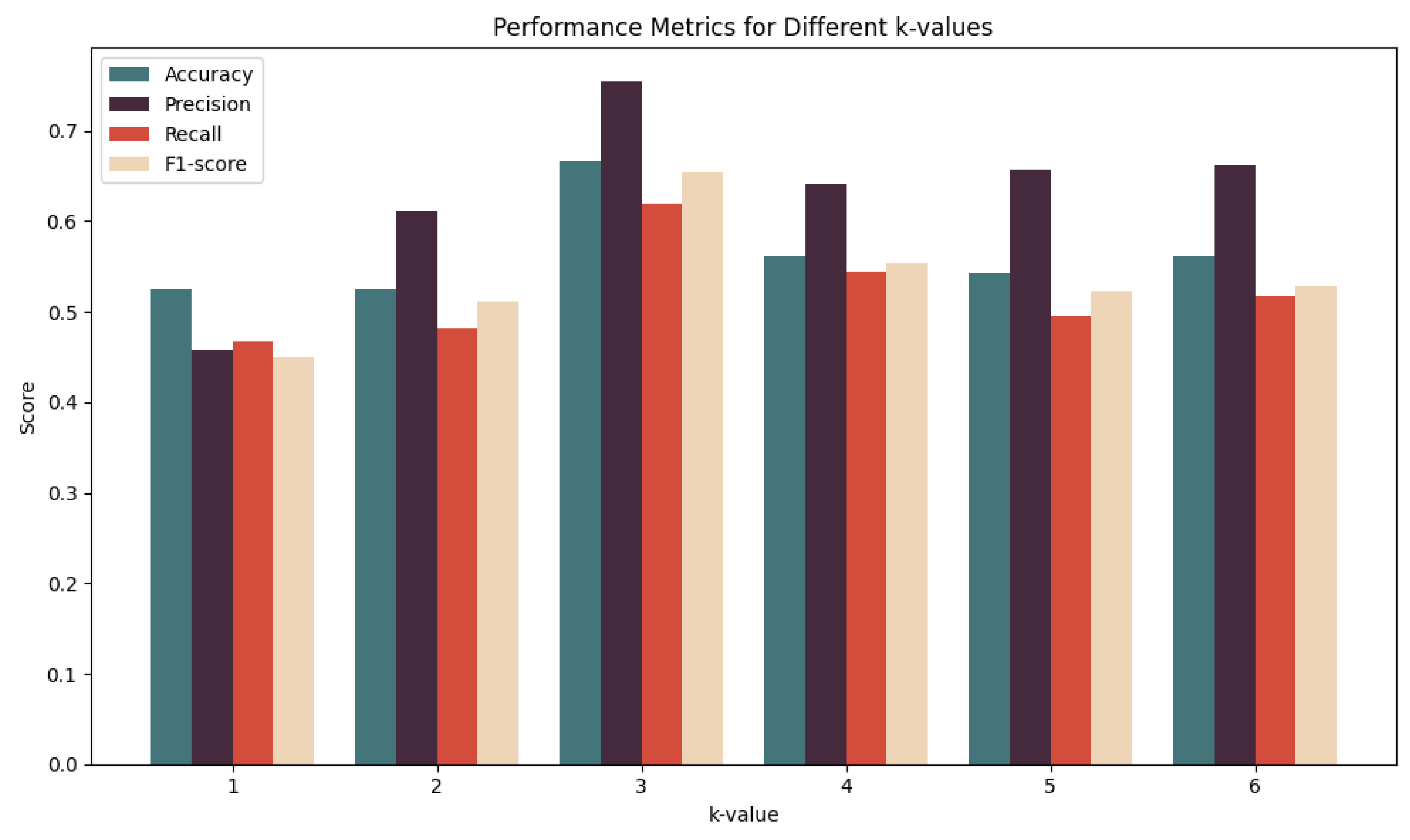
In this paper, we used a 5-fold cross-validation method to reliably estimate the performance of our PreSubLncR predictor. Specifically, the benchmark dataset was divided into five equally sized subsets, four of which were used for model training and the remaining one was used as test data for model performance evaluation. The process was repeated five times, with each repeat selecting a different subset. In the experiment, it was found that the k value of k-mer had a significant impact on the results. To determine the most suitable k-mer value, a series of experiments was conducted using different k-mer lengths of k = 1, 2, 3, 4, 5, and 6 for encoding. The experimental results indicate that different k-mer values have different effects on the predictive performance of RNA subcellular localization. In Figure 2, the experimental results are shown, and the predictive performance of the model is relatively poor when using smaller k values, such as k = 1 and k = 2. This may be because these shorter k-mers are unable to fully capture key information in RNA sequences, resulting in the lower accuracy of the model. However, the optimal predictive performance was observed at k = 3. This indicates that a k-mer length of k = 3 can better capture contextual information of RNA sequences, thereby improving the accuracy of the model. Therefore, k = 3 was selected as the optimal k-mer value for further analysis. It is worth noting that using longer k-mer values, such as k = 4, 5, and 6, does not further improve the predictive performance of the model. This may be because longer k-mer values may introduce noise or excessive features, reducing the model’s generalization ability. Therefore, in this study, a k-mer length value of k = 3 was ultimately determined as the optimal choice for RNA subcellular localization prediction based on word2vec encoding. This result indicates that the k-mer length with k = 3 performs best in balancing information capture and feature dimension control.
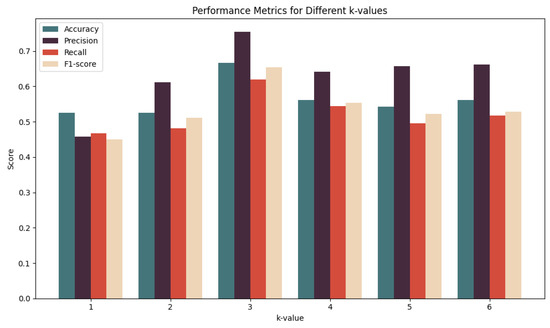
Figure 2.
Effects of different k-mer values on the predictive performance of RNA subcellular localization on the benchmark dataset using 5-fold cross-validation.
3.2. Ablation Experiment
To verify the effectiveness of the model and its components, we performed a five-fold cross-validation experiment on a benchmark dataset, including a benchmark model and different ablation models. Firstly, one-dimensional convolution was used as the benchmark model. Subsequently, attention mechanisms and bidirectional LSTM were gradually added to form different ablation models, namely, CNN, CNN attention, and CNN attention BiLSTM. The experimental results are listed in Table 2, and it is observed that the model incorporating the attention mechanism outperforms the baseline model in terms of overall performance. The precision reached 0.736, indicating that the introduction of the attention mechanism significantly improved the performance of RNA subcellular localization models. Subsequently, a bidirectional LSTM was added to form a CNN attention BiLSTM ablation model, which further improved its performance and achieved the highest accuracy and F1 score. This once again proves that the introduction of a bidirectional LSTM layer is crucial for capturing sequence context information in this task. Therefore, the experimental results clearly show that the introduction of the attention mechanism and bidirectional LSTM layer had a positive impact on the performance of the model, making it perform well in RNA subcellular localization tasks. We analyzed the performance enhancements, which can be attributed to the following two factors. The first factor is that the PreSubLncR predictor combines the features of a CNN and BiLSTM, allowing it to efficiently capture local features and bidirectional dependencies in lncRNA sequence data. The second factor is related to the introduction of the attention mechanism to focus on key information, so CNN + attention + BiLSTM can improve prediction performance.
Table 2.
Results of ablation experiments.
3.3. Comparison with Other Predictors
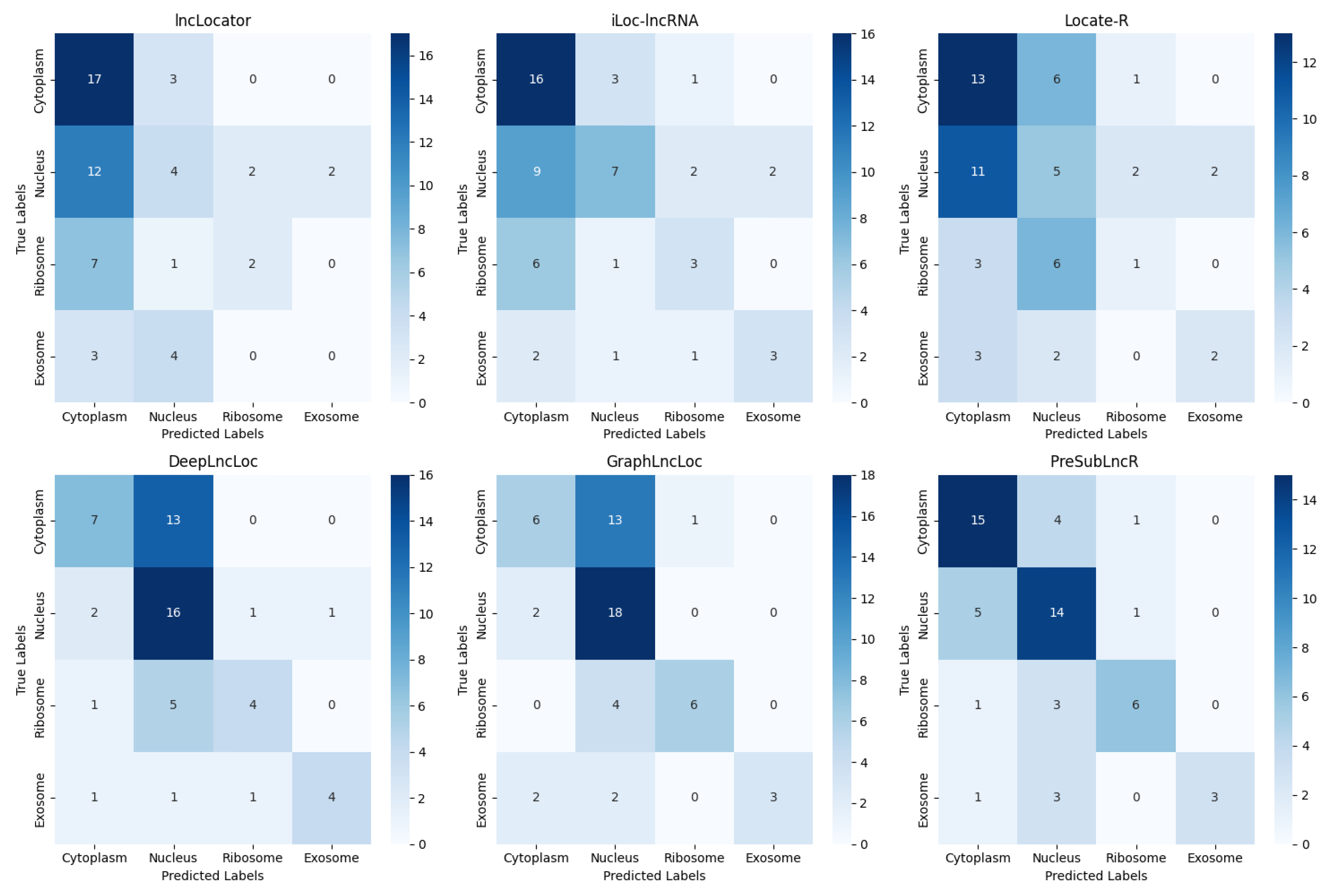
To highlight the superior performance of the PreSubLncR method, this study conducted a comprehensive comparison with the lncLocator, iLoc-lncRNA, Locate-R, DeepLncLoc, and GraphLncLoc methods on an independent test set. The independent test set was obtained from the lncSLdb database. A wide range of performance metrics were used to evaluate its performance, including accuracy, precision, recall, and F1 score. These metrics were used to evaluate the model’s classification performance, prediction accuracy, and performance in multi-class problems. In Table 3, the PreSubLncR method performs well on these performance metrics, achieving the highest accuracy and F1 scores. Figure 3 is a confusion matrix plot showing the classification results of the PreSubLncR method on different classes relative to other methods. The confusion matrix diagram clearly shows the classification accuracy and error of the PreSubLncR method relative to other methods in each category. This further highlights the excellent performance of the PreSubLncR method in RNA subcellular localization problems. Therefore, the PreSubLncR method performs well in the subcellular localization of lncRNAs, indicating that it has excellent performance in prediction accuracy and multi-class problem handling.
Table 3.
Performance comparison results of the PreSubLncR predictor based on the independent test set with other methods.
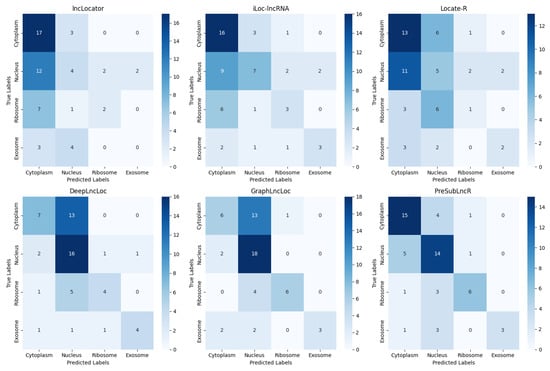
Figure 3.
Confusion matrix of PreSubLncR versus other methods on the test set.
To evaluate the performance of the method proposed in this study on each category, a series of comparisons were conducted on an independent test set, including the PreSubLncR method, iLoc-lncRNA, and DeepLncLoc. Table 4 and Table 5, respectively, show the comparison results of the PreSubLncR method with the iLoc-lncRNA and DeepLncLoc methods in terms of accuracy, recall, and F1 scores for each category. It is evident that the F1 values of the PreSubLncR model are higher than those of iLoc-lncRNA in the cytoplasm, nucleus, ribosome, and exosome categories. Still, in the exosome category, the F1 values of the DeepLncLoc model are slightly higher than those of the PreSubLncR model. This indicates that the PreSubLncR model is competitive overall, especially in multiple categories.
Table 4.
Comparison of PreSubLncR and iLoc-lncRNA methods on an independent test set.
Table 5.
Comparison of PreSubLncR and DeepLncLoc methods on an independent test set.
4. Conclusions
In this study, we introduce PreSubLncR, a novel predictor for predicting the subcellular localization of lncRNAs. Our results show the superiority of PreSubLncR over other competing methodologies. This remarkable performance stems from three pivotal factors: (1) The fusion of k-mer and word2vec technologies enables a more exhaustive exploration of the feature landscape within lncRNA sequences, thereby enhancing the richness and diversity of feature representations. This integrative approach facilitates a comprehensive understanding of sequence characteristics, augmenting the predictive power of our model. (2) The incorporation of multi-scale convolution and attention mechanisms empowers PreSubLncR to efficiently extract and assimilate crucial features embedded within the sequences. By adaptively focusing on salient regions of the input, these mechanisms facilitate enhanced feature learning, thereby improving the discriminative capacity of the model. (3) The utilization of bidirectional LSTM architecture allows for the effective capture of long-term dependencies inherent in lncRNA sequences. Through bidirectional information transfer, our model gains a more holistic understanding of sequence characteristics, thereby refining its predictive accuracy. Furthermore, when subjected to evaluation on an independent test set, PreSubLncR consistently outperformed existing methods, showcasing exceptional accuracy and robustness. These findings not only underscore the efficacy of our proposed model but also highlight its potential for accurately predicting the subcellular localization of lncRNAs in diverse biological contexts. PreSubLncR represents a significant advancement in the field of lncRNA localization prediction, offering a potent combination of advanced techniques and superior performance. We anticipate that our model will catalyze further research endeavors aimed at elucidating the functional roles of lncRNAs and their implications in various biological processes and disease contexts.
Author Contributions
X.W.: investigation, supervision, design of the PreSubLncR system and writing—review. S.W.: conceptualization, methodology, data curation, visualization, writing—original draft, implementation of the PreSubLncR system using the Python programming language and writing—review and editing. R.W.: supervision and writing—review and editing. X.G.: supervision and writing—review. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported in part by funds from Key Research Project of Colleges and Universities of Henan Province (No. 22A520013, No. 23B520004), Key Science and Technology Development Program of Henan Province (No. 232102210020, No. 202102210144), the Training Program of Young Backbone Teachers in Colleges and Universities of Henan Province (No. 2019GGJS132).
Data Availability Statement
The datasets generated and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conflicts of Interest
The authors declare there are no conflicts of interest.
References
- Kung, J.T.; Colognori, D.; Lee, J.T. Long noncoding RNAs: Past, present, and future. Genetics 2013, 193, 651–669. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, X.; Liu, L.; Deng, H.; Zhang, J.; Xu, Q.; Cen, B.; Ji, A. Regulation of lncRNA expression. Cell. Mol. Biol. Lett. 2014, 19, 561–575. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 Suppresses Target mRNA Translation. Mol. Cell 2012, 47, 648–655. [Google Scholar] [CrossRef]
- Carlevaro-Fita, J.; Johnson, R. Global Positioning System: Understanding Long Noncoding RNAs through Subcellular Localization. Mol. Cell 2019, 43, 869–883. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L. Linking Long Noncoding RNA Localization and Function. Trends Biochem. Sci. 2016, 41, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Meyer, C.; Garzia, A.; Tuschl, T. Simultaneous detection of the subcellular localization of RNAs and proteins in cultured cells by combined multicolor RNA-FISH and IF. Methods 2017, 118–119, 101–110. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Yang, M.; Luo, F.; Wu, F.X.; Li, M.; Pan, Y.; Li, Y.; Wang, J. Prediction of lncRNA-disease associations based on inductive matrix completion. Bioinformatics 2018, 34, 3357–3364. [Google Scholar] [CrossRef] [PubMed]
- Cabili, M.N.; Dunagin, M.C.; McClanahan, P.D.; Biaesch, A.; Padovan-Merhar, O.; Regev, A.; Rinn, J.L.; Raj, A. Localization and abundance analysis of human lncRNAs at single-cell and single-molecule resolution. Genome Biol. 2015, 16, 20. [Google Scholar] [CrossRef]
- Mas-Ponte, D.; Carlevaro-Fita, J.; Palumbo, E.; Pulido, T.H.; Guigo, R.; Johnson, R. LncATLAS database for subcellular localization of long noncoding RNAs. RNA 2017, 23, 1080–1087. [Google Scholar] [CrossRef]
- Chin, A.; Lécuyer, E. RNA localization: Making its way to the center stage. Biochim. Biophys. Acta Gen. Subj. 2017, 1861, 2956–2970. [Google Scholar] [CrossRef]
- Winter, J.; Jung, S.; Keller, S.; Gregory, R.I.; Diederichs, S. Many roads to maturity: MicroRNA biogenesis pathways and their regulation. Nat. Cell Biol. 2009, 11, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Liu, Y.; Li, K.; Fu, T. Prognostic value of long non-coding RNA breast cancer anti-estrogen resistance 4 in human cancers: A meta-analysis. Medicine 2019, 98, e15793. [Google Scholar] [CrossRef] [PubMed]
- Yu, B.; Shan, G. Functions of long noncoding RNAs in the nucleus. Nucleus 2016, 7, 155–166. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Valverde, A.; Ahmad, F.; Naqvi, A.R. Long Noncoding RNA in Myeloid and Lymphoid Cell Di ff erentiation, Polarization and Function. Cells 2020, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Kirk, J.M.; Kim, S.O.; Inoue, K.; Smola, M.J.; Lee, D.M.; Schertzer, M.D.; Wooten, J.S.; Baker, A.R.; Sprague, D.; Collins, D.W.; et al. Functional classification of long non-coding RNAs by k-mer content. Nat. Genet. 2018, 50, 1474–1482. [Google Scholar] [CrossRef] [PubMed]
- Feng, S.; Liang, Y.; Du, W.; Lv, W.; Li, Y. Lnclocation: Efficient subcellular location prediction of long non-coding rna-based multi-source heterogeneous feature fusion. Int. J. Mol. Sci. 2020, 21, 7271. [Google Scholar] [CrossRef] [PubMed]
- Wen, X.; Gao, L.; Guo, X.; Li, X.; Huang, X.; Wang, Y.; Xu, H.; He, R.; Jia, C.; Liang, F. LncSLdb: A resource for long non-coding RNA subcellular localization. Database 2018, 2018, bay085. [Google Scholar] [CrossRef]
- Ahmad, A.; Lin, H.; Shatabda, S. Locate-R: Subcellular localization of long non-coding RNAs using nucleotide compositions. Genomics 2020, 112, 2583–2589. [Google Scholar] [CrossRef]
- Fan, Y.; Chen, M.; Zhu, Q. LncLocPred: Predicting LncRNA Subcellular Localization Using Multiple Sequence Feature Information. IEEE Access 2020, 8, 124702–124711. [Google Scholar] [CrossRef]
- Su, Z.D.; Huang, Y.; Zhang, Z.Y.; Zhao, Y.W.; Wang, D.; Chen, W.; Chou, K.C.; Lin, H. ILoc-lncRNA: Predict the subcellular location of lncRNAs by incorporating octamer composition into general PseKNC. Bioinformatics 2018, 34, 4196–4204. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Ning, L.; Ye, X.; Yang, Y.H.; Futamura, Y.; Sakurai, T.; Lin, H. iLoc-miRNA: Extracellular/intracellular miRNA prediction using deep BiLSTM with attention mechanism. Brief. Bioinform. 2022, 23, bbac395. [Google Scholar] [CrossRef] [PubMed]
- Zuckerman, B.; Ulitsky, I. Predictive models of subcellular localization of long RNAs. RNA 2019, 25, 557–572. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, L.; Wang, R.; Wang, X. An accurate identification method of bitter peptides based on deep learning. J. Light Ind. 2023, 38, 11–16. [Google Scholar]
- Voit, E.O.; Martens, H.A.; Omholt, S.W. 150 Years of the Mass Action Law. PLoS Comput. Biol. 2015, 11, e1004012. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Pan, X.; Yang, Y.; Huang, Y.; Shen, H.B. The lncLocator: A subcellular localization predictor for long non-coding RNAs based on a stacked ensemble classifier. Bioinformatics 2018, 34, 2185–2194. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Zhao, B.; Yin, R.; Lu, C.; Guo, F.; Zeng, M. GraphLncLoc: Long non-coding RNA subcellular localization prediction using graph convolutional networks based on sequence to graph transformation. Brief. Bioinform. 2023, 24, bbac565. [Google Scholar] [CrossRef] [PubMed]
- Zeng, M.; Zhang, F.; Wu, F.X.; Li, Y.; Wang, J.; Li, M. Protein-protein interaction site prediction through combining local and global features with deep neural networks. Bioinformatics 2020, 36, 1114–1120. [Google Scholar] [CrossRef]
- Wang, J.; Li, J.; Yue, K.; Wang, L.; Ma, Y.; Li, Q. NMCMDA: Neural multicategory MiRNA-disease association prediction. Brief. Bioinform. 2021, 22, bbab074. [Google Scholar] [CrossRef]
- Zeng, M.; Wu, Y.; Lu, C.; Zhang, F.; Wu, F.X.; Li, M. DeepLncLoc: A deep learning framework for long non-coding RNA subcellular localization prediction based on subsequence embedding. Brief. Bioinform. 2022, 23, bbab360. [Google Scholar] [CrossRef]
- He, K.; Zhang, X.; Ren, S.; Sun, J. Spatial Pyramid Pooling in Deep Convolutional Networks for Visual Recognition. IEEE Trans. Pattern Anal. Mach. Intell. 2015, 37, 1904–1916. [Google Scholar] [CrossRef]
- Compeau, P.E.C.; Pevzner, P.A.; Tesler, G. How to apply de Bruijn graphs to genome assembly. Nat. Biotechnol. 2011, 29, 987–991. [Google Scholar] [CrossRef] [PubMed]
- Zhou, R.; Lu, Z.; Luo, H.; Xiang, J.; Zeng, M.; Li, M. NEDD: A network embedding based method for predicting drug-disease associations. BMC Bioinform. 2020, 21, 387. [Google Scholar] [CrossRef] [PubMed]
- Shibuya, Y.; Belazzougui, D.; Kucherov, G. Space-efficient representation of genomic k-mer count tables. Algorithms Mol. Biol. 2022, 17, 5. [Google Scholar] [CrossRef]
- Yu, A.; Choi, Y.H.; Tu, M. RNA drugs and RNA targets for small molecules: Principles, progress, and challenges. Pharmacol. Rev. 2020, 72, 862–898. [Google Scholar] [CrossRef] [PubMed]
- Chou, K. Some remarks on protein attribute prediction and pseudo amino acid composition. J. Theor. Biol. 2011, 273, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Cui, T.; Dou, Y.; Tan, P.; Ni, Z.; Liu, T.; Wang, D.L.; Huang, Y.; Cai, K.; Zhao, X.; Xu, D.; et al. RNALocate v2.0: An updated resource for RNA subcellular localization with increased coverage and annotation. Nucleic. Acids Res. 2022, 50, D333–D339. [Google Scholar] [CrossRef] [PubMed]
- Taliaferro, J.M. Transcriptome-scale methods for uncovering subcellular RNA localization mechanisms. Biochim. Biophys. Acta Mol. Cell Res. 2022, 1869, 119–202. [Google Scholar] [CrossRef]
- Zhang, T.; Tan, P.; Wang, L.; Jin, N.; Li, Y.; Zhang, L.; Yang, H.; Hu, Z.; Zhang, L.; Hu, C.; et al. RNALocate: A resource for RNA subcellular localizations. Nucleic. Acids Res. 2017, 45, D135–D138. [Google Scholar]
- Huang, Y.; Niu, B.; Gao, Y.; Fu, L.; Li, W. CD-HIT Suite: A web server for clustering and comparing biological sequences. Bioinformatics 2010, 26, 680–682. [Google Scholar] [CrossRef]
- Xu, M.; Chen, Y.; Xu, Z.; Zhang, L.; Jiang, H.; Pian, C. MiRLoc: Predicting miRNA subcellular localization by incorporating miRNA-mRNA interactions and mRNA subcellular localization. Brief. Bioinform. 2022, 23, bbac044. [Google Scholar] [CrossRef]
- Ameen, Z.S.; Mostafa, H.; Ozsahin, D.U.; Mubarak, A.S. Accelerating SARS-CoV-2 Vaccine Development: Leveraging Novel Hybrid Deep Learning Models and Bioinformatics Analysis for Epitope Selection and Classification. Processes 2023, 11, 1829. [Google Scholar] [CrossRef]
- Eze, M.C.; Vafaei, L.E.; Eze, C.T.; Tursoy, T.; Ozsahin, D.U.; Mustapha, M.T. Development of a Novel Multi-Modal Contextual Fusion Model for Early Detection of Varicella Zoster Virus Skin Lesions in Human Subjects. Processes 2023, 11, 2268. [Google Scholar] [CrossRef]
- Kondo, Y. Long non-coding RNAs as an epigenetic regulator in human cancers. Cancer Sci. 2017, 108, 1927–1933. [Google Scholar] [CrossRef]
- Zhang, Z.Y.; Yang, Y.H.; Ding, H.; Wang, D.; Chen, W.; Lin, H. Design powerful predictor for mRNA subcellular location prediction in Homo sapiens. Brief. Bioinform. 2021, 22, 526–535. [Google Scholar] [CrossRef] [PubMed]
- Quang, D.; Xie, X. DanQ: A hybrid convolutional and recurrent deep neural network for quantifying the function of DNA sequences. Nucleic Acids Res. 2016, 44, e107. [Google Scholar] [CrossRef] [PubMed]
- Bai, T.; Yan, K.; Liu, B. DAmiRLocGNet: miRNA subcellular localization prediction by combining miRNA–disease associations and graph convolutional networks. Brief. Bioinform. 2023, 24, bbad212. [Google Scholar] [CrossRef] [PubMed]
- Muhammod, R.; Ahmed, S.; Farid, D.; Shatabda, S.; Dehzangi, A. PyFeat: A Python-based effective feature generation tool for DNA. RNA and protein sequences. Bioinformatics 2019, 35, 3831–3833. [Google Scholar] [CrossRef]
- Rna, N.; Quinn, J.J.; Chang, H.Y. Unique features of long non-coding RNA biogenesis and function. Nat. Rev. Genet. 2016, 17, 47–62. [Google Scholar]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).