
Reduction of Sulfoxides in Multigram Scale, an Alternative to the Use of Chlorinated Solvents



Abstract
:
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of Phenyl Propyl Sulfoxide (7)
2.3. Reduction in Sulfoxides
2.3.1. Phenyl Propyl Sulfide (8)
2.3.2. Albendazole Hydrochloride (12)
2.3.3. Reduction in Unstable Sulfoxides
2-(((3-Methyl-4-(2,2,2-trifluoroethoxy)pyridin-2-yl)methyl)thio)-1H-benzo[d]imidazole (10)
5-Methoxy-2-(((4-methoxy-3,5-dimethylpyridin-2-yl)methyl)thio)-1H-benzo[d]imidazole (14)
2.4. Characterization Techniques
3. Results and Discussion
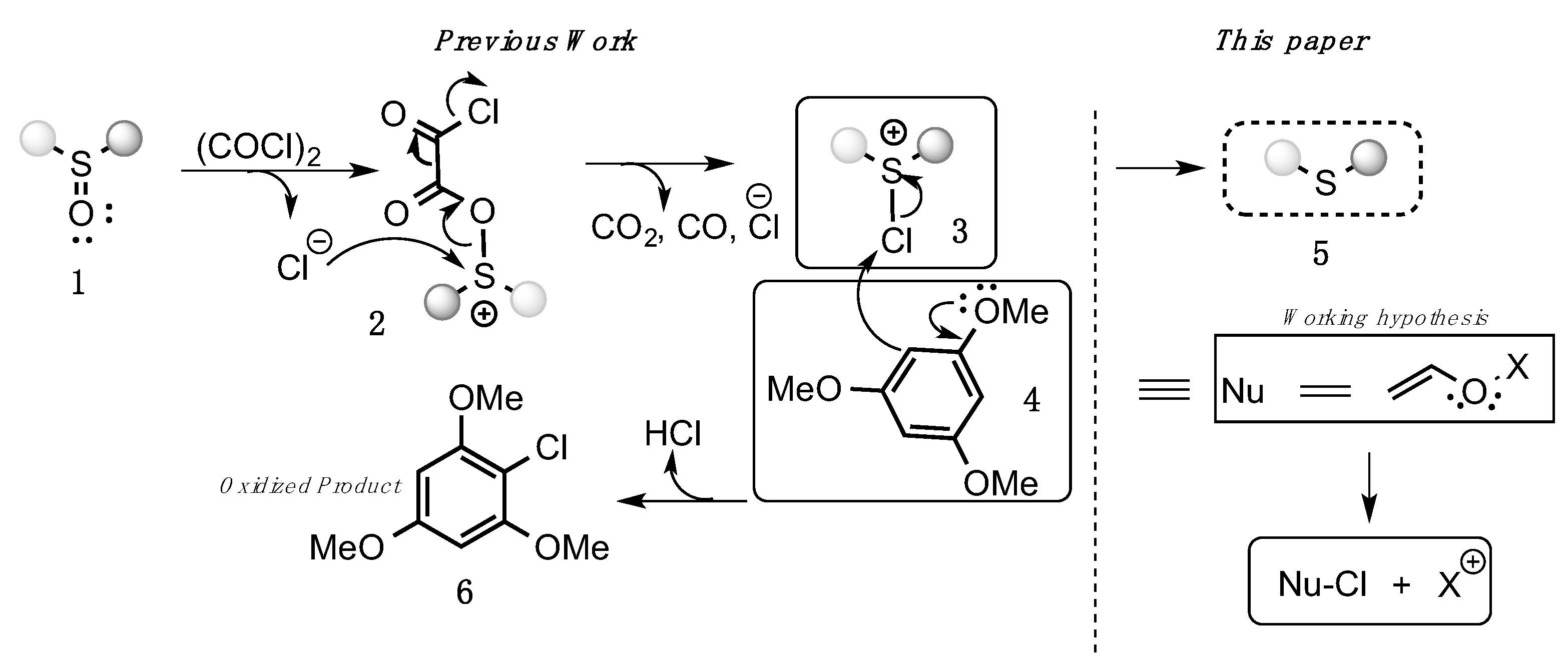
3.1. Optimization
3.2. Application to Substrates of Pharmaceutical Interest
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Witschi, C.; Doelker, E. Residual solvents in pharmaceutical products: Acceptable limits, influences on physicochemical properties, analytical methods and documented values. Eur. J. Pharm. Biopharm. 1997, 43, 215–242. [Google Scholar] [CrossRef]
- Madesclaire, M. Reduction of sulfoxides to thioethers. Tetrahedron 1988, 44, 6537–6580. [Google Scholar] [CrossRef]
- Drabowicz, J.; Togo, H.; Mikołajczyk, M.; Oae, S. Reduction of Sulfoxides. A Review. Org. Prep. Proced. Int. 1984, 16, 171–198. [Google Scholar] [CrossRef]
- Shiri, L.; Kazemi, M. Deoxygenation of sulfoxides. Res. Chem. Intermed. 2017, 43, 6007–6041. [Google Scholar] [CrossRef]
- Kong, Z.; Pan, C.; Li, M.; Wen, L.; Guo, W. Scalable electrochemical reduction of sulfoxides to sulfides. Green Chem. 2021, 23, 2773–2777. [Google Scholar] [CrossRef]
- Kuwahara, Y.; Yoshimura, Y.; Haematsu, K.; Yamashita, H. Mild Deoxygenation of Sulfoxides over Plasmonic Molybdenum Oxide Hybrid with Dramatic Activity Enhancement under Visible Light. J. Am. Chem. Soc. 2018, 140, 9203–9210. [Google Scholar] [CrossRef] [PubMed]
- García, N.; García-García, P.; Fernández-Rodríguez, M.A.; Rubio, R.; Pedrosa, M.R.; Arnáiz, F.J.; Sanz, R. Pinacol as a New Green Reducing Agent: Molybdenum-Catalyzed Chemoselective Reduction of Sulfoxides and Nitroaromatics. Adv. Synth. Catal. 2012, 354, 321–327. [Google Scholar] [CrossRef] [Green Version]
- Ma, R.; Liu, A.-H.; Huang, C.-B.; Li, X.-D.; He, L.-N. Reduction of sulfoxides and pyridine-N-oxides over iron powder with water as hydrogen source promoted by carbon dioxide. Green Chem. 2013, 15, 1274–1279. [Google Scholar] [CrossRef]
- Gevorgyan, A.; Mkrtchyan, S.; Grigoryan, T.; Iaroshenko, V.O. Application of Silicon-Initiated Water Splitting for the Reduction of Organic Substrates. ChemPlusChem 2018, 83, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Guzmán, P.; Mahecha-Mahecha, C.; Gamba-Sánchez, D. Electrophilic Chlorine from Chlorosulfonium Salts: A Highly Chemoselective Reduction of Sulfoxides. Chem. Eur. J. 2020, 26, 10348–10354. [Google Scholar] [CrossRef]
- Kodama, K.; Morita, R.; Hirose, T. Formation of Ternary Inclusion Crystal and Enantioseparation of Alkyl Aryl Sulfoxides by the Salt of Urea-Modified l-Phenylalanine and an Achiral Amine. Cryst. Growth Des. 2016, 16, 5206–5213. [Google Scholar] [CrossRef]
- Dawar, P.; Bhagavan Raju, M.; Ramakrishna, R.A. One-pot esterification and Ritter reaction: Chemo- and regioselectivity from tert-butyl methyl ether. Tetrahedron Lett. 2011, 52, 4262–4265. [Google Scholar] [CrossRef]
- Mallesha, N.; Prahlada Rao, S.; Suhas, R.; Channe Gowda, D. An efficient synthesis of tert-butyl ethers/esters of alcohols/amino acids using methyl tert-butyl ether. Tetrahedron Lett. 2012, 53, 641–645. [Google Scholar] [CrossRef]
- Vrbanec, T.; Šket, P.; Merzel, F.; Smrkolj, M.; Grdadolnik, J. Spectroscopic Characterization of Omeprazole and Its Salts. J. Spectrosc. 2017, 2017, 6505706. [Google Scholar] [CrossRef] [Green Version]

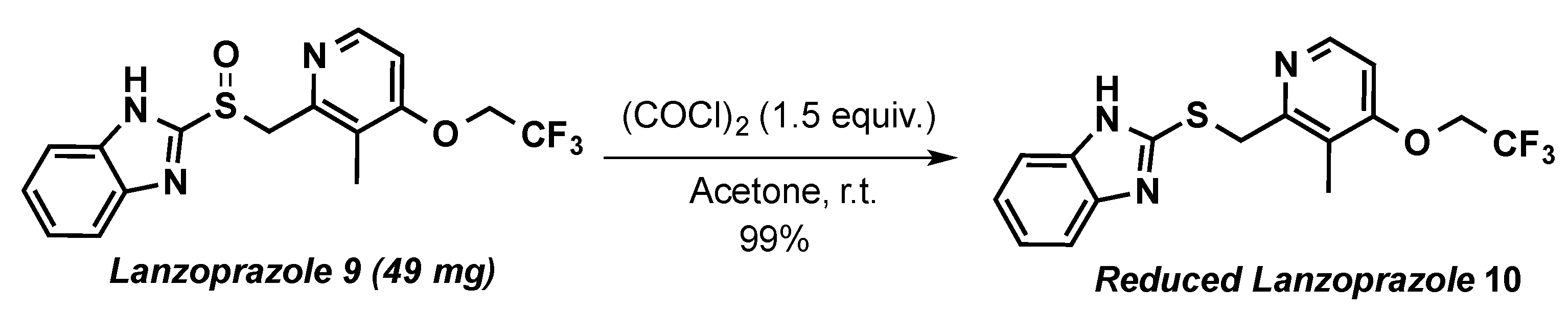

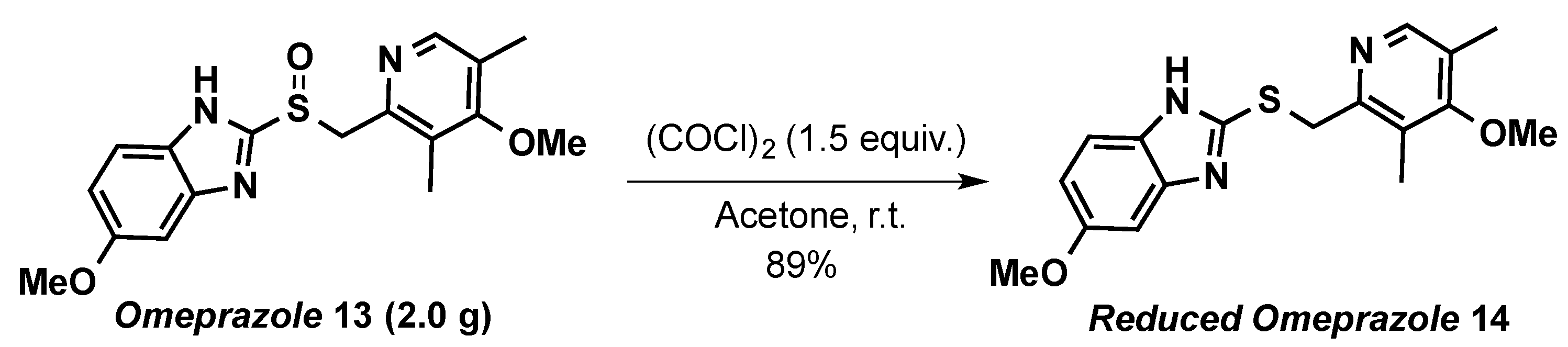

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Nucleophile | Solvent | Concentration | Yield (%) | Observations |
| 1 | Vinyl acetate | N.A. | 0.15 M 1 | N.D. | Sulfide + unidentified mixture of polar products 2 |
| 2 | Ethyl vinyl ether | N.A. | 0.15 M 1 | N.D. | Sulfide + unidentified mixture of polar products 2 |
| 3 | Butyl vinyl ether | N.A. | 0.15 M 1 | N.D. | Sulfide + unidentified mixture of polar products 2 |
| 4 | Ethyl vinyl ether | tBuOMe | 0.15 M | N.D. | No conversion |
| 5 | Ethyl vinyl ether | DCM | 0.15 M | N.D. | Complete conversion, small impurities |
| 6 | Ethyl vinyl ether | Acetone | 0.15 M | N.D. | Complete conversion, clean product |
| 7 | Ethyl vinyl ether | Acetone | 0.50 M | N.D. | Complete conversion, medium impurities |
| 8 | Ethyl vinyl ether | Acetone | 1.00 M | N.D. | Complete conversion, dirty crude |
| 9 | Ethyl vinyl ether | Acetone | 0.15 M 3 | 93 | Product clean without further purification |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adarve-Cardona, L.; Gamba-Sánchez, D. Reduction of Sulfoxides in Multigram Scale, an Alternative to the Use of Chlorinated Solvents. Processes 2022, 10, 1115. https://doi.org/10.3390/pr10061115
Adarve-Cardona L, Gamba-Sánchez D. Reduction of Sulfoxides in Multigram Scale, an Alternative to the Use of Chlorinated Solvents. Processes. 2022; 10(6):1115. https://doi.org/10.3390/pr10061115
Chicago/Turabian StyleAdarve-Cardona, Laura, and Diego Gamba-Sánchez. 2022. "Reduction of Sulfoxides in Multigram Scale, an Alternative to the Use of Chlorinated Solvents" Processes 10, no. 6: 1115. https://doi.org/10.3390/pr10061115
APA StyleAdarve-Cardona, L., & Gamba-Sánchez, D. (2022). Reduction of Sulfoxides in Multigram Scale, an Alternative to the Use of Chlorinated Solvents. Processes, 10(6), 1115. https://doi.org/10.3390/pr10061115