
CrossTx: Cross-Cell-Line Transcriptomic Signature Predictions


Abstract
1. Introduction
2. Materials and Methods
2.1. Prediction of Drug Signatures by CrossTx
2.2. Application to the CMap Dataset
2.3. Performance Scoring
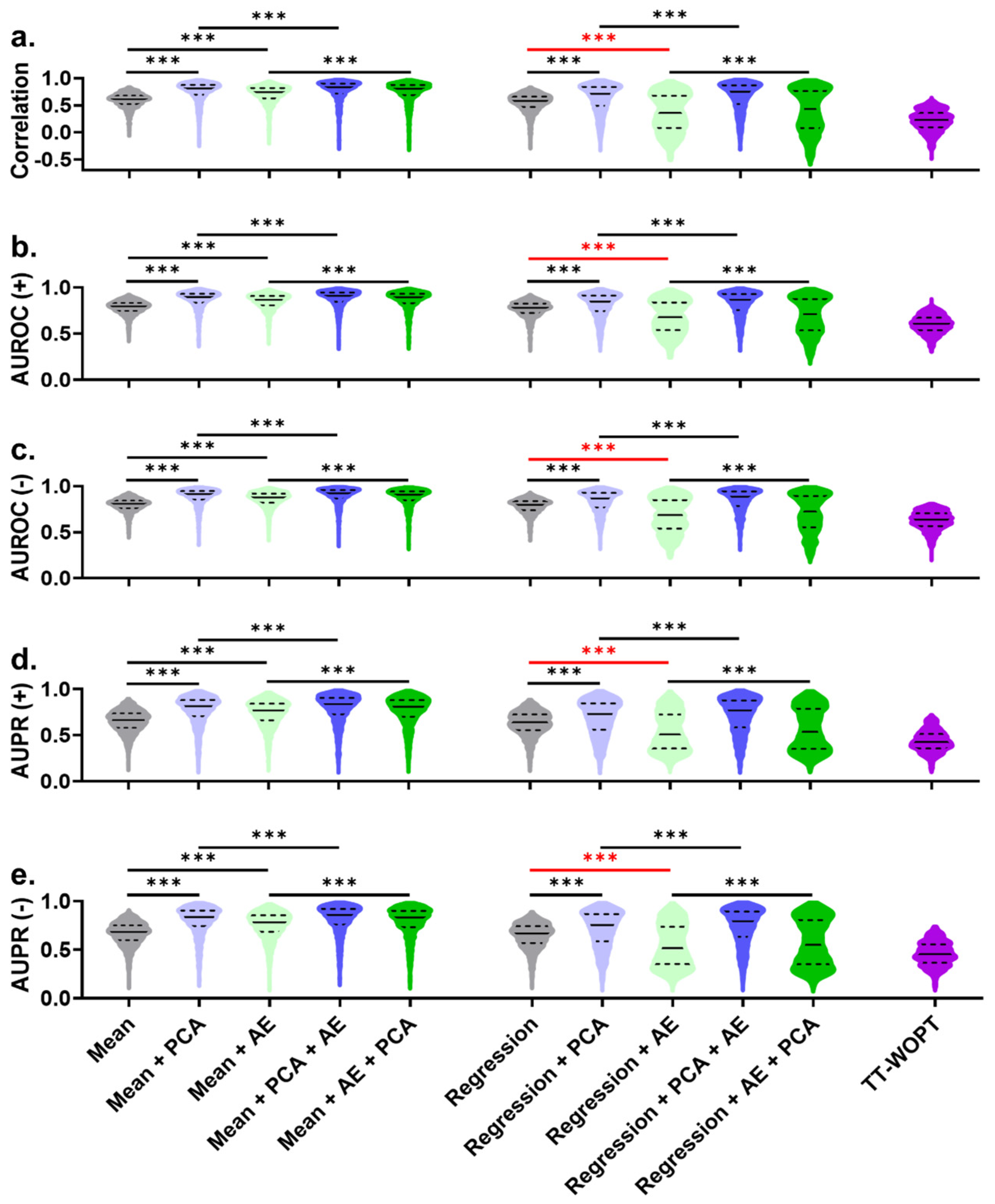
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Louhimo, R.; Laakso, M.; Belitskin, D.; Klefstrom, J.; Lehtonen, R.; Hautaniemi, S. Data integration to prioritize drugs using genomics and curated data. BioData Min. 2016, 9, 21. [Google Scholar] [CrossRef] [PubMed]
- Dudley, J.T.; Deshpande, T.; Butte, A.J. Exploiting drug-disease relationships for computational drug repositioning. Brief. Bioinform. 2011, 12, 303–311. [Google Scholar] [CrossRef] [PubMed]
- Jin, G.; Wong, S.T. Toward better drug repositioning: Prioritizing and integrating existing methods into efficient pipelines. Drug Discov. Today 2014, 19, 637–644. [Google Scholar] [CrossRef] [PubMed]
- Kim, R.S.; Goossens, N.; Hoshida, Y. Use of big data in drug development for precision medicine. Expert Rev. Precis. Med. Drug Dev. 2016, 1, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Qian, T.; Zhu, S.; Hoshida, Y. Use of big data in drug development for precision medicine: An update. Expert Rev. Precis. Med. Drug Dev. 2019, 4, 189–200. [Google Scholar] [CrossRef]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452e1417. [Google Scholar] [CrossRef]
- Wang, Y.-Y.; Kang, H.; Xu, T.; Hao, L.; Bao, Y.; Jia, P. CeDR Atlas: A knowledgebase of cellular drug response. Nucleic Acids Res. 2021, 50, D1164–D1171. [Google Scholar] [CrossRef]
- Zhao, W.; Dovas, A.; Spinazzi, E.F.; Levitin, H.M.; Banu, M.A.; Upadhyayula, P.; Sudhakar, T.; Marie, T.; Otten, M.L.; Sisti, M.B.; et al. Deconvolution of cell type-specific drug responses in human tumor tissue with single-cell RNA-seq. Genome Med. 2021, 13, 82. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Hodos, R.; Zhang, P.; Lee, H.C.; Duan, Q.; Wang, Z.; Clark, N.R.; Ma’ayan, A.; Wang, F.; Kidd, B.; Hu, J.; et al. Cell-specific prediction and application of drug-induced gene expression profiles. In Biocomputing 2018: Proceedings of the Pacific Symposium, 2018; World Scientific Publishing Company: Singapore, 2018; Volume 23, pp. 32–43. [Google Scholar]
- Iwata, M.; Yuan, L.; Zhao, Q.; Tabei, Y.; Berenger, F.; Sawada, R.; Akiyoshi, S.; Hamano, M.; Yamanishi, Y. Predicting drug-induced transcriptome responses of a wide range of human cell lines by a novel tensor-train decomposition algorithm. Bioinformatics 2019, 35, i191–i199. [Google Scholar] [CrossRef]
- Mancuso, C.A.; Canfield, J.L.; Singla, D.; Krishnan, A. A flexible, interpretable, and accurate approach for imputing the expression of unmeasured genes. Nucleic Acids Res. 2020, 48, e125. [Google Scholar] [CrossRef]
- Tibshirani, R. Regression shrinkage and selection via the Lasso. J. R. Stat. Soc. Ser. B 1996, 58, 267–288. [Google Scholar] [CrossRef]
- Liu, Y.; Jun, E.; Li, Q.; Heer, J. Latent Space Cartography: Visual Analysis of Vector Space Embeddings. Comput. Graph. Forum 2019, 38, 67–78. [Google Scholar] [CrossRef]
- Beck, J.V.; Arnold, K.J.; Arnold, K.J. Parameter Estimation in Engineering and Science; Wiley: New York, NY, USA, 1977; pp. 213–327, 501. [Google Scholar]
- Arisdakessian, C.; Poirion, O.; Yunits, B.; Zhu, X.; Garmire, L.X. DeepImpute: An accurate, fast, and scalable deep neural network method to impute single-cell RNA-seq data. Genome Biol. 2019, 20, 211. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Gu, J. VASC: Dimension Reduction and Visualization of Single-cell RNA-seq Data by Deep Variational Autoencoder. Genom. Proteom. Bioinform. 2018, 16, 320–331. [Google Scholar] [CrossRef] [PubMed]
- Way, G.P.; Greene, C.S. Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. In Biocomputing 2018: Proceedings of the Pacific Symposium, 2018; World Scientific Publishing Company: Singapore, 2018; Volume 23, pp. 80–91. [Google Scholar]
- Pham, T.H.; Qiu, Y.; Zeng, J.; Xie, L.; Zhang, P. A deep learning framework for high-throughput mechanism-driven phenotype compound screening and its application to COVID-19 drug repurposing. Nat. Mach. Intell. 2021, 3, 247–257. [Google Scholar] [CrossRef] [PubMed]
- Lotfollahi, M.; Wolf, F.A.; Theis, F.J. scGen predicts single-cell perturbation responses. Nat. Methods 2019, 16, 715–721. [Google Scholar] [CrossRef]
- Xie, R.; Wen, J.; Quitadamo, A.; Cheng, J.; Shi, X. A deep auto-encoder model for gene expression prediction. BMC Genom. 2017, 18, 845. [Google Scholar] [CrossRef] [PubMed]
- Dincer, A.B.; Janizek, J.D.; Lee, S.-I. Adversarial deconfounding autoencoder for learning robust gene expression embeddings. Bioinformatics 2020, 36, i573–i582. [Google Scholar] [CrossRef]
- Qiu, Y.; Lu, T.; Lim, H.; Xie, L. A Bayesian approach to accurate and robust signature detection on LINCS L1000 data. Bioinformatics 2020, 36, 2787–2795. [Google Scholar] [CrossRef]
- Snoek, J.; Larochelle, H.; Adams, R.P. Practical Bayesian Optimization of Machine Learning Algorithms. arXiv 2012, arXiv:1206.2944. [Google Scholar]
- Kingma, D.P.; Ba, J. Adam: A Method for Stochastic Optimization. arXiv 2014, arXiv:1412.6980. [Google Scholar]
- Chollet, F. Keras. 2015. Available online: https://keras.io (accessed on 5 December 2021).
- Abadi, M.; Agarwal, A.; Barham, P.; Brevdo, E.; Chen, Z.; Citro, C.; Greg, S.; Davis, A.; Dean, J.; Devin, M.; et al. TensorFlow: Large-Scale Machine Learning on Heterogeneous Distributed Systems. arXiv 2016, arXiv:1603.04467. [Google Scholar]
- Saito, T.; Rehmsmeier, M. The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS ONE 2015, 10, e0118432. [Google Scholar] [CrossRef] [PubMed]
- Marbach, D.; Lamparter, D.; Quon, G.; Kellis, M.; Kutalik, Z.; Bergmann, S. Tissue-specific regulatory circuits reveal variable modular perturbations across complex diseases. Nat. Methods 2016, 13, 366–370. [Google Scholar] [CrossRef] [PubMed]
- Schultz, A.; Qutub, A.A. Reconstruction of Tissue-Specific Metabolic Networks Using CORDA. PLoS Comput. Biol. 2016, 12, e1004808. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Petsalaki, E. Large-scale datasets uncovering cell signalling networks in cancer: Context matters. Curr. Opin. Genet. Dev. 2019, 54, 118–124. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Size | MCF7 | A375 | HT29 | PC3 | HA1E | YAPC | HELA |
|---|---|---|---|---|---|---|---|
| Total: | 1000 | 817 | 596 | 833 | 792 | 456 | 800 |
| Test 1: | |||||||
| Mean/Regression | 250/253 | 354/377 | 263/272 | 240/246 | 267/280 | 209/210 | 173/174 |
| Cell | MCF7 | A375 | HT29 | PC3 | HA1E | YAPC | HELA | |
|---|---|---|---|---|---|---|---|---|
| Method | ||||||||
| Predictor: Mean (μ) + Corrector: PCA, AE, PCA + AE, AE + PCA | ||||||||
| μ | ρ | 0.59 | 0.58 | 0.6 | 0.59 | 0.58 | 0.58 | 0.62 |
| AUROC | 0.78/0.80 | 0.78/0.79 | 0.79/0.81 | 0.79/0.80 | 0.79/0.79 | 0.78/0.79 | 0.80/0.81 | |
| AUPR | 0.66/0.65 | 0.65/0.67 | 0.66/0.67 | 0.65/0.65 | 0.65/0.66 | 0.65/0.68 | 0.68/0.69 | |
| μ + PCA | 0.76 * | 0.77 * | 0.77 * | 0.75 * | 0.74 * | 0.72 * | 0.76 * | |
| 0.87 */0.89 * | 0.88 */0.89 * | 0.87 */0.89 * | 0.87 */0.88 * | 0.86 */0.87 * | 0.85 */0.87 * | 0.87 */0.89 * | ||
| 0.78 */0.79 * | 0.79 */0.82 * | 0.79 */0.81 * | 0.77 */0.78 * | 0.77 */0.79 * | 0.75 */0.79 * | 0.79 */0.81 * | ||
| μ + AE | 0.72 * | 0.67 * | 0.73 * | 0.71 * | 0.67 * | 0.69 * | 0.76 * | |
| 0.85 */0.87 * | 0.84 */0.84 * | 0.86 */0.87 * | 0.85 */0.86 * | 0.84 */0.84 * | 0.84 */0.86 * | 0.88 */0.89 * | ||
| 0.75 */0.76 * | 0.73 */0.74 * | 0.76 */0.78 * | 0.74 */0.74 * | 0.72 */0.72 * | 0.73 */0.76 * | 0.78 */0.80 * | ||
| μ + PCA + AE | 0.78 * | 0.78 * | 0.78 * | 0.77 * | 0.75 * | 0.74 * | 0.79 * | |
| 0.88 */0.90 * | 0.89 */0.90 * | 0.88 */0.90 * | 0.88 */0.89 * | 0.87 */0.88 * | 0.86 */0.88 * | 0.89 */0.90 * | ||
| 0.79 */0.81 * | 0.80 */0.83 * | 0.80 */0.82 * | 0.79 */0.80 * | 0.78 */0.80 * | 0.78 */0.81 * | 0.81 */0.83 * | ||
| μ + AE + PCA | 0.75 * | 0.76 * | 0.76 * | 0.73 * | 0.73 * | 0.69 | 0.76 | |
| 0.87 */0.88 * | 0.87 */0.88 * | 0.87 */0.89 * | 0.86/0.87 * | 0.86 */0.87 * | 0.84/0.86 | 0.87/0.89 | ||
| 0.77 */0.79 * | 0.78 */0.80 * | 0.78 */0.81 * | 0.76 */0.77 * | 0.76 */0.78 * | 0.74/0.77 | 0.79/0.80 | ||
| Predictor: Regression + Corrector: PCA, AE, PCA + AE, AE + PCA | ||||||||
| Regression | 0.55 | 0.56 | 0.56 | 0.56 | 0.54 | 0.52 | 0.54 | |
| 0.77/0.79 | 0.77/0.78 | 0.77/0.79 | 0.77/0.79 | 0.76/0.77 | 0.75/0.77 | 0.76/0.78 | ||
| 0.64/0.63 | 0.64/0.67 | 0.63/0.65 | 0.63/0.64 | 0.62/0.64 | 0.61/0.65 | 0.63/0.64 | ||
| Regression + PCA | 0.62 * | 0.69 * | 0.66 * | 0.61 * | 0.60 * | 0.62 * | 0.63 * | |
| 0.80 */0.83 * | 0.84 */0.86 * | 0.82 */0.84 * | 0.80 */0.82 * | 0.8 */0.81 * | 0.80 */0.83 * | 0.81 */0.83 * | ||
| 0.67 */0.69 * | 0.74 */0.76 * | 0.71 */0.73 * | 0.66 */0.68 * | 0.66 */0.69 * | 0.68 */0.72 * | 0.69 */0.71 * | ||
| Regression + AE | 0.46 * | 0.25 * | 0.44 * | 0.35 * | 0.23 * | 0.38 * | 0.49 * | |
| 0.73 */0.75 * | 0.63 */0.61 * | 0.72 */0.73 * | 0.67 */0.68 * | 0.62 */0.60 * | 0.69 */0.70 * | 0.74 */0.77 * | ||
| 0.58 */0.59 * | 0.51 */0.50 * | 0.58 */0.58 * | 0.52 */0.52 * | 0.49 */0.48 * | 0.55 */0.56 * | 0.60 */0.63 * | ||
| Regression + PCA + AE | 0.66 * | 0.72 * | 0.68 * | 0.63 * | 0.61 * | 0.66 * | 0.66 * | |
| 0.82 */0.85 * | 0.86 */0.87 * | 0.83 */0.85 * | 0.81 */0.83 * | 0.80 */0.82 * | 0.82 */0.85 * | 0.82 */0.85 * | ||
| 0.71 */0.73 * | 0.77 */0.78 * | 0.73 */0.76 * | 0.69 */0.70 * | 0.69 */0.71 * | 0.71 */0.75 * | 0.72 */0.74 * | ||
| Regress + AE + PCA | 0.45 | 0.28 * | 0.55 * | 0.39 * | 0.23 | 0.47 * | 0.47 | |
| 0.73/0.75 | 0.64 */0.63 * | 0.76 */0.79 * | 0.69 */0.71 * | 0.62/0.61 | 0.73 */0.74 * | 0.73/0.77 | ||
| 0.58/0.58 * | 0.54 */0.53 * | 0.64 */0.66 * | 0.54 */0.55 * | 0.50 */0.49 * | 0.59 */0.61 * | 0.59/0.63 | ||
| TT-WOPT | ||||||||
| TT-WOPT | 0.31 | 0.19 | 0.18 | 0.05 | 0.36 | 0.18 | 0.24 | |
| 0.65/0.69 | 0.59/0.60 | 0.59/0.61 | 0.52/0.55 | 0.68/0.69 | 0.57/0.62 | 0.62/0.64 | ||
| 0.48/0.50 | 0.43/0.46 | 0.43/0.43 | 0.35/0.37 | 0.52/0.53 | 0.41/0.45 | 0.44/0.48 | ||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chrysinas, P.; Chen, C.; Gunawan, R. CrossTx: Cross-Cell-Line Transcriptomic Signature Predictions. Processes 2024, 12, 332. https://doi.org/10.3390/pr12020332
Chrysinas P, Chen C, Gunawan R. CrossTx: Cross-Cell-Line Transcriptomic Signature Predictions. Processes. 2024; 12(2):332. https://doi.org/10.3390/pr12020332
Chicago/Turabian StyleChrysinas, Panagiotis, Changyou Chen, and Rudiyanto Gunawan. 2024. "CrossTx: Cross-Cell-Line Transcriptomic Signature Predictions" Processes 12, no. 2: 332. https://doi.org/10.3390/pr12020332
APA StyleChrysinas, P., Chen, C., & Gunawan, R. (2024). CrossTx: Cross-Cell-Line Transcriptomic Signature Predictions. Processes, 12(2), 332. https://doi.org/10.3390/pr12020332