Differential Diagnosis of Pediatric Multiple Sclerosis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
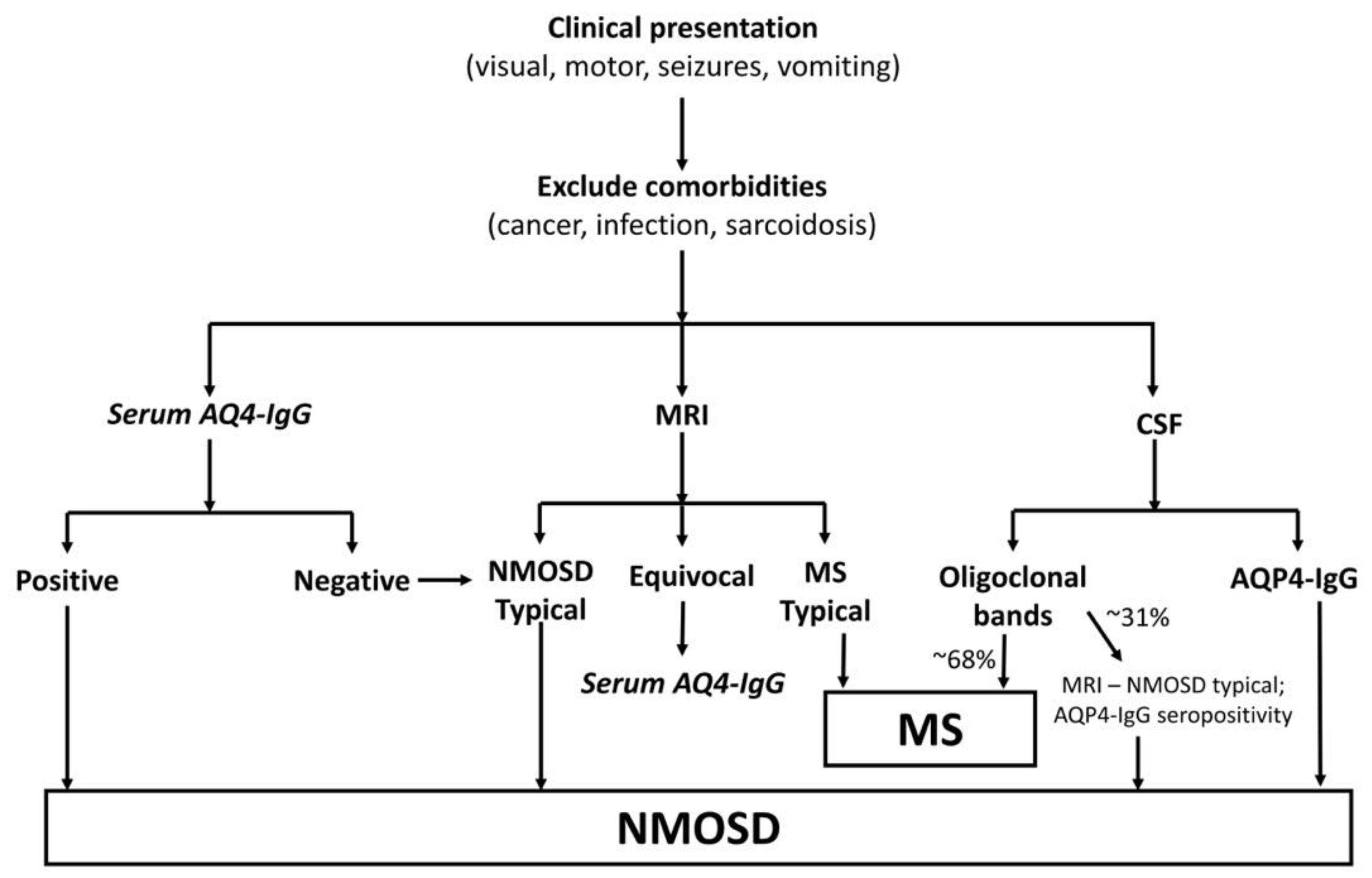
2. Neuromyelitis Optica Spectrum Disorders
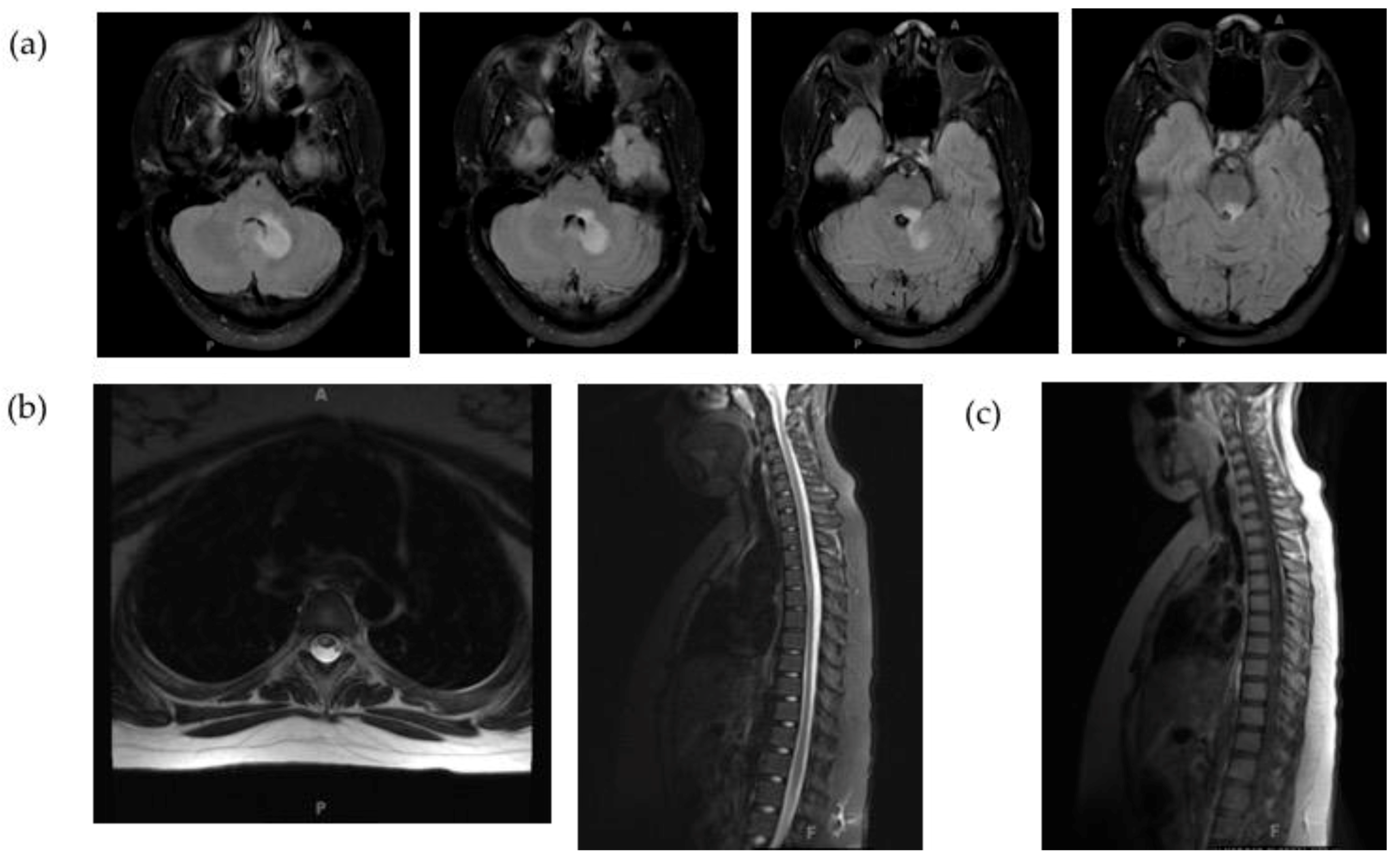
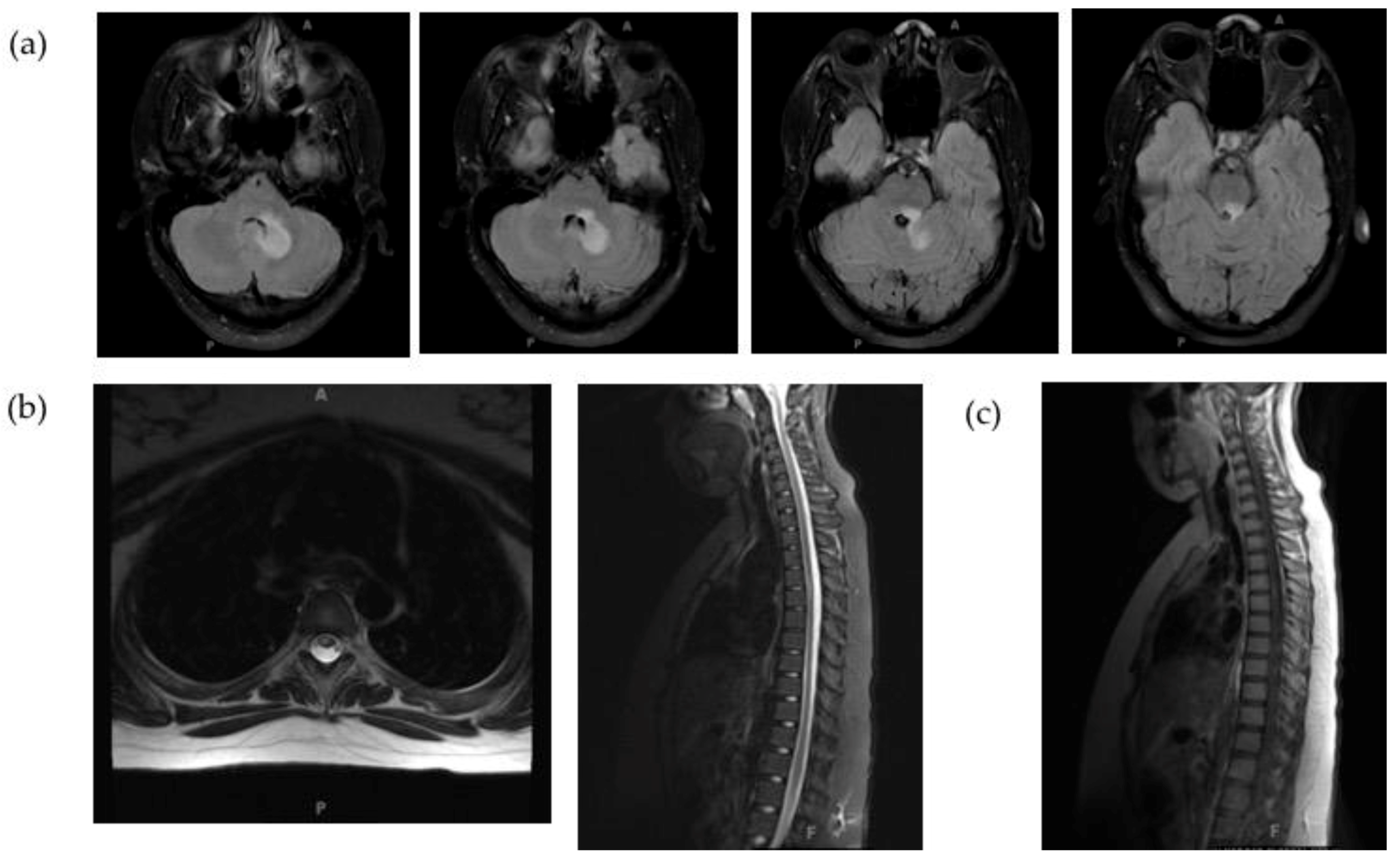
2.1. Clinical Case
2.2. Epidemiology and Pathophysiology
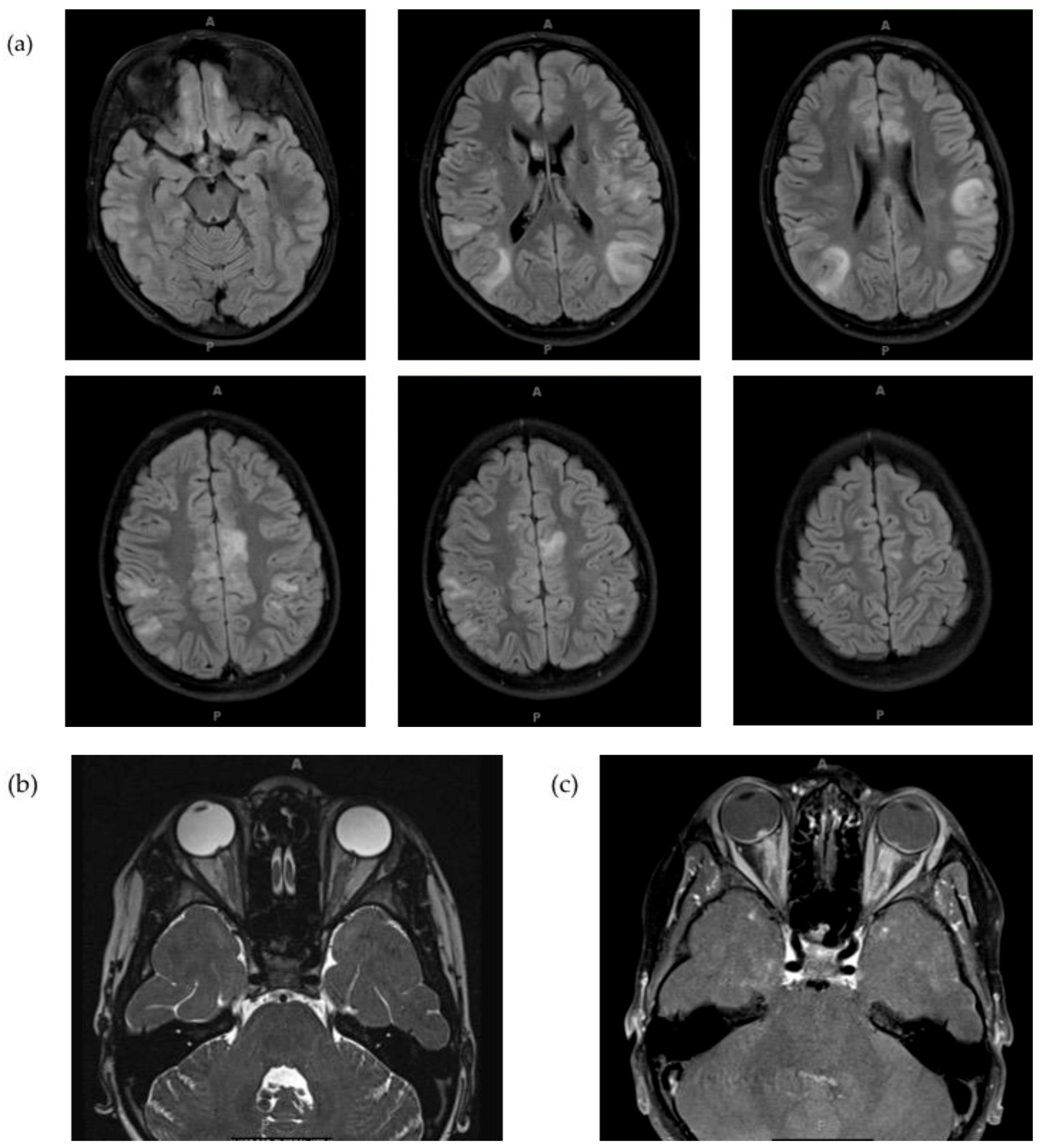
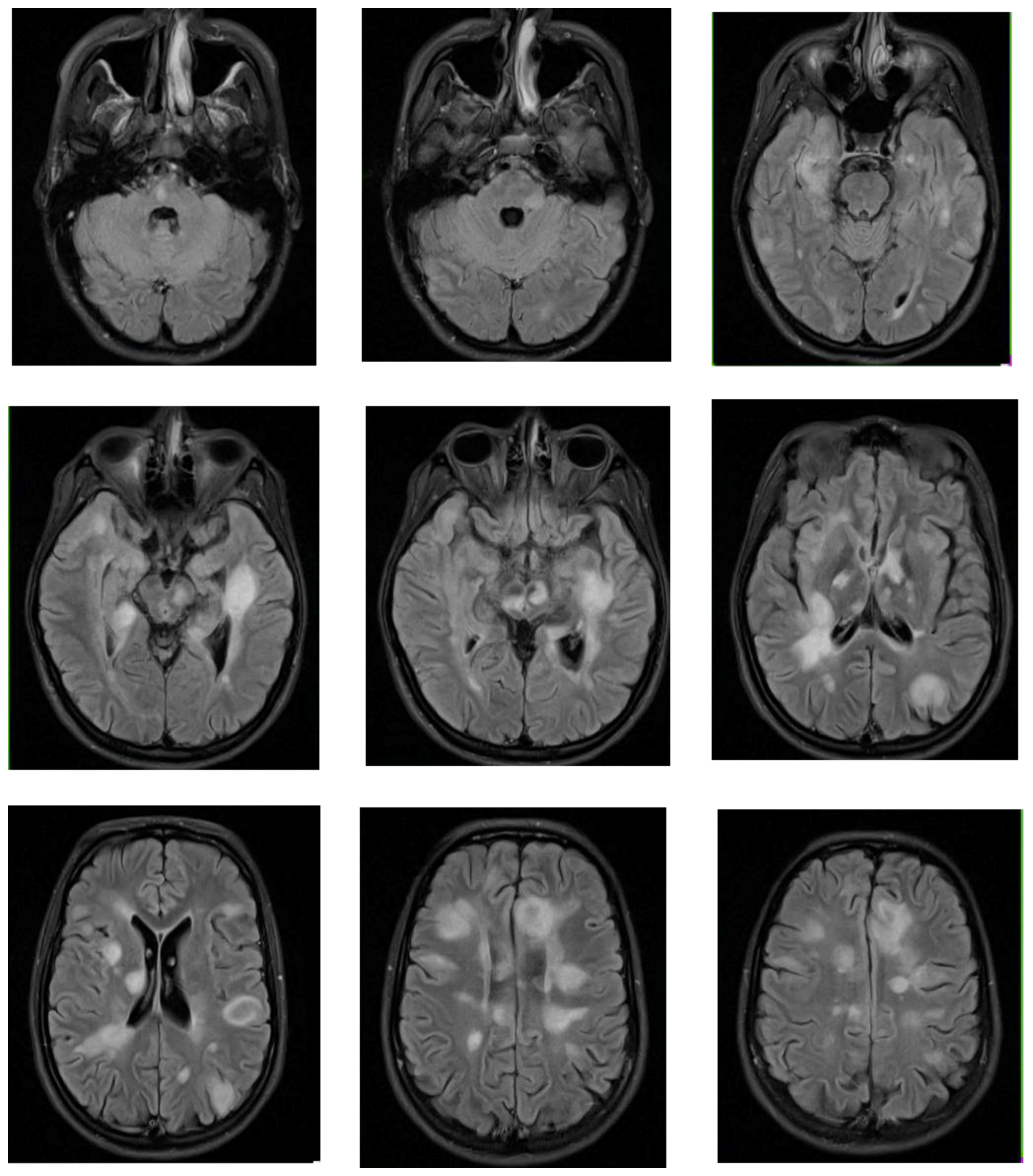
2.3. Clinical Presentation
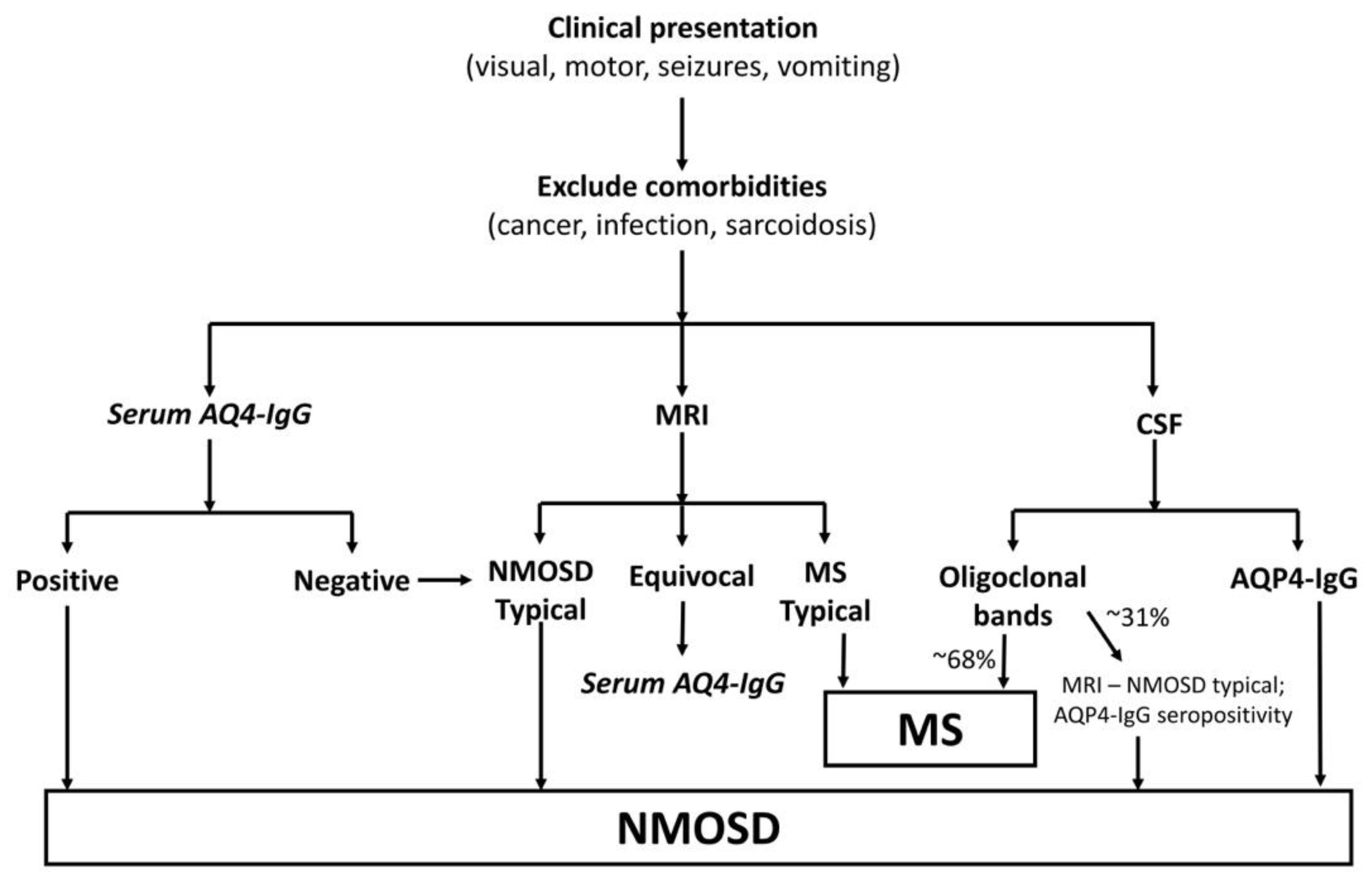
2.4. Diagnosis
2.5. Treatment and Prognosis
3. MOG Antibody-Associated Disorders
3.1. Clinical Case
3.2. Epidemiology and Pathophysiology
3.3. Clinical Presentation
3.4. Diagnosis
3.5. Treatment and Prognosis
4. Acute Disseminated Encephalomyelitis
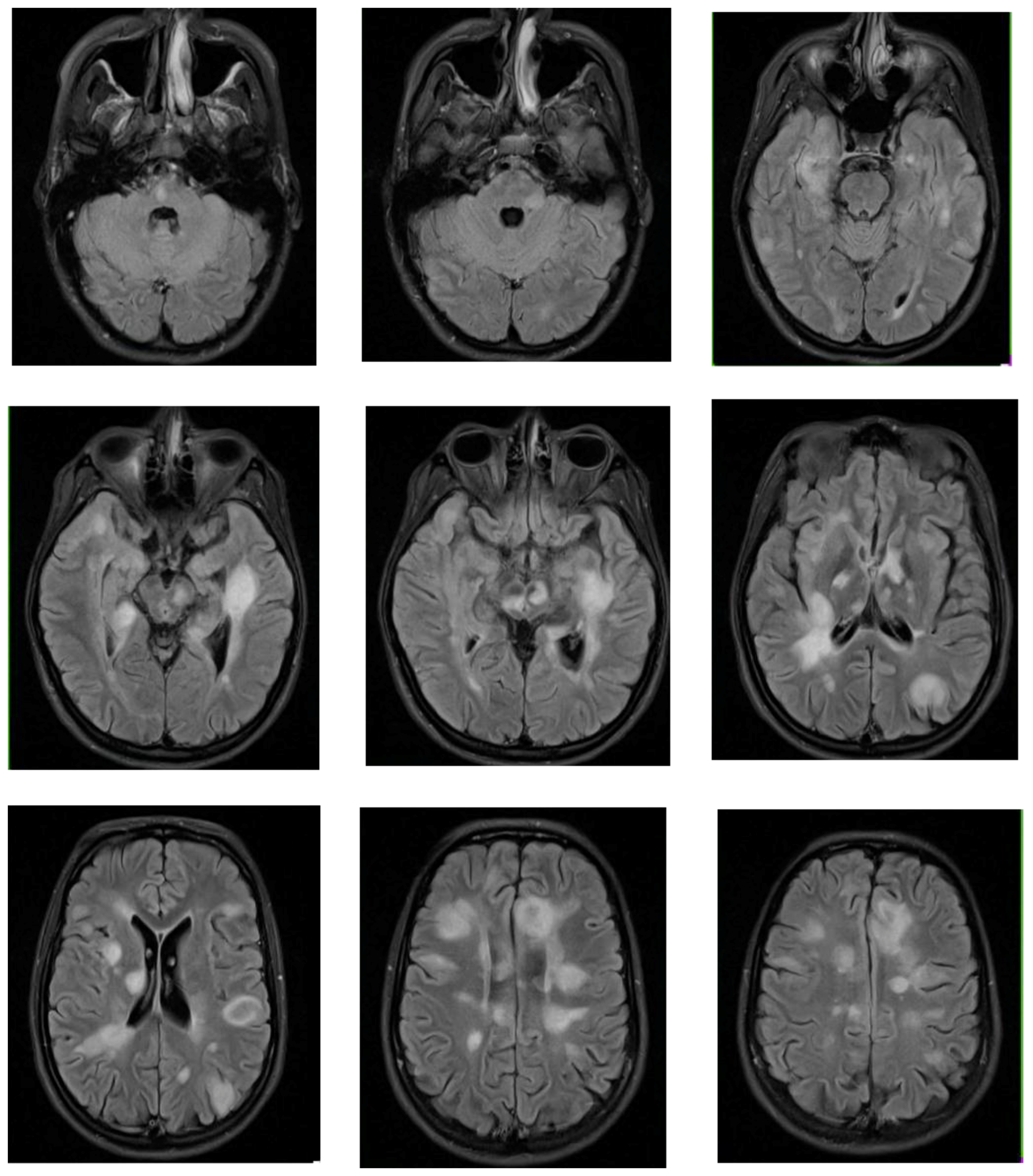
4.1. Clinical Case
4.2. Epidemiology and Pathophysiology
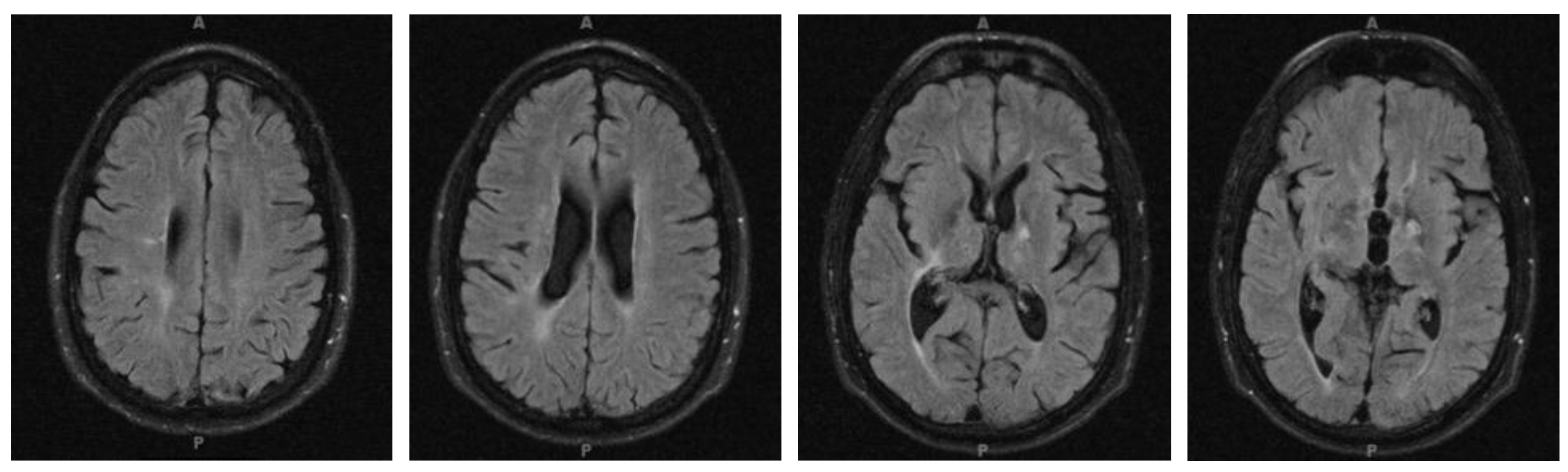
4.3. Clinical Presentation
4.4. Diagnosis
4.5. Treatment and Prognosis
5. Metabolic and Mitochondrial Disorders
5.1. Clinical Case
5.2. Epidemiology and Pathophysiology
5.3. Clinical Presentation
5.4. Diagnosis
5.5. Treatment and Prognosis
6. Leukodystrophies
6.1. Clinical Case
6.2. Epidemiology and Pathophysiology
6.3. Clinical Presentation
6.4. Diagnosis
6.5. Treatment and Prognosis
7. Conclusions
Conflicts of Interest
References
- Chitnis, T.; Ness, J.; Krupp, L.; Waubant, E.; Hunt, T.; Olsen, C.S.; Rodriguez, M.; Lotze, T.; Gorman, M.; Benson, L.; et al. Clinical features of neuromyelitis optica in children: US Network of Pediatric MS Centers report. Neurology 2016, 86, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Fragoso, Y.D.; Adoni, T.; Bichuetti, D.B.; Brooks, J.B.; Ferreira, M.L.; Oliveira, E.M.; Oliveira, C.L.; Ribeiro, S.B.; Silva, A.E.; Siquineli, F. Neuromyelitis optica and pregnancy. J. Neurol. 2013, 260, 2614–2619. [Google Scholar] [CrossRef] [PubMed]
- Bourre, B.; Marignier, R.; Zephir, H.; Papeix, C.; Brassat, D.; Castelnovo, G.; Collongues, N.; Vukusic, S.; Labauge, P.; Outteryck, O.; et al. Neuromyelitis optica and pregnancy. Neurology 2012, 78, 875–879. [Google Scholar] [CrossRef] [PubMed]
- Confavreux, C.; Hutchinson, M.; Hours, M.M.; Cortinovis-Tourniaire, P.; Moreau, T. Rate of pregnancy-related relapse in multiple sclerosis. Pregnancy in Multiple Sclerosis Group. N. Engl. J. Med. 1998, 339, 285–291. [Google Scholar] [CrossRef]
- Kim, S.H.; Mealy, M.A.; Levy, M.; Schmidt, F.; Ruprecht, K.; Paul, F.; Ringelstein, M.; Aktas, O.; Hartung, H.P.; Asgari, N.; et al. Racial differences in neuromyelitis optica spectrum disorder. Neurology 2018, 91, e2089–e2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, V.A.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A serum autoantibody marker of neuromyelitis optica: Distinction from multiple sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Fujihara, K.; Nakashima, I.; Misu, T.; Miyazawa, I.; Nakamura, M.; Watanabe, S.; Shiga, Y.; Kanaoka, C.; Fujimori, J.; et al. Anti-aquaporin-4 antibody is involved in the pathogenesis of NMO: A study on antibody titre. Brain 2007, 130, 1235–1243. [Google Scholar] [CrossRef]
- Lucchinetti, C.F.; Guo, Y.; Popescu, B.F.; Fujihara, K.; Itoyama, Y.; Misu, T. The pathology of an autoimmune astrocytopathy: Lessons learned from neuromyelitis optica. Brain Pathol. 2014, 24, 83–97. [Google Scholar] [CrossRef]
- Takano, R.; Misu, T.; Takahashi, T.; Sato, S.; Fujihara, K.; Itoyama, Y. Astrocytic damage is far more severe than demyelination in NMO: A clinical CSF biomarker study. Neurology 2010, 75, 208–216. [Google Scholar] [CrossRef]
- Weinshenker, B.G.; Wingerchuk, D.M. Neuromyelitis Spectrum Disorders. Mayo Clin. Proc. 2017, 92, 663–679. [Google Scholar] [CrossRef] [Green Version]
- Keegan, M.; Konig, F.; McClelland, R.; Bruck, W.; Morales, Y.; Bitsch, A.; Panitch, H.; Lassmann, H.; Weinshenker, B.; Rodriguez, M.; et al. Relation between humoral pathological changes in multiple sclerosis and response to therapeutic plasma exchange. Lancet 2005, 366, 579–582. [Google Scholar] [CrossRef]
- Palace, J.; Leite, M.I.; Nairne, A.; Vincent, A. Interferon Beta treatment in neuromyelitis optica: Increase in relapses and aquaporin 4 antibody titers. Arch. Neurol. 2010, 67, 1016–1017. [Google Scholar] [CrossRef] [PubMed]
- Gelfand, J.M.; Cotter, J.; Klingman, J.; Huang, E.J.; Cree, B.A. Massive CNS monocytic infiltration at autopsy in an alemtuzumab-treated patient with NMO. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e34. [Google Scholar] [CrossRef]
- Min, J.H.; Kim, B.J.; Lee, K.H. Development of extensive brain lesions following fingolimod (FTY720) treatment in a patient with neuromyelitis optica spectrum disorder. Mult. Scler. 2012, 18, 113–115. [Google Scholar] [CrossRef]
- Kleiter, I.; Hellwig, K.; Berthele, A.; Kumpfel, T.; Linker, R.A.; Hartung, H.P.; Paul, F.; Aktas, O. Failure of natalizumab to prevent relapses in neuromyelitis optica. Arch. Neurol. 2012, 69, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Wildemann, B.; Paul, F. Neuromyelitis optica: Clinical features, immunopathogenesis and treatment. Clin. Exp. Immunol. 2014, 176, 149–164. [Google Scholar] [CrossRef]
- Brum, D.G.; Barreira, A.A.; dos Santos, A.C.; Kaimen-Maciel, D.R.; Matiello, M.; Costa, R.M.; Deghaide, N.H.; Costa, L.S.; Louzada-Junior, P.; Diniz, P.R.; et al. HLA-DRB association in neuromyelitis optica is different from that observed in multiple sclerosis. Mult. Scler. 2010, 16, 21–29. [Google Scholar] [CrossRef]
- Cree, B.A.; Spencer, C.M.; Varrin-Doyer, M.; Baranzini, S.E.; Zamvil, S.S. Gut microbiome analysis in neuromyelitis optica reveals overabundance of Clostridium perfringens. Ann. Neurol. 2016, 80, 443–447. [Google Scholar] [CrossRef]
- Varrin-Doyer, M.; Spencer, C.M.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; Cree, B.A.; Zamvil, S.S. Aquaporin 4-specific T cells in neuromyelitis optica exhibit a Th17 bias and recognize Clostridium ABC transporter. Ann. Neurol. 2012, 72, 53–64. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Lennon, V.A.; Pittock, S.J.; Lucchinetti, C.F.; Weinshenker, B.G. Revised diagnostic criteria for neuromyelitis optica. Neurology 2006, 66, 1485–1489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devic, E. Myélite subaiguë compliquée de névrite optique. Le Bulletin Médicale 1894, 8, 1033–1034. [Google Scholar]
- Popescu, B.F.; Lennon, V.A.; Parisi, J.E.; Howe, C.L.; Weigand, S.D.; Cabrera-Gomez, J.A.; Newell, K.; Mandler, R.N.; Pittock, S.J.; Weinshenker, B.G.; et al. Neuromyelitis optica unique area postrema lesions: Nausea, vomiting, and pathogenic implications. Neurology 2011, 76, 1229–1237. [Google Scholar] [CrossRef]
- McKeon, A.; Lennon, V.A.; Lotze, T.; Tenenbaum, S.; Ness, J.M.; Rensel, M.; Kuntz, N.L.; Fryer, J.P.; Homburger, H.; Hunter, J.; et al. CNS aquaporin-4 autoimmunity in children. Neurology 2008, 71, 93–100. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; de Seze, J.; Fujihara, K.; Greenberg, B.; Jacob, A.; et al. International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef] [PubMed]
- Jarius, S.; Ruprecht, K.; Wildemann, B.; Kuempfel, T.; Ringelstein, M.; Geis, C.; Kleiter, I.; Kleinschnitz, C.; Berthele, A.; Brettschneider, J.; et al. Contrasting disease patterns in seropositive and seronegative neuromyelitis optica: A multicentre study of 175 patients. J. Neuroinflamm. 2012, 9, 14. [Google Scholar] [CrossRef]
- Banwell, B.; Tenembaum, S.; Lennon, V.A.; Ursell, E.; Kennedy, J.; Bar-Or, A.; Weinshenker, B.G.; Lucchinetti, C.F.; Pittock, S.J. Neuromyelitis optica-IgG in childhood inflammatory demyelinating CNS disorders. Neurology 2008, 70, 344–352. [Google Scholar] [CrossRef] [PubMed]
- Bradshaw, M.J.; Vu, N.; Hunley, T.E.; Chitnis, T. Child Neurology: Neuromyelitis optica spectrum disorders. Neurology 2017, 88, e10–e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarius, S.; Franciotta, D.; Paul, F.; Ruprecht, K.; Bergamaschi, R.; Rommer, P.S.; Reuss, R.; Probst, C.; Kristoferitsch, W.; Wandinger, K.P.; et al. Cerebrospinal fluid antibodies to aquaporin-4 in neuromyelitis optica and related disorders: Frequency, origin, and diagnostic relevance. J. Neuroinflamm. 2010, 7, 52. [Google Scholar] [CrossRef]
- Poser, C.M.; Paty, D.W.; Scheinberg, L.; McDonald, W.I.; Davis, F.A.; Ebers, G.C.; Johnson, K.P.; Sibley, W.A.; Silberberg, D.H.; Tourtellotte, W.W. New diagnostic criteria for multiple sclerosis: Guidelines for research protocols. Ann. Neurol. 1983, 13, 227–231. [Google Scholar] [CrossRef]
- Jarius, S.; Paul, F.; Franciotta, D.; Ruprecht, K.; Ringelstein, M.; Bergamaschi, R.; Rommer, P.; Kleiter, I.; Stich, O.; Reuss, R.; et al. Cerebrospinal fluid findings in aquaporin-4 antibody positive neuromyelitis optica: Results from 211 lumbar punctures. J. Neurol. Sci. 2011, 306, 82–90. [Google Scholar] [CrossRef] [PubMed]
- Klawiter, E.C.; Alvarez, E., 3rd; Xu, J.; Paciorkowski, A.R.; Zhu, L.; Parks, B.J.; Cross, A.H.; Naismith, R.T. NMO-IgG detected in CSF in seronegative neuromyelitis optica. Neurology 2009, 72, 1101–1103. [Google Scholar] [CrossRef] [PubMed]
- McKeon, A.; Pittock, S.J.; Lennon, V.A. CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG. Neurology 2011, 76, 1108–1110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kira, J. Neuromyelitis optica and opticospinal multiple sclerosis: Mechanisms and pathogenesis. Pathophysiology 2011, 18, 69–79. [Google Scholar] [CrossRef]
- Kurtzke, J.F. Rating neurologic impairment in multiple sclerosis: An expanded disability status scale (EDSS). Neurology 1983, 33, 1444–1452. [Google Scholar] [CrossRef]
- Warabi, Y.; Matsumoto, Y.; Hayashi, H. Interferon beta-1b exacerbates multiple sclerosis with severe optic nerve and spinal cord demyelination. J. Neurol. Sci. 2007, 252, 57–61. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Pittock, S.J.; Lucchinetti, C.F.; Lennon, V.A.; Weinshenker, B.G. A secondary progressive clinical course is uncommon in neuromyelitis optica. Neurology 2007, 68, 603–605. [Google Scholar] [CrossRef]
- Kessler, R.A.; Mealy, M.A.; Levy, M. Treatment of Neuromyelitis Optica Spectrum Disorder: Acute, Preventive, and Symptomatic. Curr. Treat. Options Neurol. 2016, 18, 2. [Google Scholar] [CrossRef]
- Abboud, H.; Petrak, A.; Mealy, M.; Sasidharan, S.; Siddique, L.; Levy, M. Treatment of acute relapses in neuromyelitis optica: Steroids alone versus steroids plus plasma exchange. Mult. Scler. 2016, 22, 185–192. [Google Scholar] [CrossRef]
- Kleiter, I.; Gahlen, A.; Borisow, N.; Fischer, K.; Wernecke, K.D.; Wegner, B.; Hellwig, K.; Pache, F.; Ruprecht, K.; Havla, J.; et al. Neuromyelitis optica: Evaluation of 871 attacks and 1,153 treatment courses. Ann. Neurol. 2016, 79, 206–216. [Google Scholar] [CrossRef]
- Cree, B.A.; Lamb, S.; Morgan, K.; Chen, A.; Waubant, E.; Genain, C. An open label study of the effects of rituximab in neuromyelitis optica. Neurology 2005, 64, 1270–1272. [Google Scholar] [CrossRef]
- Nosadini, M.; Alper, G.; Riney, C.J.; Benson, L.A.; Mohammad, S.S.; Ramanathan, S.; Nolan, M.; Appleton, R.; Leventer, R.J.; Deiva, K.; et al. Rituximab monitoring and redosing in pediatric neuromyelitis optica spectrum disorder. Neurol. Neuroimmunol. Neuroinflamm. 2016, 3, e188. [Google Scholar] [CrossRef] [PubMed]
- Costanzi, C.; Matiello, M.; Lucchinetti, C.F.; Weinshenker, B.G.; Pittock, S.J.; Mandrekar, J.; Thapa, P.; McKeon, A. Azathioprine: Tolerability, efficacy, and predictors of benefit in neuromyelitis optica. Neurology 2011, 77, 659–666. [Google Scholar] [CrossRef] [PubMed]
- Jacob, A.; Matiello, M.; Weinshenker, B.G.; Wingerchuk, D.M.; Lucchinetti, C.; Shuster, E.; Carter, J.; Keegan, B.M.; Kantarci, O.H.; Pittock, S.J. Treatment of neuromyelitis optica with mycophenolate mofetil: Retrospective analysis of 24 patients. Arch. Neurol. 2009, 66, 1128–1133. [Google Scholar] [CrossRef] [PubMed]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019. [Google Scholar] [CrossRef]
- Collongues, N.; Marignier, R.; Zephir, H.; Papeix, C.; Fontaine, B.; Blanc, F.; Rodriguez, D.; Fleury, M.; Vukusic, S.; Pelletier, J.; et al. Long-term follow-up of neuromyelitis optica with a pediatric onset. Neurology 2010, 75, 1084–1088. [Google Scholar] [CrossRef]
- Collongues, N.; Marignier, R.; Jacob, A.; Leite, M.I.; Siva, A.; Paul, F.; Zephir, H.; Akman-Demir, G.; Elsone, L.; Jarius, S.; et al. Characterization of neuromyelitis optica and neuromyelitis optica spectrum disorder patients with a late onset. Mult. Scler. 2014, 20, 1086–1094. [Google Scholar] [CrossRef]
- Kang, H.; Chen, T.; Li, H.; Xu, Q.; Cao, S.; Wei, S. Prognostic factors and disease course in aquaporin-4 antibody-positive Chinese patients with acute optic neuritis. J. Neurol. 2017, 264, 2130–2140. [Google Scholar] [CrossRef] [PubMed]
- Sellner, J.; Boggild, M.; Clanet, M.; Hintzen, R.Q.; Illes, Z.; Montalban, X.; Du Pasquier, R.A.; Polman, C.H.; Sorensen, P.S.; Hemmer, B. EFNS guidelines on diagnosis and management of neuromyelitis optica. Eur. J. Neurol. 2010, 17, 1019–1032. [Google Scholar] [CrossRef]
- Kim, S.H.; Jeong, I.H.; Hyun, J.W.; Joung, A.; Jo, H.J.; Hwang, S.H.; Yun, S.; Joo, J.; Kim, H.J. Treatment Outcomes with Rituximab in 100 Patients With Neuromyelitis Optica: Influence of FCGR3A Polymorphisms on the Therapeutic Response to Rituximab. JAMA Neurol. 2015, 72, 989–995. [Google Scholar] [CrossRef]
- Tradtrantip, L.; Zhang, H.; Saadoun, S.; Phuan, P.W.; Lam, C.; Papadopoulos, M.C.; Bennett, J.L.; Verkman, A.S. Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann. Neurol. 2012, 71, 314–322. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, M.C.; Verkman, A.S. Aquaporin 4 and neuromyelitis optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Boesen, M.S.; Blinkenberg, M.; Koch-Henriksen, N.; Thygesen, L.C.; Uldall, P.V.; Magyari, M.; Born, A.P. Implications of the International Paediatric Multiple Sclerosis Study Group consensus criteria for paediatric acute disseminated encephalomyelitis: A nationwide validation study. Dev. Med. Child Neurol. 2018, 60, 1123–1131. [Google Scholar] [CrossRef] [PubMed]
- Hacohen, Y.; Absoud, M.; Deiva, K.; Hemingway, C.; Nytrova, P.; Woodhall, M.; Palace, J.; Wassmer, E.; Tardieu, M.; Vincent, A.; et al. Myelin oligodendrocyte glycoprotein antibodies are associated with a non-MS course in children. Neurol. Neuroimmunol. Neuroinflamm. 2015, 2, e81. [Google Scholar] [CrossRef]
- Reindl, M.; Di Pauli, F.; Rostasy, K.; Berger, T. The spectrum of MOG autoantibody-associated demyelinating diseases. Nat. Rev. Neurol. 2013, 9, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Rostasy, K.; Mader, S.; Schanda, K.; Huppke, P.; Gartner, J.; Kraus, V.; Karenfort, M.; Tibussek, D.; Blaschek, A.; Bajer-Kornek, B.; et al. Anti-myelin oligodendrocyte glycoprotein antibodies in pediatric patients with optic neuritis. Arch. Neurol. 2012, 69, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Tenembaum, S.; Chitnis, T.; Nakashima, I.; Collongues, N.; McKeon, A.; Levy, M.; Rostasy, K. Neuromyelitis optica spectrum disorders in children and adolescents. Neurology 2016, 87, S59–66. [Google Scholar] [CrossRef]
- Fernandez-Carbonell, C.; Vargas-Lowy, D.; Musallam, A.; Healy, B.; McLaughlin, K.; Wucherpfennig, K.W.; Chitnis, T. Clinical and MRI phenotype of children with MOG antibodies. Mult. Scler. 2016, 22, 174–184. [Google Scholar] [CrossRef]
- Ketelslegers, I.A.; Van Pelt, D.E.; Bryde, S.; Neuteboom, R.F.; Catsman-Berrevoets, C.E.; Hamann, D.; Hintzen, R.Q. Anti-MOG antibodies plead against MS diagnosis in an Acquired Demyelinating Syndromes cohort. Mult. Scler. 2015, 21, 1513–1520. [Google Scholar] [CrossRef]
- Hacohen, Y.; Wong, Y.Y.; Lechner, C.; Jurynczyk, M.; Wright, S.; Konuskan, B.; Kalser, J.; Poulat, A.L.; Maurey, H.; Ganelin-Cohen, E.; et al. Disease Course and Treatment Responses in Children with Relapsing Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease. JAMA Neurol. 2018, 75, 478–487. [Google Scholar] [CrossRef]
- Brunner, C.; Lassmann, H.; Waehneldt, T.V.; Matthieu, J.M.; Linington, C. Differential ultrastructural localization of myelin basic protein, myelin/oligodendroglial glycoprotein, and 2’,3’-cyclic nucleotide 3’-phosphodiesterase in the CNS of adult rats. J. Neurochem. 1989, 52, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Johns, T.G.; Bernard, C.C. The structure and function of myelin oligodendrocyte glycoprotein. J. Neurochem. 1999, 72, 1–9. [Google Scholar] [CrossRef]
- Lebar, R.; Lubetzki, C.; Vincent, C.; Lombrail, P.; Boutry, J.M. The M2 autoantigen of central nervous system myelin, a glycoprotein present in oligodendrocyte membrane. Clin. Exp. Immunol. 1986, 66, 423–434. [Google Scholar] [PubMed]
- Linington, C.; Lassmann, H. Antibody responses in chronic relapsing experimental allergic encephalomyelitis: Correlation of serum demyelinating activity with antibody titre to the myelin/oligodendrocyte glycoprotein (MOG). J. Neuroimmunol. 1987, 17, 61–69. [Google Scholar] [CrossRef]
- Peschl, P.; Bradl, M.; Hoftberger, R.; Berger, T.; Reindl, M. Myelin Oligodendrocyte Glycoprotein: Deciphering a Target in Inflammatory Demyelinating Diseases. Front. Immunol. 2017, 8, 529. [Google Scholar] [CrossRef]
- Lalive, P.H.; Hausler, M.G.; Maurey, H.; Mikaeloff, Y.; Tardieu, M.; Wiendl, H.; Schroeter, M.; Hartung, H.P.; Kieseier, B.C.; Menge, T. Highly reactive anti-myelin oligodendrocyte glycoprotein antibodies differentiate demyelinating diseases from viral encephalitis in children. Mult. Scler. 2011, 17, 297–302. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Sahin, K.; Lechner, C.; Hennes, E.M.; Schanda, K.; Mader, S.; Karenfort, M.; Selch, C.; Hausler, M.; Eisenkolbl, A.; et al. Clinical and neuroradiological differences of paediatric acute disseminating encephalomyelitis with and without antibodies to the myelin oligodendrocyte glycoprotein. J. Neurol. Neurosurg Psychiatr. 2015, 86, 265–272. [Google Scholar] [CrossRef]
- Probstel, A.K.; Dornmair, K.; Bittner, R.; Sperl, P.; Jenne, D.; Magalhaes, S.; Villalobos, A.; Breithaupt, C.; Weissert, R.; Jacob, U.; et al. Antibodies to MOG are transient in childhood acute disseminated encephalomyelitis. Neurology 2011, 77, 580–588. [Google Scholar] [CrossRef]
- Rostasy, K.; Mader, S.; Hennes, E.M.; Schanda, K.; Gredler, V.; Guenther, A.; Blaschek, A.; Korenke, C.; Pritsch, M.; Pohl, D.; et al. Persisting myelin oligodendrocyte glycoprotein antibodies in aquaporin-4 antibody negative pediatric neuromyelitis optica. Mult. Scler. 2013, 19, 1052–1059. [Google Scholar] [CrossRef]
- Hennes, E.M.; Baumann, M.; Schanda, K.; Anlar, B.; Bajer-Kornek, B.; Blaschek, A.; Brantner-Inthaler, S.; Diepold, K.; Eisenkölbl, A.; Gotwald, T.; et al. Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome. Neurology 2017, 89, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Brilot, F.; Dale, R.C.; Selter, R.C.; Grummel, V.; Kalluri, S.R.; Aslam, M.; Busch, V.; Zhou, D.; Cepok, S.; Hemmer, B. Antibodies to native myelin oligodendrocyte glycoprotein in children with inflammatory demyelinating central nervous system disease. Ann. Neurol. 2009, 66, 833–842. [Google Scholar] [CrossRef]
- Thulasirajah, S.; Pohl, D.; Davila-Acosta, J.; Venkateswaran, S. Myelin Oligodendrocyte Glycoprotein-Associated Pediatric Central Nervous System Demyelination: Clinical Course, Neuroimaging Findings, and Response to Therapy. Neuropediatrics 2016, 47, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Pohl, D.; Rostasy, K.; Reiber, H.; Hanefeld, F. CSF characteristics in early-onset multiple sclerosis. Neurology 2004, 63, 1966–1967. [Google Scholar] [CrossRef] [PubMed]
- Dale, R.C.; Tantsis, E.M.; Merheb, V.; Kumaran, R.Y.; Sinmaz, N.; Pathmanandavel, K.; Ramanathan, S.; Booth, D.R.; Wienholt, L.A.; Prelog, K.; et al. Antibodies to MOG have a demyelination phenotype and affect oligodendrocyte cytoskeleton. Neurol. Neuroimmunol. Neuroinflamm. 2014, 1, e12. [Google Scholar] [CrossRef]
- Rostasy, K.; Reindl, M. Role of autoantibodies in acquired inflammatory demyelinating diseases of the central nervous system in children. Neuropediatrics 2013, 44, 297–301. [Google Scholar] [CrossRef] [PubMed]
- von Budingen, H.C.; Hauser, S.L.; Ouallet, J.C.; Tanuma, N.; Menge, T.; Genain, C.P. Frontline: Epitope recognition on the myelin/oligodendrocyte glycoprotein differentially influences disease phenotype and antibody effector functions in autoimmune demyelination. Eur. J. Immunol. 2004, 34, 2072–2083. [Google Scholar] [CrossRef] [PubMed]
- Dos Passos, G.R.; Oliveira, L.M.; da Costa, B.K.; Apostolos-Pereira, S.L.; Callegaro, D.; Fujihara, K.; Sato, D.K. MOG-IgG-Associated Optic Neuritis, Encephalitis, and Myelitis: Lessons Learned from Neuromyelitis Optica Spectrum Disorder. Front. Neurol. 2018, 9, 217. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.; Hennes, E.M.; Schanda, K.; Karenfort, M.; Kornek, B.; Seidl, R.; Diepold, K.; Lauffer, H.; Marquardt, I.; Strautmanis, J.; et al. Children with multiphasic disseminated encephalomyelitis and antibodies to the myelin oligodendrocyte glycoprotein (MOG): Extending the spectrum of MOG antibody positive diseases. Mult. Scler. 2016, 22, 1821–1829. [Google Scholar] [CrossRef]
- Absoud, M.; Lim, M.J.; Chong, W.K.; De Goede, C.G.; Foster, K.; Gunny, R.; Hemingway, C.; Jardine, P.E.; Kneen, R.; Likeman, M.; et al. Paediatric acquired demyelinating syndromes: Incidence, clinical and magnetic resonance imaging features. Mult. Scler. 2013, 19, 76–86. [Google Scholar] [CrossRef]
- Pohl, D.; Hennemuth, I.; von Kries, R.; Hanefeld, F. Paediatric multiple sclerosis and acute disseminated encephalomyelitis in Germany: Results of a nationwide survey. Eur. J. Pediatr. 2007, 166, 405–412. [Google Scholar] [CrossRef]
- Torisu, H.; Kira, R.; Ishizaki, Y.; Sanefuji, M.; Yamaguchi, Y.; Yasumoto, S.; Murakami, Y.; Shimono, M.; Nagamitsu, S.; Masuzaki, M.; et al. Clinical study of childhood acute disseminated encephalomyelitis, multiple sclerosis, and acute transverse myelitis in Fukuoka Prefecture, Japan. Brain Dev. 2010, 32, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.H.; Yan, Y.; Liao, Z.; Peng, S.H.; Wen, H.R.; Zhang, Y.X.; Chen, S.H.; Li, J.; Chen, H.Y.; Feng, X.W.; et al. Epidemiological characteristics of acute disseminated encephalomyelitis in Nanchang, China: A retrospective study. BMC Public Health 2014, 14, 111. [Google Scholar] [CrossRef] [PubMed]
- Langer-Gould, A.; Zhang, J.L.; Chung, J.; Yeung, Y.; Waubant, E.; Yao, J. Incidence of acquired CNS demyelinating syndromes in a multiethnic cohort of children. Neurology 2011, 77, 1143–1148. [Google Scholar] [CrossRef]
- Dale, R.C.; de Sousa, C.; Chong, W.K.; Cox, T.C.; Harding, B.; Neville, B.G. Acute disseminated encephalomyelitis, multiphasic disseminated encephalomyelitis and multiple sclerosis in children. Brain 2000, 123 Pt 12, 2407–2422. [Google Scholar] [CrossRef] [Green Version]
- Erol, I.; Ozkale, Y.; Alkan, O.; Alehan, F. Acute disseminated encephalomyelitis in children and adolescents: A single center experience. Pediatr. Neurol. 2013, 49, 266–273. [Google Scholar] [CrossRef]
- Hviid, A.; Svanstrom, H.; Scheller, N.M.; Gronlund, O.; Pasternak, B.; Arnheim-Dahlstrom, L. Human papillomavirus vaccination of adult women and risk of autoimmune and neurological diseases. J. Intern. Med. 2018, 283, 154–165. [Google Scholar] [CrossRef]
- Langer-Gould, A.; Qian, L.; Tartof, S.Y.; Brara, S.M.; Jacobsen, S.J.; Beaber, B.E.; Sy, L.S.; Chao, C.; Hechter, R.; Tseng, H.F. Vaccines and the risk of multiple sclerosis and other central nervous system demyelinating diseases. JAMA Neurol. 2014, 71, 1506–1513. [Google Scholar] [CrossRef] [PubMed]
- Scheller, N.M.; Svanstrom, H.; Pasternak, B.; Arnheim-Dahlstrom, L.; Sundstrom, K.; Fink, K.; Hviid, A. Quadrivalent HPV vaccination and risk of multiple sclerosis and other demyelinating diseases of the central nervous system. JAMA 2015, 313, 54–61. [Google Scholar] [CrossRef]
- Fujinami, R.S.; Oldstone, M.B. Amino acid homology between the encephalitogenic site of myelin basic protein and virus: Mechanism for autoimmunity. Science 1985, 230, 1043–1045. [Google Scholar] [CrossRef]
- Mader, S.; Gredler, V.; Schanda, K.; Rostasy, K.; Dujmovic, I.; Pfaller, K.; Lutterotti, A.; Jarius, S.; Di Pauli, F.; Kuenz, B.; et al. Complement activating antibodies to myelin oligodendrocyte glycoprotein in neuromyelitis optica and related disorders. J. Neuroinflamm. 2011, 8, 184. [Google Scholar] [CrossRef]
- Krupp, L.B.; Tardieu, M.; Amato, M.P.; Banwell, B.; Chitnis, T.; Dale, R.C.; Ghezzi, A.; Hintzen, R.; Kornberg, A.; Pohl, D.; et al. International Pediatric Multiple Sclerosis Study Group criteria for pediatric multiple sclerosis and immune-mediated central nervous system demyelinating disorders: Revisions to the 2007 definitions. Mult. Scler. 2013, 19, 1261–1267. [Google Scholar] [CrossRef]
- Krupp, L.B.; Banwell, B.; Tenembaum, S. Consensus definitions proposed for pediatric multiple sclerosis and related disorders. Neurology 2007, 68, S7–S12. [Google Scholar] [CrossRef] [PubMed]
- Pavone, P.; Pettoello-Mantovano, M.; Le Pira, A.; Giardino, I.; Pulvirenti, A.; Giugno, R.; Parano, E.; Polizzi, A.; Distefano, A.; Ferro, A.; et al. Acute disseminated encephalomyelitis: A long-term prospective study and meta-analysis. Neuropediatrics 2010, 41, 246–255. [Google Scholar] [CrossRef]
- Yamaguchi, Y.; Torisu, H.; Kira, R.; Ishizaki, Y.; Sakai, Y.; Sanefuji, M.; Ichiyama, T.; Oka, A.; Kishi, T.; Kimura, S.; et al. A nationwide survey of pediatric acquired demyelinating syndromes in Japan. Neurology 2016, 87, 2006–2015. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Chiriboga, A.S.; Majed, M.; Fryer, J.; Dubey, D.; McKeon, A.; Flanagan, E.P.; Jitprapaikulsan, J.; Kothapalli, N.; Tillema, J.M.; Chen, J.; et al. Association of MOG-IgG Serostatus With Relapse After Acute Disseminated Encephalomyelitis and Proposed Diagnostic Criteria for MOG-IgG-Associated Disorders. JAMA Neurol. 2018, 75, 1355–1363. [Google Scholar] [CrossRef] [PubMed]
- de Mol, C.L.; Wong, Y.Y.M.; van Pelt, E.D.; Ketelslegers, I.A.; Bakker, D.P.; Boon, M.; Braun, K.P.J.; van Dijk, K.G.J.; Eikelenboom, M.J.; Engelen, M.; et al. Incidence and outcome of acquired demyelinating syndromes in Dutch children: Update of a nationwide and prospective study. J. Neurol. 2018, 265, 1310–1319. [Google Scholar] [CrossRef] [PubMed]
- Kothur, K.; Wienholt, L.; Mohammad, S.S.; Tantsis, E.M.; Pillai, S.; Britton, P.N.; Jones, C.A.; Angiti, R.R.; Barnes, E.H.; Schlub, T.; et al. Utility of CSF Cytokine/Chemokines as Markers of Active Intrathecal Inflammation: Comparison of Demyelinating, Anti-NMDAR and Enteroviral Encephalitis. PLoS ONE 2016, 11, e0161656. [Google Scholar] [CrossRef]
- Duignan, S.; Wright, S.; Rossor, T.; Cazabon, J.; Gilmour, K.; Ciccarelli, O.; Wassmer, E.; Lim, M.; Hemingway, C.; Hacohen, Y. Myelin oligodendrocyte glycoprotein and aquaporin-4 antibodies are highly specific in children with acquired demyelinating syndromes. Dev. Med. Child Neurol. 2018, 60, 958–962. [Google Scholar] [CrossRef] [Green Version]
- Huppke, P.; Rostasy, K.; Karenfort, M.; Huppke, B.; Seidl, R.; Leiz, S.; Reindl, M.; Gartner, J. Acute disseminated encephalomyelitis followed by recurrent or monophasic optic neuritis in pediatric patients. Mult. Scler. 2013, 19, 941–946. [Google Scholar] [CrossRef]
- Kim, S.M.; Waters, P.; Woodhall, M.; Yang, J.W.; Yang, H.; Kim, J.E.; Sung, J.J.; Park, K.S.; Lee, K.W. Characterization of the spectrum of Korean inflammatory demyelinating diseases according to the diagnostic criteria and AQP4-Ab status. BMC Neurol. 2014, 14, 93. [Google Scholar] [CrossRef]
- Hynson, J.L.; Kornberg, A.J.; Coleman, L.T.; Shield, L.; Harvey, A.S.; Kean, M.J. Clinical and neuroradiologic features of acute disseminated encephalomyelitis in children. Neurology 2001, 56, 1308–1312. [Google Scholar] [CrossRef]
- Tenembaum, S.; Chamoles, N.; Fejerman, N. Acute disseminated encephalomyelitis: A long-term follow-up study of 84 pediatric patients. Neurology 2002, 59, 1224–1231. [Google Scholar] [CrossRef]
- Anlar, B.; Basaran, C.; Kose, G.; Guven, A.; Haspolat, S.; Yakut, A.; Serdaroglu, A.; Senbil, N.; Tan, H.; Karaagaoglu, E.; et al. Acute disseminated encephalomyelitis in children: Outcome and prognosis. Neuropediatrics 2003, 34, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Kleiman, M.; Brunquell, P. Acute disseminated encephalomyelitis: Response to intravenous immunoglobulin. J. Child Neurol. 1995, 10, 481–483. [Google Scholar] [CrossRef]
- Pradhan, S.; Gupta, R.P.; Shashank, S.; Pandey, N. Intravenous immunoglobulin therapy in acute disseminated encephalomyelitis. J. Neurol. Sci. 1999, 165, 56–61. [Google Scholar] [CrossRef]
- Sahlas, D.J.; Miller, S.P.; Guerin, M.; Veilleux, M.; Francis, G. Treatment of acute disseminated encephalomyelitis with intravenous immunoglobulin. Neurology 2000, 54, 1370–1372. [Google Scholar] [CrossRef]
- Nishikawa, M.; Ichiyama, T.; Hayashi, T.; Ouchi, K.; Furukawa, S. Intravenous immunoglobulin therapy in acute disseminated encephalomyelitis. Pediatr. Neurol. 1999, 21, 583–586. [Google Scholar] [CrossRef]
- Stricker, R.B.; Miller, R.G.; Kiprov, D.D. Role of plasmapheresis in acute disseminated (postinfectious) encephalomyelitis. J. Clin. Apher. 1992, 7, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Balestri, P.; Grosso, S.; Acquaviva, A.; Bernini, M. Plasmapheresis in a child affected by acute disseminated encephalomyelitis. Brain Dev. 2000, 22, 123–126. [Google Scholar] [CrossRef]
- Miyazawa, R.; Hikima, A.; Takano, Y.; Arakawa, H.; Tomomasa, T.; Morikawa, A. Plasmapheresis in fulminant acute disseminated encephalomyelitis. Brain Dev. 2001, 23, 424–426. [Google Scholar] [CrossRef]
- Khurana, D.S.; Melvin, J.J.; Kothare, S.V.; Valencia, I.; Hardison, H.H.; Yum, S.; Faerber, E.N.; Legido, A. Acute disseminated encephalomyelitis in children: Discordant neurologic and neuroimaging abnormalities and response to plasmapheresis. Pediatrics 2005, 116, 431–436. [Google Scholar] [CrossRef] [PubMed]
- Suppiej, A.; Cainelli, E.; Casara, G.; Cappellari, A.; Nosadini, M.; Sartori, S. Long-term neurocognitive outcome and quality of life in pediatric acute disseminated encephalomyelitis. Pediatr. Neurol. 2014, 50, 363–367. [Google Scholar] [CrossRef] [PubMed]
- Neuteboom, R.F.; Boon, M.; Catsman Berrevoets, C.E.; Vles, J.S.; Gooskens, R.H.; Stroink, H.; Vermeulen, R.J.; Rotteveel, J.J.; Ketelslegers, I.A.; Peeters, E.; et al. Prognostic factors after a first attack of inflammatory CNS demyelination in children. Neurology 2008, 71, 967–973. [Google Scholar] [CrossRef] [PubMed]
- El-Hattab, A.W. Inborn errors of metabolism. Clin. Perinatol. 2015, 42, 413–439. [Google Scholar] [CrossRef] [PubMed]
- Bottcher, T.; Rolfs, A.; Tanislav, C.; Bitsch, A.; Kohler, W.; Gaedeke, J.; Giese, A.K.; Kolodny, E.H.; Duning, T. Fabry disease—Underestimated in the differential diagnosis of multiple sclerosis? PLoS ONE 2013, 8, e71894. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Ito, H.; Mori, K.; Kagami, S. Neuroimaging of stroke-like episodes in MELAS. Brain Dev. 2011, 33, 283–288. [Google Scholar] [CrossRef]
- Cocozza, S.; Russo, C.; Pisani, A.; Olivo, G.; Riccio, E.; Cervo, A.; Pontillo, G.; Feriozzi, S.; Veroux, M.; Battaglia, Y.; et al. Redefining the Pulvinar Sign in Fabry Disease. AJNR Am. J. Neuroradiol. 2017, 38, 2264–2269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanderver, A.; Hussey, H.; Schmidt, J.L.; Pastor, W.; Hoffman, H.J. Relative incidence of inherited white matter disorders in childhood to acquired pediatric demyelinating disorders. Semin. Pediatr. Neurol. 2012, 19, 219–223. [Google Scholar] [CrossRef]
- van der Knaap, M.S.; Bugiani, M. Leukodystrophies: A proposed classification system based on pathological changes and pathogenetic mechanisms. Acta Neuropathol. 2017, 134, 351–382. [Google Scholar] [CrossRef]
- Parikh, S.; Bernard, G.; Leventer, R.J.; van der Knaap, M.S.; van Hove, J.; Pizzino, A.; McNeill, N.H.; Helman, G.; Simons, C.; Schmidt, J.L.; et al. A clinical approach to the diagnosis of patients with leukodystrophies and genetic leukoencephelopathies. Mol. Genet. Metab. 2015, 114, 501–515. [Google Scholar] [CrossRef] [PubMed]
- Kohler, W. Diagnostic algorithm for the differentiation of leukodystrophies in early MS. J. Neurol. 2008, 255 (Suppl. 6), 123–126. [Google Scholar] [CrossRef]
- Vanderver, A.; Prust, M.; Tonduti, D.; Mochel, F.; Hussey, H.M.; Helman, G.; Garbern, J.; Eichler, F.; Labauge, P.; Aubourg, P.; et al. Case definition and classification of leukodystrophies and leukoencephalopathies. Mol. Genet. Metab. 2015, 114, 494–500. [Google Scholar] [CrossRef] [Green Version]
- Schiffmann, R.; van der Knaap, M.S. Invited article: An MRI-based approach to the diagnosis of white matter disorders. Neurology 2009, 72, 750–759. [Google Scholar] [CrossRef]
- Eichler, F.; Grodd, W.; Grant, E.; Sessa, M.; Biffi, A.; Bley, A.; Kohlschuetter, A.; Loes, D.J.; Kraegeloh-Mann, I. Metachromatic leukodystrophy: A scoring system for brain MR imaging observations. AJNR Am. J. Neuroradiol. 2009, 30, 1893–1897. [Google Scholar] [CrossRef] [PubMed]
- Gordon-Lipkin, E.; Fatemi, A. Current Therapeutic Approaches in Leukodystrophies: A Review. J. Child Neurol. 2018, 33, 861–868. [Google Scholar] [CrossRef] [PubMed]
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Galardi, M.M.; Gaudioso, C.; Ahmadi, S.; Evans, E.; Gilbert, L.; Mar, S. Differential Diagnosis of Pediatric Multiple Sclerosis. Children 2019, 6, 75. https://doi.org/10.3390/children6060075
Galardi MM, Gaudioso C, Ahmadi S, Evans E, Gilbert L, Mar S. Differential Diagnosis of Pediatric Multiple Sclerosis. Children. 2019; 6(6):75. https://doi.org/10.3390/children6060075
Chicago/Turabian StyleGalardi, Maria Milagros, Cristina Gaudioso, Saumel Ahmadi, Emily Evans, Laura Gilbert, and Soe Mar. 2019. "Differential Diagnosis of Pediatric Multiple Sclerosis" Children 6, no. 6: 75. https://doi.org/10.3390/children6060075
APA StyleGalardi, M. M., Gaudioso, C., Ahmadi, S., Evans, E., Gilbert, L., & Mar, S. (2019). Differential Diagnosis of Pediatric Multiple Sclerosis. Children, 6(6), 75. https://doi.org/10.3390/children6060075