
Ileal Bile Acid Transporter Inhibition Reduces Post-Transplant Diarrhea and Growth Failure in FIC1 Disease—A Case Report

 , ,
, ,  , , ,
, , ,
Abstract
:1. Introduction
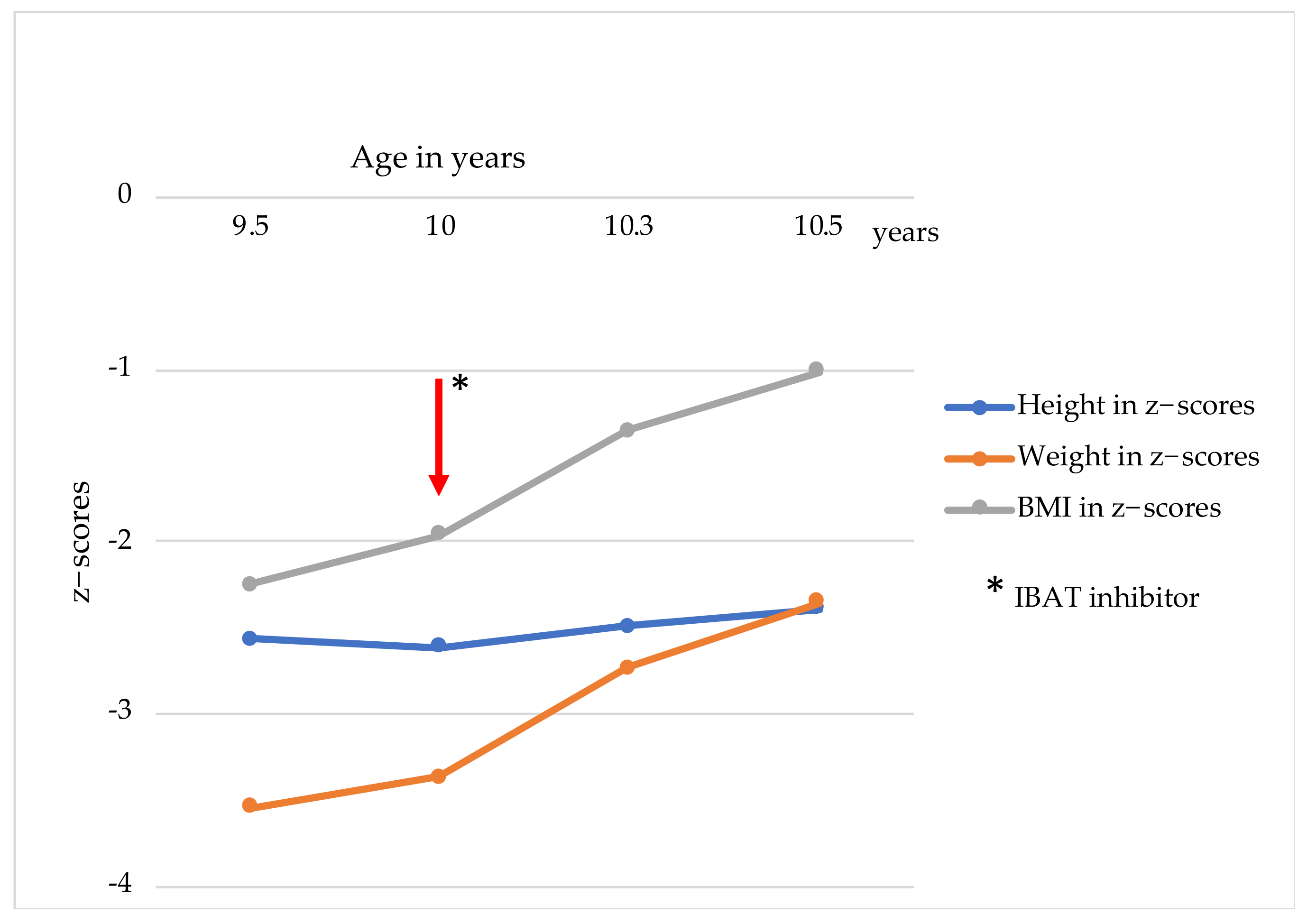
2. Case Report
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Laboratory Parameters | Age of 1 Year | Before PEBD | Before LT | Normal Range |
|---|---|---|---|---|
| Bilirubin (µmol/L) | 71 | 170 | 70 | 3–17 |
| ALAT (U/I) | 39 | 72 | 56 | <35 |
| ASAT (U/I) | 94 | 147 | 113 | <45 |
| INR | 1.11 | 1.20 | 1.25 | 0.90–1.25 |
| Vitamin A (mg/L) | 0.39 | 0.17 | 0.33 | 0.3–0.06 |
| Vitamin E (mg/L) | 13.34 | 7.36 | 5.65 | 3–9 |
| 25-OH-D3 (ng/mL) | n.d. | 13.3 | 32.0 | 20–70 |
References
- Chen, H.-L.; Wu, S.-H.; Hsu, S.-H.; Liou, B.-Y.; Chen, H.-L.; Chang, M.-H. Jaundice revisited: Recent advances in the diagnosis and treatment of inherited cholestatic liver diseases. J. Biomed. Sci. 2018, 25, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bull, L.N.; Van Eijk, M.J.T.; Pawlikowska, L.; Deyoung, J.A.; Juijn, J.A.; Liao, M.; Klomp, L.W.J.; Lomri, N.; Berger, R.; Scharschmidt, B.R.; et al. A gene encoding a P-type ATPase mutated in two forms of hereditary cholestasis. Nat. Genet. 1998, 18, 219–224. [Google Scholar] [CrossRef]
- Verhulst, P.M.; van der Velden, L.M.; Oorschot, V.; van Faassen, E.E.; Klumperman, J.; Houwen, R.H.J.; Pomorski, T.G.; Holthuis, J.C.M.; Klomp, L.W.J. A flippase-independent function of ATP8B1, the protein affected in familial intrahepatic cholestasis type 1, is required for apical protein expression and microvillus formation in polarized epithelial cells. Hepatology 2010, 51, 2049–2060. [Google Scholar] [CrossRef]
- Knisely, A.; Houwen, R.H. Liver Steatosis and Diarrhea After Liver Transplantation for Progressive Familial Intrahepatic Cholestasis Type 1: Can Biliary Diversion Solve These Problems? J. Pediatr. Gastroenterol. Nutr. 2020, 72, 341–342. [Google Scholar] [CrossRef]
- Okamoto, T.; Sonoda, M.; Ogawa, E.; Ito, S.; Togawa, T.; Hayashi, H.; Okajima, H.; Uemoto, S. Long-term Outcomes of Living-donor Liver Transplantation for Progressive Familial Intrahepatic Cholestasis Type 1. J. Pediatr. Gastroenterol. Nutr. 2020, 72, 425–429. [Google Scholar] [CrossRef]
- Miyagawa-Hayashino, A.; Egawa, H.; Yorifuji, T.; Hasegawa, M.; Haga, H.; Tsuruyama, T.; Wen, M.-C.; Sumazaki, R.; Manabe, T.; Uemoto, S. Allograft steatohepatitis in progressive familial intrahepatic cholestasis type 1 after living donor liver transplantation. Liver Transplant. 2009, 15, 610–618. [Google Scholar] [CrossRef] [PubMed]
- Bull, L.N.; Thompson, R. Progressive Familial Intrahepatic Cholestasis. Clin. Liver Dis. 2018, 22, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Jankowska, I.; Pawłowska, J.; Szymczak, M.; Ismail, H.; Broniszczak, D.; Cielecka-Kuszyk, J.; Socha, P.; Jarzębicka, D.; Czubkowski, P. A Report of 2 Infant Siblings with Progressive Intrahepatic Familial Cholestasis Type 1 and a Novel Homozygous Mutation in the ATP8B1 Gene Treated with Partial External Biliary Diversion and Liver Transplant. Am. J. Case Rep. 2021, 22, e932374-1–e932374-7. [Google Scholar] [CrossRef]
- Alrabadi, L.S.; Morotti, R.A.; Valentino, P.L.; Rodriguez-Davalos, M.I.; Ekong, U.D.; Emre, S.H. Biliary drainage as treatment for allograft steatosis following liver transplantation for PFIC-1 disease: A single-center experience. Pediatr. Transplant. 2018, 22, e13184. [Google Scholar] [CrossRef] [PubMed]
- Mali, V.P.; Fukuda, A.; Shigeta, T.; Uchida, H.; Hirata, Y.; Rahayatri, T.H.; Kanazawa, H.; Sasaki, K.; Goyet, J.D.V.D.; Kasahara, M. Total internal biliary diversion during liver transplantation for type 1 progressive familial intrahepatic cholestasis: A novel approach. Pediatr. Transplant. 2016, 20, 981–986. [Google Scholar] [CrossRef]
- Wong, B.S.; Camilleri, M. Elobixibat for the treatment of constipation. Expert Opin. Investig. Drugs 2012, 22, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Graffner, H.; Gillberg, P.; Rikner, L.; Marschall, H.-U. The ileal bile acid transporter inhibitor A4250 decreases serum bile acids by interrupting the enterohepatic circulation. Aliment. Pharmacol. Ther. 2015, 43, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Shirley, M. Maralixibat: First Approval. Drugs 2021, 82, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E.D. Odevixibat: First Approval. Drugs 2021, 81, 1781–1786. [Google Scholar] [CrossRef]
- Reshetnyak, V.I. Physiological and molecular biochemical mechanisms of bile formation. World J. Gastroenterol. 2013, 19, 7341–7360. [Google Scholar] [CrossRef]
- Bajor, A.; Gillberg, P.-G.; Abrahamsson, H. Bile acids: Short and long term effects in the intestine. Scand. J. Gastroenterol. 2010, 45, 645–664. [Google Scholar] [CrossRef]
- Acosta, A.; Camilleri, M. Elobixibat and its potential role in chronic idiopathic constipation. Ther. Adv. Gastroenterol. 2014, 7, 167–175. [Google Scholar] [CrossRef] [Green Version]
- Mosińska, P.; Fichna, J.; Storr, M. Inhibition of ileal bile acid transporter: An emerging therapeutic strategy for chronic idiopathic constipation. World J. Gastroenterol. 2015, 21, 7436–7442. [Google Scholar] [CrossRef]
- Wong, B.S.; Camilleri, M.; McKinzie, S.; Burton, D.; Graffner, H.; Zinsmeister, A.R. Effects of A3309, an Ileal Bile Acid Transporter Inhibitor, on Colonic Transit and Symptoms in Females with Functional Constipation. Am. J. Gastroenterol. 2011, 106, 2154–2164. [Google Scholar] [CrossRef]
- Chey, W.D.; Camilleri, M.; Chang, L.; Rikner, L.; Graffner, H. A Randomized Placebo-Controlled Phase IIb Trial of A3309, A Bile Acid Transporter Inhibitor, for Chronic Idiopathic Constipation. Am. J. Gastroenterol. 2011, 106, 1803–1812. [Google Scholar] [CrossRef] [Green Version]
- di Gregorio, M.; Cautela, J.; Galantini, L. Physiology and Physical Chemistry of Bile Acids. Int. J. Mol. Sci. 2021, 22, 1780. [Google Scholar] [CrossRef]
- Van De Peppel, I.P.; Verkade, H.J.; Jonker, J.W. Metabolic consequences of ileal interruption of the enterohepatic circulation of bile acids. Am. J. Physiol. Liver Physiol. 2020, 319, G619–G625. [Google Scholar] [CrossRef]
- Gonzales, E.; Hardikar, W.; Stormon, M.; Baker, A.; Hierro, L.; Gliwicz, D.; Lacaille, F.; Lachaux, A.; Sturm, E.; Setchell, K.D.R.; et al. Efficacy and safety of maralixibat treatment in patients with Alagille syndrome and cholestatic pruritus (ICONIC): A randomised phase 2 study. Lancet 2021, 398, 1581–1592. [Google Scholar] [CrossRef]
- Ooba, N.; Takahashi, Y.; Nagamura, M.; Takahashi, M.; Ushida, M.; Kawakami, E.; Kimura, M.; Sato, T.; Tokuyoshi, J.; Miyazaki, C.; et al. Safety of elobixibat and lubiprostone in Japanese patients with chronic constipation: A retrospective cohort study. Expert Opin. Drug Saf. 2021, 20, 1553–1558. [Google Scholar] [CrossRef]
- Al Shaer, D.; Al Musaimi, O.; Albericio, F.; de la Torre, B.G. 2021 FDA TIDES (Peptides and Oligonucleotides) Harvest. Pharmaceuticals 2022, 15, 222. [Google Scholar] [CrossRef]
- Ishak, K.; Baptista, A.; Bianchi, L.; Callea, F.; De Groote, J.; Gudat, F.; Denk, H.; Desmet, V.; Korb, G.; Macsween, R.N.; et al. Histological grading and staging of chronic hepatitis. J. Hepatol. 1995, 22, 696–699. [Google Scholar] [CrossRef]
- Nicastro, E.; Stephenne, X.; Smets, F.; Fusaro, F.; De Magnée, C.; Reding, R.; Sokal, E.M. Recovery of graft steatosis and protein-losing enteropathy after biliary diversion in a PFIC 1 liver transplanted child. Pediatr. Transplant. 2011, 16, E177–E182. [Google Scholar] [CrossRef]
- Kamath, B.M.; Stein, P.; Houwen, R.H.J.; Verkade, H.J. Potential of ileal bile acid transporter inhibition as a therapeutic target in Alagille syndrome and progressive familial intrahepatic cholestasis. Liver Int. 2020, 40, 1812–1822. [Google Scholar] [CrossRef]
- Baumann, U.; Sturm, E.; Lacaille, F.; Gonzalès, E.; Arnell, H.; Fischler, B.; Jørgensen, M.H.; Thompson, R.J.; Mattsson, J.P.; Ekelund, M.; et al. Effects of odevixibat on pruritus and bile acids in children with cholestatic liver disease: Phase 2 study. Clin. Res. Hepatol. Gastroenterol. 2021, 45, 101751. [Google Scholar] [CrossRef]
- Sanchez, P.; Farkhondeh, A.; Pavlinov, I.; Baumgaertel, K.; Rodems, S.; Zheng, W. Therapeutics Development for Alagille Syndrome. Front. Pharmacol. 2021, 12, 704586. [Google Scholar] [CrossRef]
- Karpen, S.J.; Kelly, D.; Mack, C.; Stein, P. Ileal bile acid transporter inhibition as an anticholestatic therapeutic target in biliary atresia and other cholestatic disorders. Hepatol. Int. 2020, 14, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Slavetinsky, C.; Sturm, E. Odevixibat and partial external biliary diversion showed equal improvement of cholestasis in a patient with progressive familial intrahepatic cholestasis. BMJ Case Rep. 2020, 13, e234185. [Google Scholar] [CrossRef] [PubMed]





| Laboratory Parameters | Before Treatment | After 3 Months IBAT Inhibitor | After 7 Months IBAT Inhibitor | Normal Range |
|---|---|---|---|---|
| Bilirubin (µmol/L) | 3 | 3 | 3 | 3–17 |
| ALAT (U/I) | 33 | 42 | 37 | <35 |
| ASAT (U/I) | 29 | 50 | 45 | <45 |
| INR | 1.01 | 0.97 | 0.98 | 0.90–1.25 |
| Vitamin A (mg/L) | 0.21 | 0.36 | 0.43 | 0.3–0.06 |
| Vitamin E (mg/L) | 10.3 | 11.67 | 13.82 | 3–9 |
| 25-OH-D3 (ng/mL) | 32.8 | 40.3 | 45 | 20–70 |
| Tacrolimus (µg/L) | 3.3 | 1.5 | 1.8 | 2–5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ohlendorf, J.; Goldschmidt, I.; Junge, N.; Laue, T.; Nasser, H.; Jäckel, E.; Mutschler, F.; Pfister, E.-D.; Herebian, D.; Keitel, V.; et al. Ileal Bile Acid Transporter Inhibition Reduces Post-Transplant Diarrhea and Growth Failure in FIC1 Disease—A Case Report. Children 2022, 9, 669. https://doi.org/10.3390/children9050669
Ohlendorf J, Goldschmidt I, Junge N, Laue T, Nasser H, Jäckel E, Mutschler F, Pfister E-D, Herebian D, Keitel V, et al. Ileal Bile Acid Transporter Inhibition Reduces Post-Transplant Diarrhea and Growth Failure in FIC1 Disease—A Case Report. Children. 2022; 9(5):669. https://doi.org/10.3390/children9050669
Chicago/Turabian StyleOhlendorf, Johanna, Imeke Goldschmidt, Norman Junge, Tobias Laue, Hamoud Nasser, Elmar Jäckel, Frauke Mutschler, Eva-Doreen Pfister, Diran Herebian, Verena Keitel, and et al. 2022. "Ileal Bile Acid Transporter Inhibition Reduces Post-Transplant Diarrhea and Growth Failure in FIC1 Disease—A Case Report" Children 9, no. 5: 669. https://doi.org/10.3390/children9050669
APA StyleOhlendorf, J., Goldschmidt, I., Junge, N., Laue, T., Nasser, H., Jäckel, E., Mutschler, F., Pfister, E.-D., Herebian, D., Keitel, V., & Baumann, U. (2022). Ileal Bile Acid Transporter Inhibition Reduces Post-Transplant Diarrhea and Growth Failure in FIC1 Disease—A Case Report. Children, 9(5), 669. https://doi.org/10.3390/children9050669