
Molecular Mechanisms of Functional Adrenocortical Adenoma and Carcinoma: Genetic Characterization and Intracellular Signaling Pathway


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
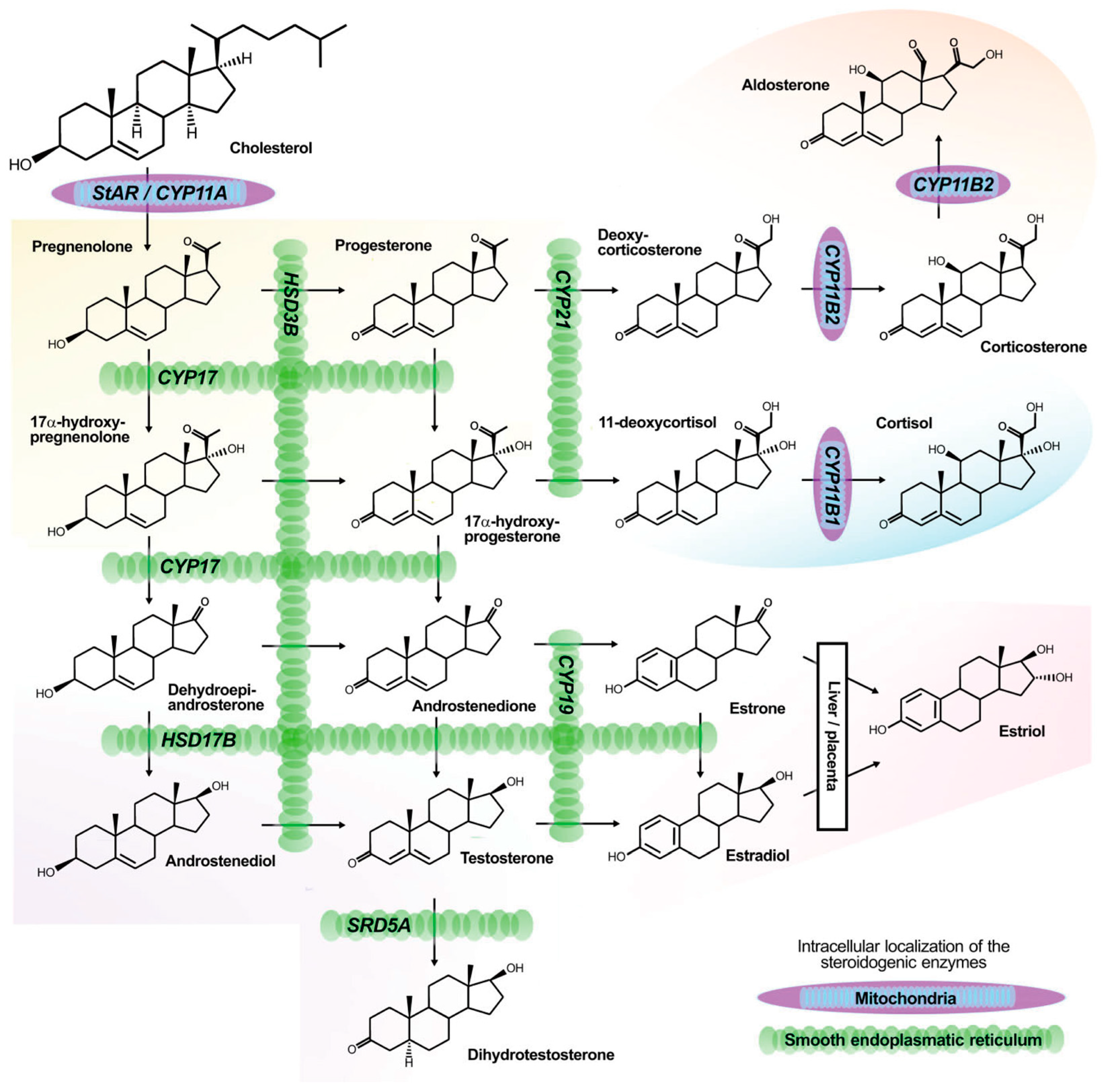
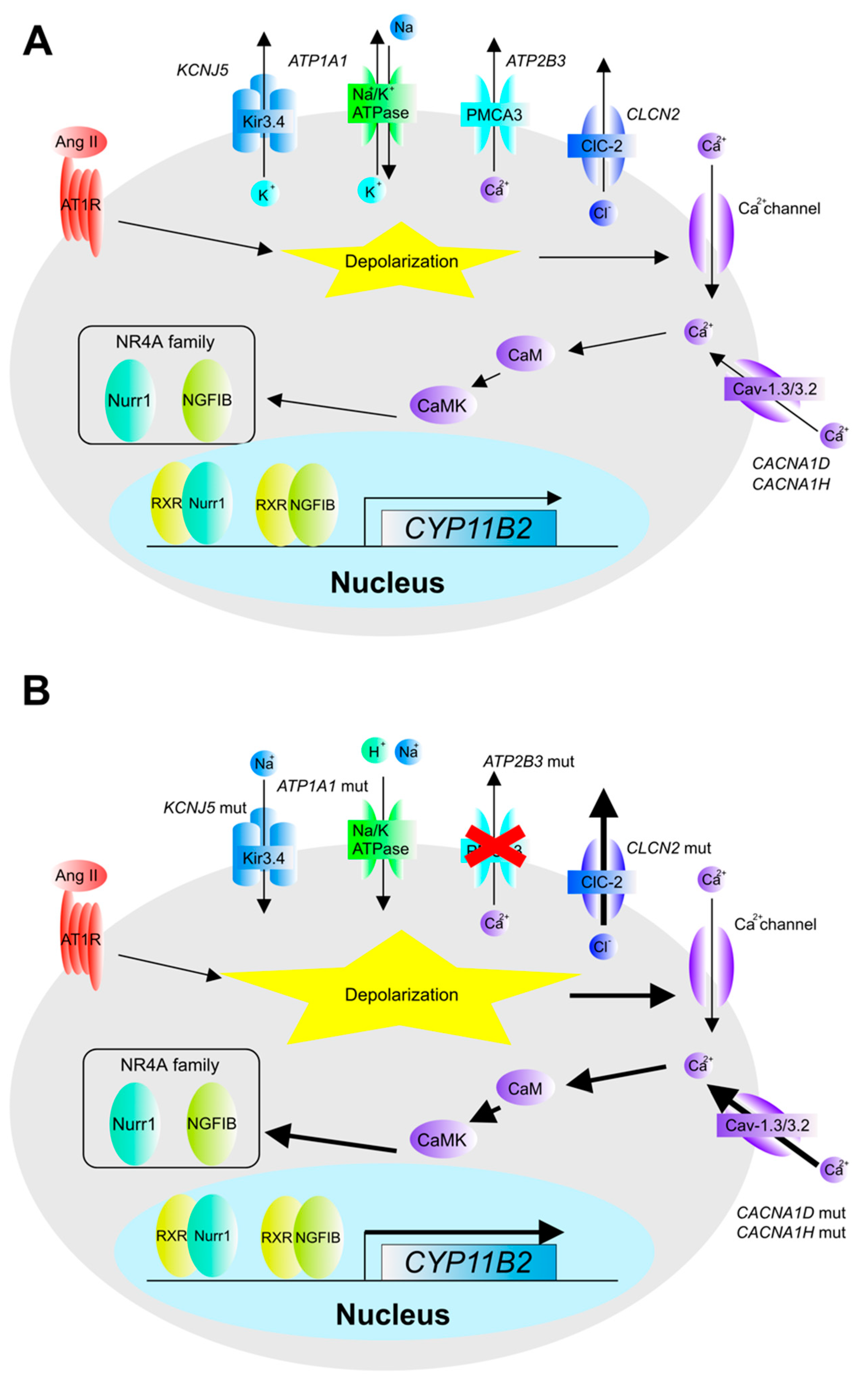
2. The Pathogenesis and Molecular Mechanisms of Aldosterone Overproduction in Aldosterone-Producing Adenoma (APA)
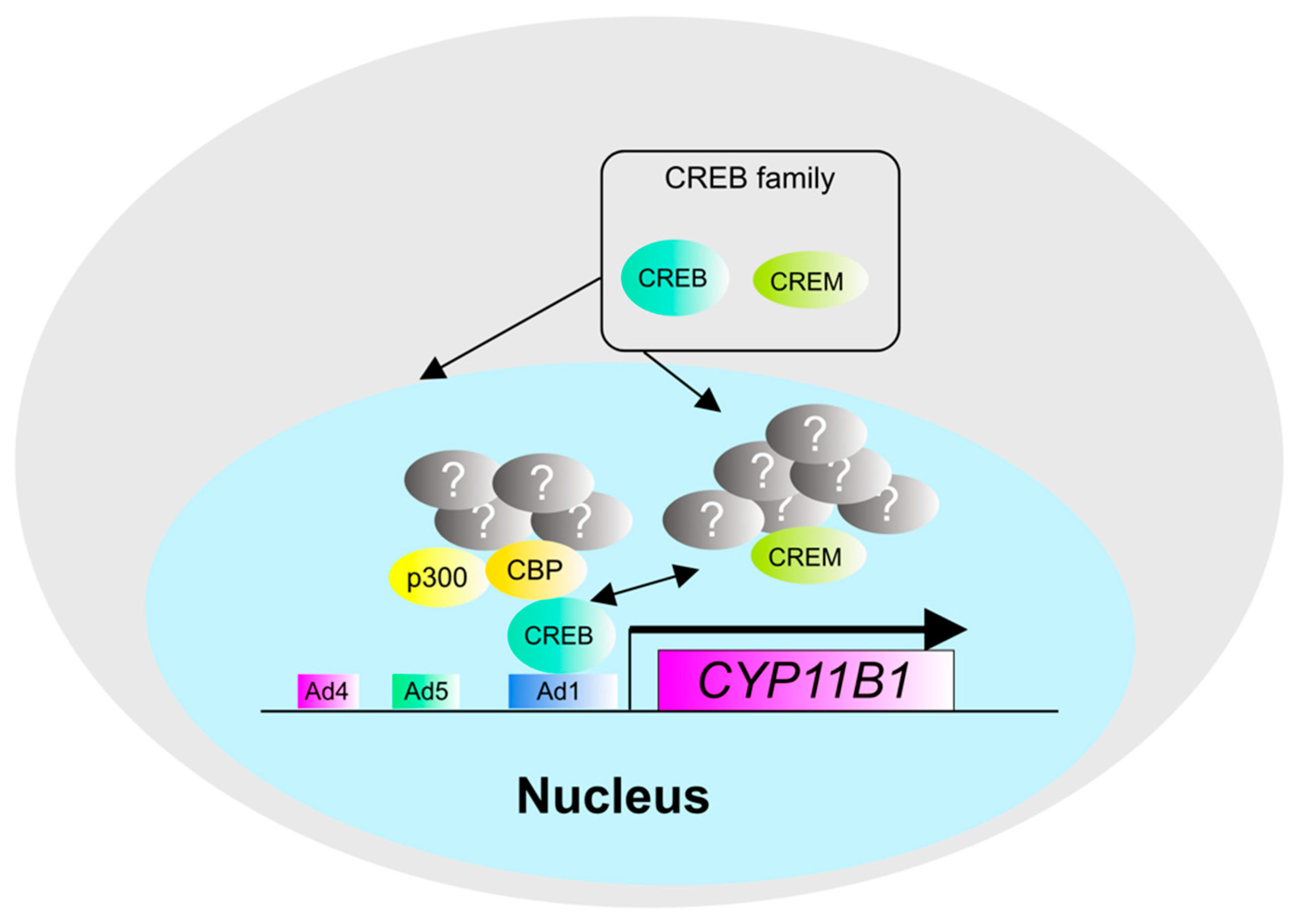
3. The Pathogenesis and Molecular Mechanisms of Cortisol Production in Cortisol-Producing Adenoma (CPA)
4. The Pathogenesis and Molecular Mechanisms in Adrenocortical Carcinoma (ACC)
5. Conclusions
Funding
Conflicts of Interest
Abbreviations
| KCNJ5 | potassium inwardly rectifying channel subfamily J member 5 |
| ATP1A1 | ATPase Na+/K+ transporting subunit alpha 1 |
| ATP2B3 | ATPase plasma membrane Ca2+ transporting 3 |
| CACNA1D | calcium voltage-gated channel subunit alpha1 D |
| CACNA1H | calcium voltage-gated channel subunit alpha1 H |
| CLCN2 | chloride voltage-gated channel 2 |
| CTNNB1 | catenin beta 1 |
| NR4A1 | nuclear receptor subfamily 4, group A, member 1 |
| NR4A2 | nuclear receptor subfamily 4, group A, member 2 |
| NR4A3 | nuclear receptor subfamily 4, group A, member 3 |
| CYP11B2 | cytochrome P450 family 11 subfamily B member 2 |
| HSD3B1 | hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 1 |
| HSD3B2 | hydroxy-delta-5-steroid dehydrogenase, 3 beta- and steroid delta-isomerase 2 |
| GSK3B | glycogen synthase kinase 3 beta |
| TASK1 | potassium two pore domain channel subfamily K member 3 |
| PCP4 | Purkinje cell protein 4 |
| SRC-1 | nuclear receptor coactivator 1 |
| SF-1 | nuclear receptor subfamily 5 group A member 1, Steroidogenic factor-1 |
| COUP-TF | nuclear receptor subfamily 2 group F member 1 |
| PRKACA | protein kinase cAMP-activated catalytic subunit alpha |
| PRKACB | protein kinase cAMP-activated catalytic subunit beta |
| PRKAR1A | protein kinase cAMP-dependent type I regulatory subunit alpha |
| PRKAR1B | protein kinase cAMP-dependent type I regulatory subunit beta |
| CYP11B1 | cytochrome P450 family 11 subfamily B member 1 |
| PDE8B | phosphodiesterase 8B |
| PDE11A | phosphodiesterase 11A |
| GNAS | guanine nucleotide binding protein, alpha stimulating complex locus |
| TP53 | tumor protein p53 |
| RB1 | retinoblastoma transcriptional corepressor 1 |
| ZNRF3 | zinc and ring finger 3 |
| CDKN2A | cyclin dependent kinase inhibitor 2A |
| DAXX | death domain associated protein |
| MED12 | mediator complex subunit 12 |
| MEN1 | menin 1 |
| IGF2 | insulin like growth factor 2 |
| RPL22 | ribosomal protein L22 |
| CCNE1 | cyclin E1 |
| CDK4 | cyclin dependent kinase 4 |
| TERT | telomerase reverse transcriptase |
| TERF2 | telomeric repeat binding factor 2 |
| MLL1 | lysine methyltransferase 2A |
| MLL2 | lysine methyltransferase 2D |
References
- Nanba, K.; Blinder, A.R.; Rege, J.; Hattangady, N.G.; Else, T.; Liu, C.-J.; Tomlins, S.A.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; et al. Somatic CACNA1H Mutation as a Cause of Aldosterone-Producing Adenoma. Hypertens 2020, 75, 645–649. [Google Scholar] [CrossRef]
- Williams, T.A.; Monticone, S.; Schack, V.R.; Stindl, J.; Burrello, J.; Buffolo, F.; Annaratone, L.; Castellano, I.; Beuschlein, F.; Reincke, M.; et al. Somatic ATP1A1, ATP2B3, and KCNJ5 mutations in aldosterone-producing adenomas. Hypertens 2014, 63, 188–195. [Google Scholar] [CrossRef]
- Scholl, U.I.; Goh, G.; Stölting, G.; De Oliveira, R.C.; Choi, M.; Overton, J.D.; Fonseca, A.L.; Korah, R.; Starker, L.F.; Kunstman, J.; et al. Somatic and germline CACNA1D calcium channel mutations in aldosterone-producing adenomas and primary aldosteronism. Nat. Genet. 2013, 45, 1050–1054. [Google Scholar] [CrossRef] [PubMed]
- Dutta, R.K.; Arnesen, T.; Heie, A.; Walz, M.; Alesina, P.; Söderkvist, P.; Gimm, O. A somatic mutation in CLCN2 identified in a sporadic aldosterone-producing adenoma. Eur. J. Endocrinol. 2019, 181, K37–K41. [Google Scholar] [CrossRef]
- Rege, J.; Nanba, K.; Blinder, A.R.; Plaska, S.; Udager, A.M.; Vats, P.; Kumar-Sinha, C.; Giordano, T.J.; Rainey, W.E.; Else, T. Identification of Somatic Mutations in CLCN2 in Aldosterone-Producing Adenomas. J. Endocr. Soc. 2020, 4, bvaa123. [Google Scholar] [CrossRef]
- Espiard, S.; Knape, M.J.; Bathon, K.; Assié, G.; Rizk-Rabin, M.; Faillot, S.; Luscap-Rondof, W.; Abid, D.; Guignat, L.; Calebiro, D.; et al. Activating PRKACB somatic mutation in cortisol-producing adenomas. JCI Insight. 2018, 3, e98296. [Google Scholar] [CrossRef]
- Libé, R.; Mantovani, G.; Bondioni, S.; Lania, A.G.; Pedroni, C.; Beck-Peccoz, P.; Spada, A. Mutational Analysis of PRKAR1A and Gsα in Sporadic Adrenocortical Tumors. Exp. Clin. Endocrinol. Diabetes. 2005, 113, 248–251. [Google Scholar] [CrossRef]
- Vincent-Dejean, C.; Cazabat, L.; Groussin, L.; Perlemoine, K.; Fumey, G.; Tissier, F.; Bertagna, X.; Bertherat, J. Identification of a clinically homogenous subgroup of benign cortisol-secreting adrenocortical tumors characterized by alterations of the protein kinase A (PKA) subunits and high PKA activity. Eur. J. Endocrinol. 2008, 158, 829–839. [Google Scholar] [CrossRef]
- Leal, L.F.; Szarek, E.; Faucz, F.; Stratakis, C.A. Phosphodiesterase 8B and cyclic AMP signaling in the adrenal cortex. Endocrine 2015, 50, 27–31. [Google Scholar] [CrossRef]
- Bimpaki, E.I.; Nesterova, M.; Stratakis, C.A. Abnormalities of cAMP signaling are present in adrenocortical lesions associated with ACTH-independent Cushing syndrome despite the absence of mutations in known genes. Eur. J. Endocrinol. 2009, 161, 153–161. [Google Scholar] [CrossRef][Green Version]
- Horvath, A.; Giatzakis, C.; Tsang, K.; Greene, E.; Osorio, P.; Boikos, S.; Libè, R.; Patronas, Y.; Robinson-White, A.; Remmers, E.; et al. A cAMP-specific phosphodiesterase (PDE8B) that is mutated in adrenal hyperplasia is expressed widely in human and mouse tissues: A novel PDE8B isoform in human adrenal cortex. Eur. J. Hum. Genet. 2008, 16, 1245–1253. [Google Scholar] [CrossRef]
- Vezzosi, D.; Libe, R.; Baudry, C.; Rizk-Rabin, M.; Horvath, A.; Levy, I.; René-Corail, F.; Ragazzon, B.; Stratakis, C.A.; Vandecasteele, G.; et al. Phosphodiesterase 11A (PDE11A) gene defects in patients with acth-independent macronodular adrenal hyperplasia (AIMAH): Functional variants may contribute to genetic susceptibility of bilateral adrenal tumors. J. Clin. Endocrinol. Metab. 2012, 97, E2063–E2069. [Google Scholar] [CrossRef] [PubMed]
- Roussel, H.W.; Vezzosi, D.; Rizk-Rabin, M.; Barreau, O.; Ragazzon, B.; René-Corail, F.; de Reynies, A.; Bertherat, J.; Assié, G. Identification of Gene Expression Profiles Associated with Cortisol Secretion in Adrenocortical Adenomas. J. Clin. Endocrinol. Metab. 2013, 98, E1109–E1121. [Google Scholar] [CrossRef]
- Beuschlein, F.; Fassnacht, M.; Assié, G.; Calebiro, D.; Stratakis, C.A.; Osswald, A.; Ronchi, C.L.; Wieland, T.; Sbiera, S.; Faucz, F.R.; et al. Constitutive activation of PKA catalytic subunit in adrenal Cushing’s syndrome. N. Engl. J. Med. 2014, 370, 1019–1028. [Google Scholar] [CrossRef] [PubMed]
- Thiel, A.; Reis, A.-C.; Haase, M.; Goh, G.; Schott, M.; Willenberg, H.S.; Scholl, U.I. PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: A single-center study of 60 cases. Eur. J. Endocrinol. 2015, 172, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Rahane, C.; Kutzner, A.; Heese, K. Establishing a human adrenocortical carcinoma (ACC)-specific gene mutation signature. Cancer Genet. 2019, 230, 1–12. [Google Scholar] [CrossRef]
- Assié, G.; Letouzé, E.; Fassnacht, M.; Jouinot, A.; Luscap, W.; Barreau, O.; Omeiri, H.; Rodriguez, S.; Perlemoine, K.; René-Corail, F.; et al. Integrated genomic characterization of adrenocortical carcinoma. Nat. Genet. 2014, 46, 607–612. [Google Scholar] [CrossRef]
- Crona, J.; Beuschlein, F. Adrenocortical carcinoma—Towards genomics guided clinical care. Nat. Rev. Endocrinol. 2019, 15, 548–560. [Google Scholar] [CrossRef]
- Duan, K.; Hernandez, K.G.; Mete, O. Clinicopathological correlates of adrenal Cushing’s syndrome. J. Clin. Pathol. 2015, 68, 175–186. [Google Scholar] [CrossRef]
- Zheng, S.; Cherniack, A.D.; Dewal, N.; Moffitt, R.A.; Danilova, L.; Murray, B.A.; Lerario, A.M.; Else, T.; Knijnenburg, T.A.; Ciriello, G.; et al. Comprehensive Pan-Genomic Characterization of Adrenocortical Carcinoma. Cancer Cell 2016, 29, 723–736. [Google Scholar] [CrossRef]
- Pinto, E.M.; Chen, X.; Easton, J.; Finkelstein, D.; Liu, Z.; Pounds, S.; Rodriguez-Galindo, C.; Lund, T.C.; Mardis, E.R.; Wilson, R.K.; et al. Genomic landscape of paediatric adrenocortical tumours. Nat. Commun. 2015, 6, 6302. [Google Scholar] [CrossRef] [PubMed]
- Azizan, E.A.B.; Poulsen, H.; Tuluc, P.; Zhou, J.; Clausen, M.V.; Lieb, A.; Maniero, C.; Garg, S.; Bochukova, E.G.; Zhao, W.; et al. Somatic mutations in ATP1A1 and CACNA1D underlie a common subtype of adrenal hypertension. Nat. Genet. 2013, 45, 1055–1060. [Google Scholar] [CrossRef]
- Tan, G.C.; Negro, G.; Pinggera, A.; Tizen Laim, N.M.S.; Mohamed Rose, I.; Ceral, J.; Ryska, A.; Chin, L.K.; Kamaruddin, N.A.; Mohd Mokhtar, N.; et al. Aldosterone-Producing Adenomas: Histopathology-Genotype Correlation and Identification of a Novel CACNA1D Mutation. Hypertens 2017, 70, 129–136. [Google Scholar] [CrossRef]
- Byrd, J.B.; Turcu, A.F.; Auchus, R.J. Primary Aldosteronism. Circulation 2018, 138, 823–835. [Google Scholar] [CrossRef] [PubMed]
- Fernandes-Rosa, F.L.; Daniil, G.; Orozco, I.J.; Göppner, C.; El Zein, R.; Jain, V.; Boulkroun, S.; Jeunemaitre, X.; Amar, L.; Lefebvre, H.; et al. A gain-of-function mutation in the CLCN2 chloride channel gene causes primary aldosteronism. Nat. Genet. 2018, 50, 355–361. [Google Scholar] [CrossRef]
- Zennaro, M.-C.; Boulkroun, S.; Fernandes-Rosa, F. Genetic Causes of Functional Adrenocortical Adenomas. Endocr. Rev. 2017, 38, 516–537. [Google Scholar] [CrossRef]
- Choi, M.; Scholl, U.I.; Yue, P.; Björklund, P.; Zhao, B.; Nelson-Williams, C.; Ji, W.; Cho, Y.; Patel, A.; Men, C.J.; et al. Lifton, K+ channel mutations in adrenal aldosterone-producing adenomas and hereditary hypertension. Science 2011, 331, 768–772. [Google Scholar] [CrossRef]
- Bassett, M.H.; Suzuki, T.; Sasano, H.; White, P.C.; Rainey, W.E. The Orphan Nuclear Receptors NURR1 and NGFIB Regulate Adrenal Aldosterone Production. Mol. Endocrinol. 2004, 18, 279–290. [Google Scholar] [CrossRef]
- Romero, D.G.; Rilli, S.; Plonczynski, M.W.; Yanes, L.L.; Zhou, M.Y.; Gomez-Sanchez, E.P.; Gomez-Sanchez, C.E. Adrenal transcription regulatory genes modulated by angiotensin II and their role in steroidogenesis. Physiol. Genom. 2007, 30, 26–34. [Google Scholar] [CrossRef]
- Zennaro, M.-C.; Jeunemaitre, X.; Boulkroun, S. Integrating genetics and genomics in primary aldosteronism. Hypertens 2012, 60, 580–588. [Google Scholar] [CrossRef]
- Hattangady, N.G.; Karashima, S.; Yuan, L.; Ponce-Balbuena, D.; Jalife, J.; Gomez-Sanchez, C.E.; Auchus, R.J.; Rainey, W.E.; Else, T. Else, Mutated KCNJ5 activates the acute and chronic regulatory steps in aldosterone production. J. Mol. Endocrinol. 2016, 57, 1–11. [Google Scholar] [CrossRef]
- Nakamura, Y.; Felizola, S.J.A.; Satoh, F.; Konosu-Fukaya, S.; Sasano, H. Dissecting the molecular pathways of primary aldosteronism. Pathol. Int. 2014, 64, 482–489. [Google Scholar] [CrossRef]
- Beuschlein, F.; Boulkroun, S.; Osswald, A.; Wieland, T.; Nielsen, H.N.; Lichtenauer, U.D.; Penton, D.; Schack, V.R.; Amar, L.; Fischer, E.; et al. Somatic mutations in ATP1A1 and ATP2B3 lead to aldosterone-producing adenomas and secondary hypertension. Nat. Genet. 2013, 45, 440–444. [Google Scholar] [CrossRef]
- Stindl, J.; Tauber, P.; Sterner, C.; Tegtmeier, I.; Warth, R.; Bandulik, S. Pathogenesis of Adrenal Aldosterone-Producing Adenomas Carrying Mutations of the Na(+)/K(+)-ATPase. Endocrinology 2015, 156, 4582–4591. [Google Scholar] [CrossRef]
- Kitamoto, T.; Suematsu, S.; Yamazaki, Y.; Nakamura, Y.; Sasano, H.; Matsuzawa, Y.; Saito, J.; Omura, M.; Nishikawa, T. Clinical and Steroidogenic Characteristics of Aldosterone-Producing Adenomas with ATPase or CACNA1D Gene Mutations. J. Clin. Endocrinol. Metab. 2016, 101, 494–503. [Google Scholar] [CrossRef]
- Pinggera, A.; Negro, G.; Tuluc, P.; Brown, M.J.; Lieb, A.; Striessnig, J. Gating defects of disease-causing de novo mutations in Ca v 1.3 Ca 2+ channels. Channels 2018, 12, 388–402. [Google Scholar] [CrossRef]
- Ortner, N.J.; Kaserer, T.; Copeland, J.N.; Striessnig, J. De novo CACAN1D Ca2+ channelopathies: Clinical phenotypes and molecular mechanism. Pflügers Arch. Eur. J. Physiol. 2020, 472, 755–773. [Google Scholar] [CrossRef]
- Scholl, U.I.; Stölting, G.; Schewe, J.; Thiel, A.; Tan, H.; Nelson-Williams, C.; Vichot, A.A.; Jin, S.C.; Loring, E.; Untiet, V.; et al. CLCN2 chloride channel mutations in familial hyperaldosteronism type II. Nat. Genet. 2018, 50, 349–354. [Google Scholar] [CrossRef]
- Teo, A.E.D.; Garg, S.; Haris Shaikh, L.; Zhou, J.; Karet, F.F.E.; Gurnell, M.; Happerfield, L.; Marker, A.; Bienz, M.; Azizan, E.A.B.; et al. Pregnancy, Primary Aldosteronism, and Adrenal CTNNB1 Mutations. N. Engl. J. Med. 2015, 373, 1429–1436. [Google Scholar] [CrossRef]
- Wang, J.-J.; Peng, K.-Y.; Wu, V.-C.; Tseng, F.-Y.; Wu, K.-D. CTNNB1 Mutation in Aldosterone Producing Adenoma. Endocrinol. Metab. 2017, 32, 332. [Google Scholar] [CrossRef]
- Oki, K.; Gomez-Sanchez, C.E. The landscape of molecular mechanism for aldosterone production in aldosterone-producing adenoma. Endocr. J. 2020, 67, 989–995. [Google Scholar] [CrossRef]
- Heitzmann, D.; Derand, R.; Jungbauer, S.; Bandulik, S.; Sterner, C.; Schweda, F.; El Wakil, A.; Lalli, E.; Guy, N.; Mengual, R.; et al. Invalidation of TASK1 potassium channels disrupts adrenal gland zonation and mineralocorticoid homeostasis. EMBO J. 2008, 27, 179–187. [Google Scholar] [CrossRef]
- Felizola, S.J.A.; Nakamura, Y.; Ono, Y.; Kitamura, K.; Kikuchi, K.; Onodera, Y.; Ise, K.; Takase, K.; Sugawara, A.; Hattangady, N.; et al. PCP4: A regulator of aldosterone synthesis in human adrenocortical tissues. J. Mol. Endocrinol. 2014, 52, 159–167. [Google Scholar] [CrossRef]
- Kobuke, K.; Oki, K.; Gomez-Sanchez, C.E.; Gomez-Sanchez, E.P.; Ohno, H.; Itcho, K.; Yoshii, Y.; Yoneda, M.; Hattori, N. Calneuron 1 Increased Ca2+ in the Endoplasmic Reticulum and Aldosterone Production in Aldosterone-Producing Adenoma. Hypertens 2018, 71, 125–133. [Google Scholar] [CrossRef]
- Konosu-Fukaya, S.; Nakamura, Y.; Satoh, F.; Felizola, S.J.; Maekawa, T.; Ono, Y.; Morimoto, R.; Ise, K.; Takeda, K.-I.; Katsu, K.; et al. 3β-Hydroxysteroid dehydrogenase isoforms in human aldosterone-producing adenoma. Mol. Cell. Endocrinol. 2015, 408, 205–212. [Google Scholar] [CrossRef]
- Hattangady, N.G.; Olala, L.O.; Bollag, W.B.; Rainey, W.E. Acute and chronic regulation of aldosterone production. Mol. Cell. Endocrinol. 2012, 350, 151–162. [Google Scholar] [CrossRef]
- Bassett, M.H.; White, P.C.; E Rainey, W. The regulation of aldosterone synthase expression. Mol. Cell. Endocrinol. 2004, 217, 67–74. [Google Scholar] [CrossRef]
- Shimada, H.; Kogure, N.; Noro, E.; Kudo, M.; Sugawara, K.; Sato, I.; Shimizu, K.; Kobayashi, M.; Suzuki, D.; Parvin, R.; et al. High glucose stimulates expression of aldosterone synthase (CYP11B2) and secretion of aldosterone in human adrenal cells. FEBS Open Bio 2017, 7, 1410–1421. [Google Scholar] [CrossRef]
- Wansa, K.D.S.A.; Harris, J.; Muscat, G. The activation function-1 domain of Nur77/NR4A1 mediates trans-activation, cell specificity, and coactivator recruitment. J. Biol. Chem. 2002, 277, 33001–33011. [Google Scholar] [CrossRef]
- Kelly, S.N.; McKenna, T.J.; Young, L.S. Coregulatory protein-orphan nuclear receptor interactions in the human adrenal cortex. J. Endocrinol. 2005, 186, 33–42. [Google Scholar] [CrossRef]
- Holla, V.R.; Wu, H.; Shi, Q.; Menter, D.G.; DuBois, R.N. Nuclear orphan receptor NR4A2 modulates fatty acid oxidation pathways in colorectal cancer. J. Biol. Chem. 2011, 286, 30003–30009. [Google Scholar] [CrossRef] [PubMed]
- Noro, E.; Yokoyama, A.; Kobayashi, M.; Shimada, H.; Suzuki, S.; Hosokawa, M.; Takehara, T.; Parvin, R.; Igarashi, K.; Sugawara, A.; et al. Endogenous Purification of NR4A2 (Nurr1) Identified Poly (ADP-Ribose) Polymerase 1 as a Prime Coregulator in Human Adrenocortical H295R Cells. Int. J. Mol. Sci. 2018, 19, 1406. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Ouyang, J.; Wu, Z.; Shi, T.; Wang, B.; Ma, X.; Li, H.; Wang, S.; Zhang, X. Elementary studies on elevated steroidogenic factor-1 expression in aldosterone-producing adenoma. Urol. Oncol. Semin. Orig. Investig. 2012, 30, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Otake, H.; Sato, T.; Miyabayashi, K.; Shishido, Y.; Wang, C.-Y.; Shima, Y.; Kimura, H.; Yagi, M.; Ishihara, Y.; et al. Glycolytic genes are targets of the nuclear receptor Ad4BP/SF-1. Nat. Commun. 2014, 5, 3634. [Google Scholar] [CrossRef]
- Baba, T.; Otake, H.; Inoue, M.; Sato, T.; Ishihara, Y.; Moon, J.-Y.; Tsuchiya, M.; Miyabayashi, K.; Ogawa, H.; Shima, Y.; et al. Ad4BP/SF-1 regulates cholesterol synthesis to boost the production of steroids. Commun. Biol. 2018, 1, 18. [Google Scholar] [CrossRef] [PubMed]
- Shibata, H.; Kurihara, I.; Kobayashi, S.; Yokota, K.; Suda, N.; Saito, I.; Saruta, T. Regulation of differential COUP-TF-coregulator interactions in adrenal cortical steroidogenesis. J. Steroid Biochem. Mol. Biol. 2003, 85, 449–456. [Google Scholar] [CrossRef]
- Ota, T.; Doi, M.; Yamazaki, F.; Yarimizu, D.; Okada, K.; Murai, I.; Hayashi, H.; Kunisue, S.; Nakagawa, Y.; Okamura, H. Angiotensin II triggers expression of the adrenal gland zona glomerulosa-specific 3β-hydroxysteroid dehydrogenase isoenzyme through de novo protein synthesis of the orphan nuclear receptors NGFIB and NURR1. Mol. Cell. Biol. 2014, 34, 3880–3894. [Google Scholar] [CrossRef]
- Scholl, U.I.; Abriola, L.; Zhang, C.; Reimer, E.N.; Plummer, M.; Kazmierczak, B.I.; Zhang, J.; Hoyer, D.; Merkel, J.S.; Wang, W.; et al. Macrolides selectively inhibit mutant KCNJ5 potassium channels that cause aldosterone-producing adenoma. J. Clin. Investig. 2017, 127, 2739–2750. [Google Scholar] [CrossRef]
- Ito, R.; Sato, I.; Tsujita, T.; Yokoyama, A.; Sugawara, A. A ubiquitin-proteasome inhibitor bortezomib suppresses the expression of CYP11B2, a key enzyme of aldosterone synthesis. Biochem. Biophys. Res. Commun. 2017, 489, 21–28. [Google Scholar] [CrossRef]
- Uruno, A.; Matsuda, K.; Noguchi, N.; Yoshikawa, T.; Kudo, M.; Satoh, F.; Rainey, W.E.; Hui, X.G.; Akahira, J.I.; Nakamura, Y.; et al. Peroxisome proliferator-activated receptor-γ suppresses CYP11B2 expression and aldosterone production. J. Mol. Endocrinol. 2011, 46, 37–49. [Google Scholar] [CrossRef]
- Suzuki, D.; Saito-Hakoda, A.; Ito, R.; Shimizu, K.; Parvin, R.; Shimada, H.; Noro, E.; Suzuki, S.; Fujiwara, I.; Kagechika, H.; et al. Suppressive effects of RXR agonist PA024 on adrenal CYP11B2 expression, aldosterone secretion and blood pressure. PLoS ONE 2017, 12, e0181055. [Google Scholar] [CrossRef] [PubMed]
- Sato, Y.; Maekawa, S.; Ishii, R.; Sanada, M.; Morikawa, T.; Shiraishi, Y.; Yoshida, K.; Nagata, Y.; Sato-Otsubo, A.; Yoshizato, T.; et al. Recurrent somatic mutations underlie corticotropin-independent Cushing’s syndrome. Science 2014, 344, 917–920. [Google Scholar] [CrossRef]
- Fragoso, M.C.B.V.; Domenice, S.; Latronico, A.C.; Martin, R.M.; Pereira, M.A.A.M.; Zerbini, C.N.; Lucon, A.M.; Mendonca, B.B. Cushing’s Syndrome Secondary to Adrenocorticotropin-Independent Macronodular Adrenocortical Hyperplasia due to Activating Mutations of GNAS1 Gene. J. Clin. Endocrinol. Metab. 2003, 88, 2147–2151. [Google Scholar] [CrossRef]
- Di Dalmazi, G.; Altieri, B.; Scholz, C.-J.; Sbiera, S.; Luconi, M.; Waldman, J.; Kastelan, D.; Ceccato, F.; Chiodini, I.; Arnaldi, G.; et al. RNA Sequencing and Somatic Mutation Status of Adrenocortical Tumors: Novel Pathogenetic Insights. J. Clin. Endocrinol. Metab. 2020, 105, e4459–e4473. [Google Scholar] [CrossRef]
- Drougat, L.; Settas, N.; Ronchi, C.L.; Bathon, K.; Calebiro, D.; Maria, A.G.; Haydar, S.; Voutetakis, A.; London, E.; Faucz, F.R.; et al. Genomic and sequence variants of protein kinase A regulatory subunit type 1β (PRKAR1B) in patients with adrenocortical disease and Cushing syndrome. Genet. Med. 2021, 23, 174. [Google Scholar] [CrossRef]
- Cheng, L.-C.; Pai, T.-W.; Li, L.-A. Regulation of human CYP11B1 and CYP11B2 promoters by transposable elements and conserved cis elements. Steroids 2012, 77, 100–109. [Google Scholar] [CrossRef]
- Tung, W.-H.; Hsieh, H.-L.; Lee, I.-T.; Yang, C.-M. Enterovirus 71 modulates a COX-2/PGE2/cAMP-dependent viral replication in human neuroblastoma cells: Role of the c-Src/EGFR/p42/p44 MAPK/CREB signaling pathway. J. Cell. Biochem. 2011, 112, 559–570. [Google Scholar] [CrossRef]
- Tong, Q.; Weaver, M.R.; Kosmacek, E.A.; O’Connor, B.P.; Harmacek, L.; Venkataraman, S.; Oberley-Deegan, R.E. MnTE-2-PyP reduces prostate cancer growth and metastasis by suppressing p300 activity and p300/HIF-1/CREB binding to the promoter region of the PAI-1 gene. Free Radic. Biol. Med. 2016, 94, 185–194. [Google Scholar] [CrossRef]
- Shi, Y.; Venkataraman, S.L.; Dodson, G.E.; Mabb, A.M.; LeBlanc, S.; Tibbetts, R.S. Direct regulation of CREB transcriptional activity by ATM in response to genotoxic stress. Proc. Natl. Acad. Sci. USA 2004, 101, 5898–5903. [Google Scholar] [CrossRef]
- Guo, C.; Li, J.; Myatt, L.; Zhu, X.; Sun, K. Induction of Galphas contributes to the paradoxical stimulation of cytosolic phospholipase A2alpha expression by cortisol in human amnion fibroblasts. Mol. Endocrinol. 2010, 24, 1052–1061. [Google Scholar] [CrossRef]
- Kamilaris, C.D.C.; Hannah-Shmouni, F.; Stratakis, C.A. Adrenocortical tumorigenesis: Lessons from genetics. Best Pract. Res. Clin. Endocrinol. Metab. 2020, 34, 101428. [Google Scholar] [CrossRef]
- Juhlin, C.C.; Goh, G.; Healy, J.; Fonseca, A.L.; Scholl, U.I.; Stenman, A.; Kunstman, J.; Brown, T.C.; Overton, J.D.; Mane, S.M.; et al. Whole-Exome Sequencing Characterizes the Landscape of Somatic Mutations and Copy Number Alterations in Adrenocortical Carcinoma. J. Clin. Endocrinol. Metab. 2015, 100, E493–E502. [Google Scholar] [CrossRef]
- Bieging, K.T.; Mello, S.S.; Attardi, L.D. Unravelling mechanisms of p53-mediated tumour suppression. Nat. Rev. Cancer. 2014, 14, 359–370. [Google Scholar] [CrossRef]
- Harbour, J.W.; Dean, D.C. Rb function in cell-cycle regulation and apoptosis. Nat. Cell Biol. 2000, 2, E65–E67. [Google Scholar] [CrossRef]
- Gao, C.; Xiao, G.; Hu, J. Regulation of Wnt/β-catenin signaling by posttranslational modifications. Cell Biosci. 2014, 4, 13. [Google Scholar] [CrossRef]
- Shang, S.; Hua, F.; Hu, Z.-W. The regulation of β-catenin activity and function in cancer: Therapeutic opportunities. Oncotarget 2017, 8, 33972–33989. [Google Scholar] [CrossRef]
- Tsukiyama, T.; Koo, B.; Hatakeyama, S. Post-translational Wnt receptor regulation: Is the fog slowly clearing? BioEssays 2021, 43, 2000297. [Google Scholar] [CrossRef]
- Wang, H.; Shen, Q.; Ye, L.-H.; Ye, J. MED12 mutations in human diseases. Protein Cell 2013, 4, 643–646. [Google Scholar] [CrossRef] [PubMed]
- El Andaloussi, A.; Al-Hendy, A.; Ismail, N.; Boyer, T.G.; Halder, S.K. Introduction of Somatic Mutation in MED12 Induces Wnt4/β-Catenin and Disrupts Autophagy in Human Uterine Myometrial Cell. Reprod. Sci. 2020, 27, 823–832. [Google Scholar] [CrossRef]
- Al-Hendy, A.; Laknaur, A.; Diamond, M.P.; Ismail, N.; Boyer, T.G.; Halder, S.K. Silencing Med12 Gene Reduces Proliferation of Human Leiomyoma Cells Mediated via Wnt/β-Catenin Signaling Pathway. Endocrinology 2017, 158, en.2016-1097. [Google Scholar] [CrossRef]
- Chen, G.; A, J.; Wang, M.; Farley, S.; Lee, L.-Y.; Sawicki, M.P.; Lee, L.-Y.; Lee, L.-C. Menin promotes the Wnt signaling pathway in pancreatic endocrine cells. Mol. Cancer Res. 2008, 6, 1894–1907. [Google Scholar] [CrossRef]
- Jiang, X.; Cao, Y.; Li, F.; Su, Y.; Li, Y.; Peng, Y.; Cheng, Y.; Zhang, C.; Wang, W.; Ning, G. Targeting β-catenin signaling for therapeutic intervention in MEN1-deficient pancreatic neuroendocrine tumours. Nat. Commun. 2014, 5, 5809. [Google Scholar] [CrossRef]
- Huang, Y.-S.; Shih, H.-M. Daxx positively modulates β-catenin/TCF4-mediated transcriptional potential. Biochem. Biophys. Res. Commun. 2009, 386, 762–768. [Google Scholar] [CrossRef] [PubMed]
- Baquedano, M.S.; Berensztein, E.; Saraco, N.; Dorn, G.V.; De Dávila, M.T.; A Rivarola, M.; Belgorosky, A. Expression of the IGF System in Human Adrenal Tissues from Early Infancy to Late Puberty: Implications for the Development of Adrenarche. Pediatr. Res. 2005, 58, 451–458. [Google Scholar] [CrossRef]
- Pereira, S.; Monteiro, M.; Costa, M.M.; Moreira, Â.; Alves, M.G.; Oliveira, P.F.; Jarak, I.; Pignatelli, D. IGF2 role in adrenocortical carcinoma biology. Endocrine 2019, 66, 326–337. [Google Scholar] [CrossRef]
- Sun, Y.; Kolligs, F.T.; Hottiger, M.; Mosavin, R.; Fearon, E.R.; Nabel, G.J. Regulation of beta -catenin transformation by the p300 transcriptional coactivator. Proc. Natl. Acad. Sci. USA 2000, 97, 12613–12618. [Google Scholar] [CrossRef]
- Taurin, S.; Sandbo, N.; Qin, Y.; Browning, D.; Dulin, N.O. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J. Biol. Chem. 2006, 281, 9971–9976. [Google Scholar] [CrossRef]
- Sierra, J.; Yoshida, T.; Joazeiro, C.A.; Jones, K.A. The APC tumor suppressor counteracts beta-catenin activation and H3K4 methylation at Wnt target genes. Genes Dev. 2006, 20, 586–600. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shimada, H.; Yamazaki, Y.; Sugawara, A.; Sasano, H.; Nakamura, Y. Molecular Mechanisms of Functional Adrenocortical Adenoma and Carcinoma: Genetic Characterization and Intracellular Signaling Pathway. Biomedicines 2021, 9, 892. https://doi.org/10.3390/biomedicines9080892
Shimada H, Yamazaki Y, Sugawara A, Sasano H, Nakamura Y. Molecular Mechanisms of Functional Adrenocortical Adenoma and Carcinoma: Genetic Characterization and Intracellular Signaling Pathway. Biomedicines. 2021; 9(8):892. https://doi.org/10.3390/biomedicines9080892
Chicago/Turabian StyleShimada, Hiroki, Yuto Yamazaki, Akira Sugawara, Hironobu Sasano, and Yasuhiro Nakamura. 2021. "Molecular Mechanisms of Functional Adrenocortical Adenoma and Carcinoma: Genetic Characterization and Intracellular Signaling Pathway" Biomedicines 9, no. 8: 892. https://doi.org/10.3390/biomedicines9080892
APA StyleShimada, H., Yamazaki, Y., Sugawara, A., Sasano, H., & Nakamura, Y. (2021). Molecular Mechanisms of Functional Adrenocortical Adenoma and Carcinoma: Genetic Characterization and Intracellular Signaling Pathway. Biomedicines, 9(8), 892. https://doi.org/10.3390/biomedicines9080892