Neuroblastoma: Tumor Biology and Its Implications for Staging and Treatment



{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
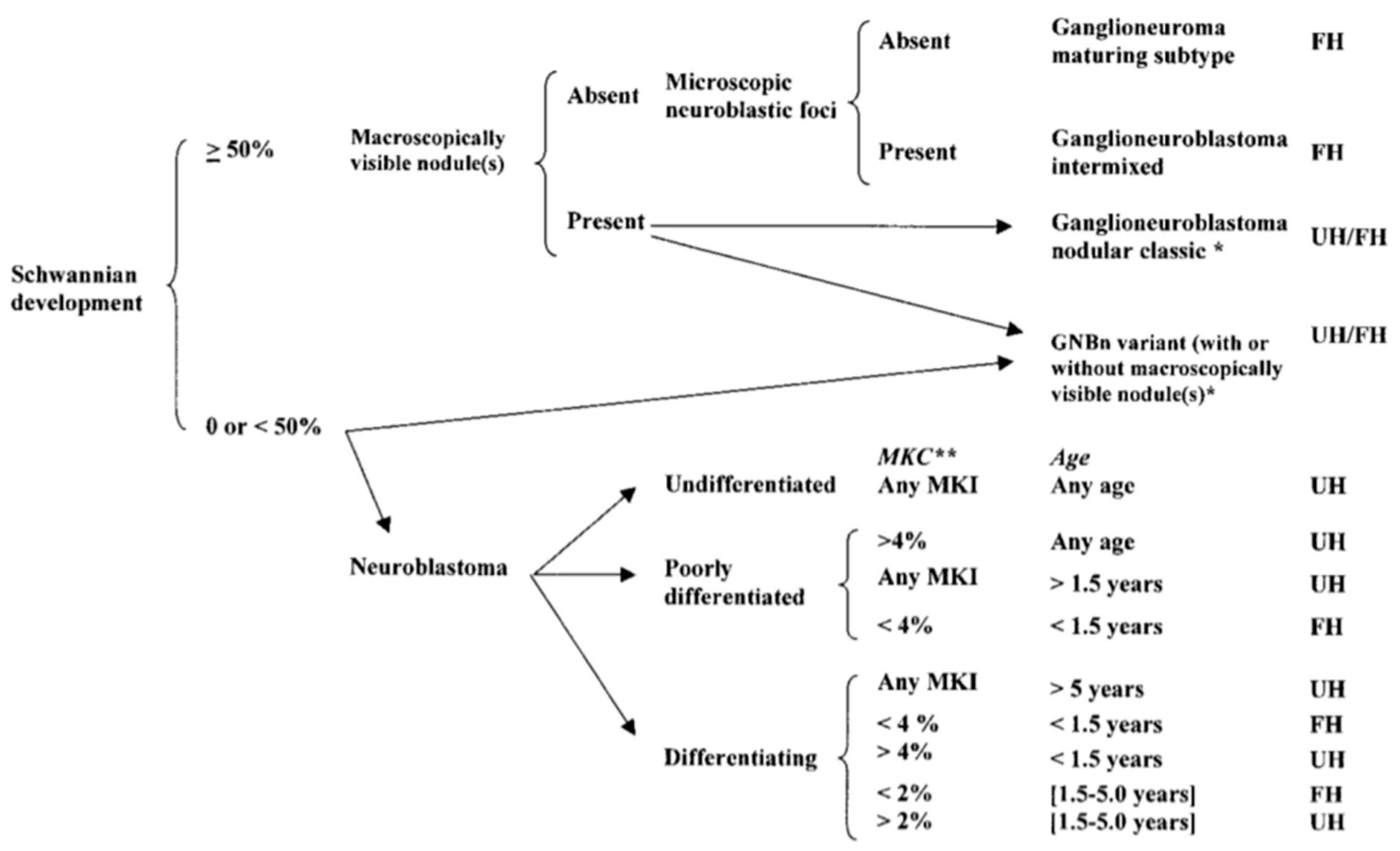
2. Tumor Histology
3. Molecular Markers
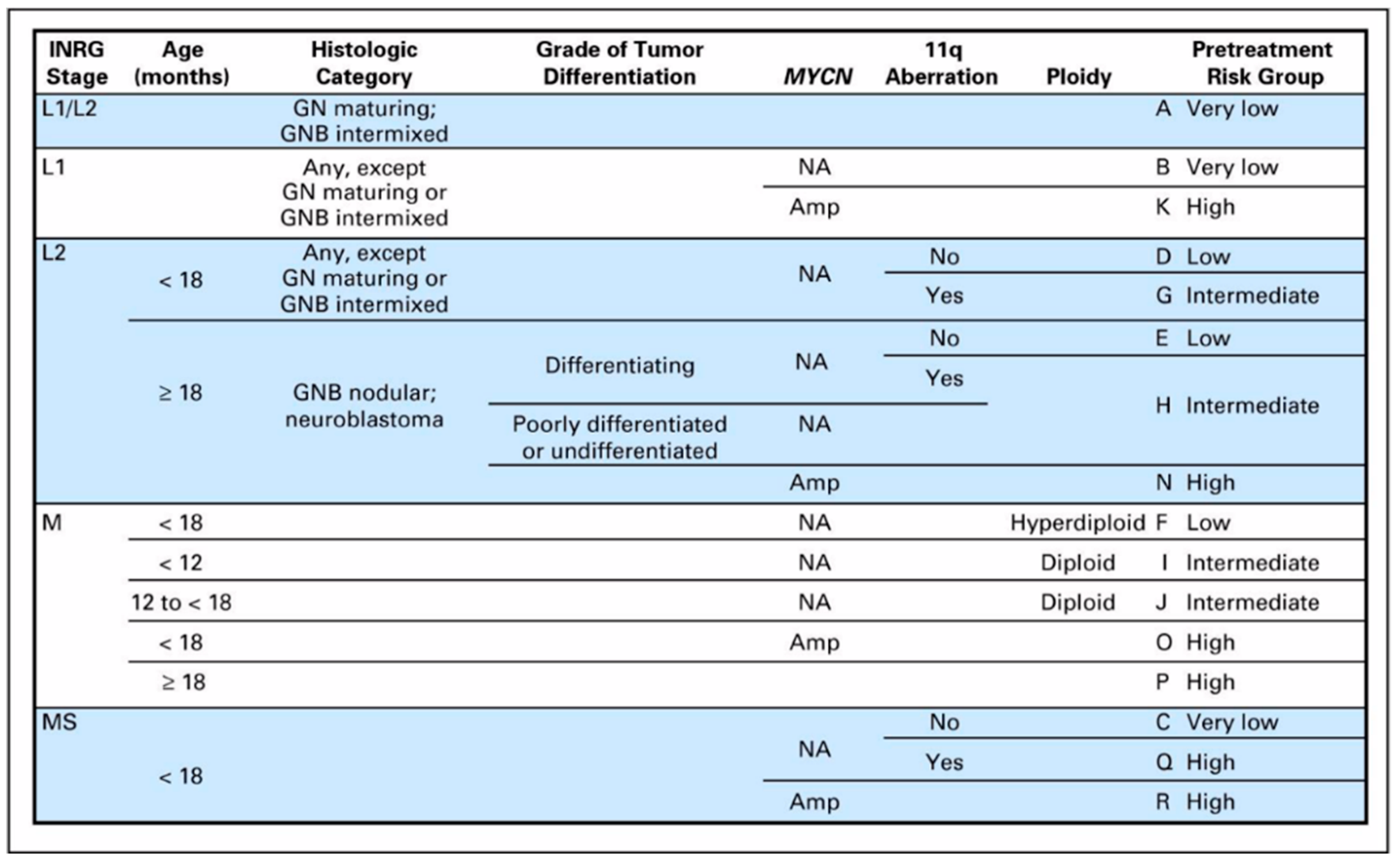
4. Current Risk Stratification
- (1)
- required that surgical resection be completed in order for staging to occur and
- (2)
- likely varied significantly amongst both surgeons and centers in terms of their judgment, skill, and aggressiveness towards complete resection and lymph node sampling.
5. Treatment by Risk Group
6. Surgical Approach
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Shimada, H.; Ambros, I.M.; Dehner, L.P.; Hata, J.; Joshi, V.V.; Roald, B.; Stram, D.O.; Gerbing, R.B.; Lukens, J.N.; Matthay, K.K.; et al. The International Neuroblastoma Pathology Classification (the Shimada system). Cancer 1999, 86, 364–372. [Google Scholar] [CrossRef]
- Shimada, H.; Umehara, S.; Monobe, Y.; Hachitanda, Y.; Nakagawa, A.; Goto, S.; Gerbing, R.B.; Stram, D.O.; Lukens, J.N.; Matthay, K.K.; et al. International neuroblastoma pathology classification for prognostic evaluation of patients with peripheral neuroblastic tumors: A report from the Children’s Cancer Group. Cancer 2001, 92, 2451–2461. [Google Scholar] [CrossRef]
- Peuchmaur, M.; d’Amore, E.S.; Joshi, V.V.; Hata, J.; Roald, B.; Dehner, L.P.; Gerbing, R.B.; Stram, D.O.; Lukens, J.N.; Matthay, K.K.; et al. Revision of the International Neuroblastoma Pathology Classification: Confirmation of favorable and unfavorable prognostic subsets in ganglioneuroblastoma, nodular. Cancer 2003, 98, 2274–2281. [Google Scholar] [CrossRef] [PubMed]
- Campbell, K.; Gastier-Foster, J.M.; Mann, M.; Naranjo, A.H.; Van Ryn, C.; Bagatell, R.; Matthay, K.K.; London, W.B.; Irwin, M.S.; Shimada, H.; et al. Association of MYCN copy number with clinical features, tumor biology, and outcomes in neuroblastoma: A report from the Children’s Oncology Group. Cancer 2017, 123, 4224–4235. [Google Scholar] [CrossRef] [PubMed]
- Look, A.T.; Hayes, F.A.; Shuster, J.J.; Douglass, E.C.; Castleberry, R.P.; Bowman, L.C.; Smith, E.I.; Brodeur, G.M. Clinical relevance of tumor cell ploidy and N-myc gene amplification in childhood neuroblastoma: A Pediatric Oncology Group study. J. Clin. Oncol. 1991, 9, 581–591. [Google Scholar] [CrossRef] [PubMed]
- Bown, N. Neuroblastoma tumour genetics: Clinical and biological aspects. J. Clin. Pathol. 2001, 54, 897–910. [Google Scholar] [CrossRef] [PubMed]
- Schleiermacher, G.; Mosseri, V.; London, W.B.; Maris, J.M.; Brodeur, G.M.; Attiyeh, E.; Haber, M.; Khan, J.; Nakagawara, A.; Speleman, F.; et al. Segmental chromosomal alterations have prognostic impact in neuroblastoma: A report from the INRG project. Br. J. Cancer 2012, 107, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Takita, J.; Sanada, M.; Hayashi, Y. Oncogenic mutations of ALK in neuroblastoma. Cancer Sci. 2011, 102, 302–308. [Google Scholar] [CrossRef] [PubMed]
- Trochet, D.; Bourdeaut, F.; Janoueix-Lerosey, I.; Deville, A.; de Pontual, L.; Schleiermacher, G.; Coze, C.; Philip, N.; Frebourg, T.; Munnich, A.; et al. Germline mutations of the paired-like homeobox 2B (PHOX2B) gene in neuroblastoma. Am. J. Hum. Genet. 2004, 74, 761–764. [Google Scholar] [CrossRef] [PubMed]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Kramer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef] [PubMed]
- Cheung, N.K.; Zhang, J.; Lu, C.; Parker, M.; Bahrami, A.; Tickoo, S.K.; Heguy, A.; Pappo, A.S.; Federico, S.; Dalton, J.; et al. Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA 2012, 307, 1062–1071. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) classification system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Monclair, T.; Brodeur, G.M.; Ambros, P.F.; Brisse, H.J.; Cecchetto, G.; Holmes, K.; Kaneko, M.; London, W.B.; Matthay, K.K.; Nuchtern, J.G.; et al. The International Neuroblastoma Risk Group (INRG) staging system: An INRG Task Force report. J. Clin. Oncol. 2009, 27, 298–303. [Google Scholar] [CrossRef] [PubMed]
- Brisse, H.J.; McCarville, M.B.; Granata, C.; Krug, K.B.; Wootton-Gorges, S.L.; Kanegawa, K.; Giammarile, F.; Schmidt, M.; Shulkin, B.L.; Matthay, K.K.; et al. Guidelines for imaging and staging of neuroblastic tumors: Consensus report from the International Neuroblastoma Risk Group Project. Radiology 2011, 261, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Nuchtern, J.G.; London, W.B.; Barnewolt, C.E.; Naranjo, A.; McGrady, P.W.; Geiger, J.D.; Diller, L.; Schmidt, M.L.; Maris, J.M.; Cohn, S.L.; et al. A prospective study of expectant observation as primary therapy for neuroblastoma in young infants: A Children’s Oncology Group study. Ann. Surg. 2012, 256, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Nishi, M.; Miyake, H.; Takeda, T.; Yonemori, H.; Hanai, J.; Kikuchi, Y.; Takasugi, N. A trial to discriminate spontaneous regression from non-regression cases during mass screening for neuroblastoma. Jpn. J. Clin. Oncol. 1994, 24, 247–251. [Google Scholar] [PubMed]
- Baker, D.L.; Schmidt, M.L.; Cohn, S.L.; Maris, J.M.; London, W.B.; Buxton, A.; Stram, D.; Castleberry, R.P.; Shimada, H.; Sandler, A.; et al. Outcome after reduced chemotherapy for intermediate-risk neuroblastoma. N. Engl. J. Med. 2010, 363, 1313–1323. [Google Scholar] [CrossRef]
- Kushner, B.H.; Cheung, N.K.; LaQuaglia, M.P.; Ambros, P.F.; Ambros, I.M.; Bonilla, M.A.; Gerald, W.L.; Ladanyi, M.; Gilbert, F.; Rosenfield, N.S.; et al. Survival from locally invasive or widespread neuroblastoma without cytotoxic therapy. J. Clin. Oncol. 1996, 14, 373–381. [Google Scholar] [CrossRef]
- Hero, B.; Simon, T.; Spitz, R.; Ernestus, K.; Gnekow, A.K.; Scheel-Walter, H.G.; Schwabe, D.; Schilling, F.H.; Benz-Bohm, G.; Berthold, F. Localized infant neuroblastomas often show spontaneous regression: Results of the prospective trials NB95-S and NB97. J. Clin. Oncol. 2008, 26, 1504–1510. [Google Scholar] [CrossRef]
- Kraal, K.C.; Tytgat, G.A.; van Eck-Smit, B.L.; Kam, B.; Caron, H.N.; van Noesel, M. Upfront treatment of high-risk neuroblastoma with a combination of 131I-MIBG and topotecan. Pediatr. Blood Cancer 2015, 62, 1886–1891. [Google Scholar] [CrossRef]
- Krytska, K.; Ryles, H.T.; Sano, R.; Raman, P.; Infarinato, N.R.; Hansel, T.D.; Makena, M.R.; Song, M.M.; Reynolds, C.P.; Mosse, Y.P.; et al. Crizotinib Synergizes with Chemotherapy in Preclinical Models of Neuroblastoma. Clin. Cancer Res. 2016, 22, 948–960. [Google Scholar] [CrossRef] [PubMed]
- Simon, T.; Haberle, B.; Hero, B.; von Schweinitz, D.; Berthold, F. Role of surgery in the treatment of patients with stage 4 neuroblastoma age 18 months or older at diagnosis. J. Clin. Oncol. 2013, 31, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Fischer, J.; Pohl, A.; Volland, R.; Hero, B.; Dubbers, M.; Cernaianu, G.; Berthold, F.; von Schweinitz, D.; Simon, T. Complete surgical resection improves outcome in INRG high-risk patients with localized neuroblastoma older than 18 months. BMC Cancer 2017, 17, 520. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Chen, J.; Wang, N.; Liu, Z.; Li, F.; Zhou, J.; Tao, B. Impact of extent of resection on survival in high-risk neuroblastoma: A systematic review and meta-analysis. J. Pediatr. Surg. 2018. [Google Scholar] [CrossRef] [PubMed]
- von Allmen, D.; Davidoff, A.M.; London, W.B.; Van Ryn, C.; Haas-Kogan, D.A.; Kreissman, S.G.; Khanna, G.; Rosen, N.; Park, J.R.; La Quaglia, M.P.; et al. Impact of Extent of Resection on Local Control and Survival in Patients from the COG A3973 Study with High-Risk Neuroblastoma. J. Clin. Oncol. 2017, 35, 208–216. [Google Scholar] [CrossRef]
- La Quaglia, M.P.; Kushner, B.H.; Su, W.; Heller, G.; Kramer, K.; Abramson, S.; Rosen, N.; Wolden, S.; Cheung, N.K. The impact of gross total resection on local control and survival in high-risk neuroblastoma. J. Pediatr. Surg. 2004, 39, 412–417. [Google Scholar] [CrossRef]
- Campagna, G.; Rosenfeld, E.; Foster, J.; Vasudevan, S.; Nuchtern, J.; Kim, E.; Commander, S.; Naik-Mathuria, B. Evolving biopsy techniques for the diagnosis of neuroblastoma in children. J. Pediatr. Surg. 2018, 53, 2235–2239. [Google Scholar] [CrossRef]
- Metz, T.; Heider, A.; Vellody, R.; Jarboe, M.D.; Gemmete, J.J.; Grove, J.J.; Smith, E.A.; Mody, R.; Newman, E.A.; Dillman, J.R.; et al. Image-guided percutaneous core needle biopsy of soft-tissue masses in the pediatric population. Pediatr. Radiol. 2016, 46, 1173–1178. [Google Scholar] [CrossRef]
- Hassan, S.F.; Mathur, S.; Magliaro, T.J.; Larimer, E.L.; Ferrell, L.B.; Vasudevan, S.A.; Patterson, D.M.; Louis, C.U.; Russell, H.V.; Nuchtern, J.G.; et al. Needle core vs open biopsy for diagnosis of intermediate- and high-risk neuroblastoma in children. J. Pediatr. Surg. 2012, 47, 1261–1266. [Google Scholar] [CrossRef]
- Kelleher, C.M.; Smithson, L.; Nguyen, L.L.; Casadiego, G.; Nasr, A.; Irwin, M.S.; Gerstle, J.T. Clinical outcomes in children with adrenal neuroblastoma undergoing open versus laparoscopic adrenalectomy. J. Pediatr. Surg. 2013, 48, 1727–1732. [Google Scholar] [CrossRef]
- Malek, M.M.; Mollen, K.P.; Kane, T.D.; Shah, S.R.; Irwin, C. Thoracic neuroblastoma: A retrospective review of our institutional experience with comparison of the thoracoscopic and open approaches to resection. J. Pediatr. Surg. 2010, 45, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Irtan, S.; Brisse, H.J.; Minard-Colin, V.; Schleiermacher, G.; Canale, S.; Sarnacki, S. Minimally invasive surgery of neuroblastic tumors in children: Indications depend on anatomical location and image-defined risk factors. Pediatr. Blood Cancer 2015, 62, 257–261. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kawashima, H.; Mori, M.; Fujiogi, M.; Suzuki, K.; Amano, H.; Morita, K.; Arakawa, Y.; Koh, K.; Oguma, E.; et al. Contraindications and image-defined risk factors in laparoscopic resection of abdominal neuroblastoma. Pediatr. Surg. Int. 2016, 32, 845–850. [Google Scholar] [CrossRef] [PubMed]
- Shirota, C.; Tainaka, T.; Uchida, H.; Hinoki, A.; Chiba, K.; Tanaka, Y. Laparoscopic resection of neuroblastomas in low- to high-risk patients without image-defined risk factors is safe and feasible. BMC Pediatr. 2017, 17, 71. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Arendonk, K.J.; Chung, D.H. Neuroblastoma: Tumor Biology and Its Implications for Staging and Treatment. Children 2019, 6, 12. https://doi.org/10.3390/children6010012
Van Arendonk KJ, Chung DH. Neuroblastoma: Tumor Biology and Its Implications for Staging and Treatment. Children. 2019; 6(1):12. https://doi.org/10.3390/children6010012
Chicago/Turabian StyleVan Arendonk, Kyle J., and Dai H. Chung. 2019. "Neuroblastoma: Tumor Biology and Its Implications for Staging and Treatment" Children 6, no. 1: 12. https://doi.org/10.3390/children6010012
APA StyleVan Arendonk, K. J., & Chung, D. H. (2019). Neuroblastoma: Tumor Biology and Its Implications for Staging and Treatment. Children, 6(1), 12. https://doi.org/10.3390/children6010012