
A Magnetic Nanocomposite Modifier for Improved Ultrasensitive Detection of Hexavalent Chromium in Water Samples



Abstract
:
1. Introduction
2. Experimental
2.1. Solutions
2.2. Preparation of MNPs@SiO2@Lys
2.3. Techniques for the Characterization of MNPs@SiO2@Lys and the Hydrophilicity of Electrodes
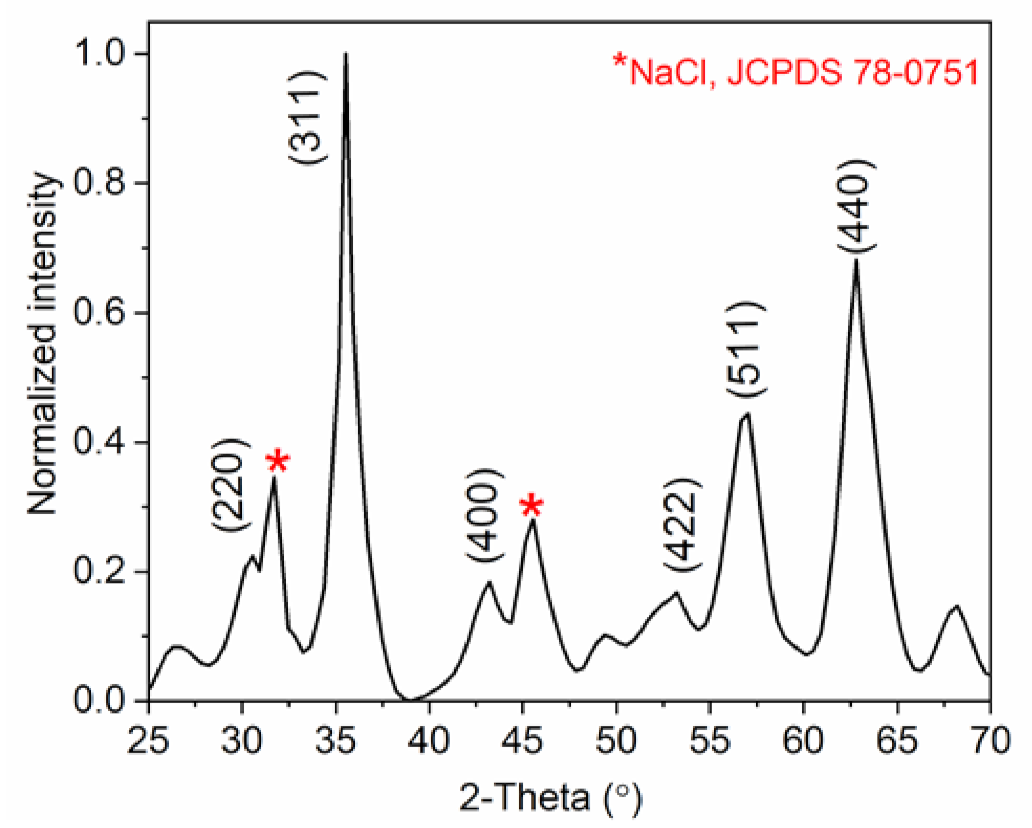
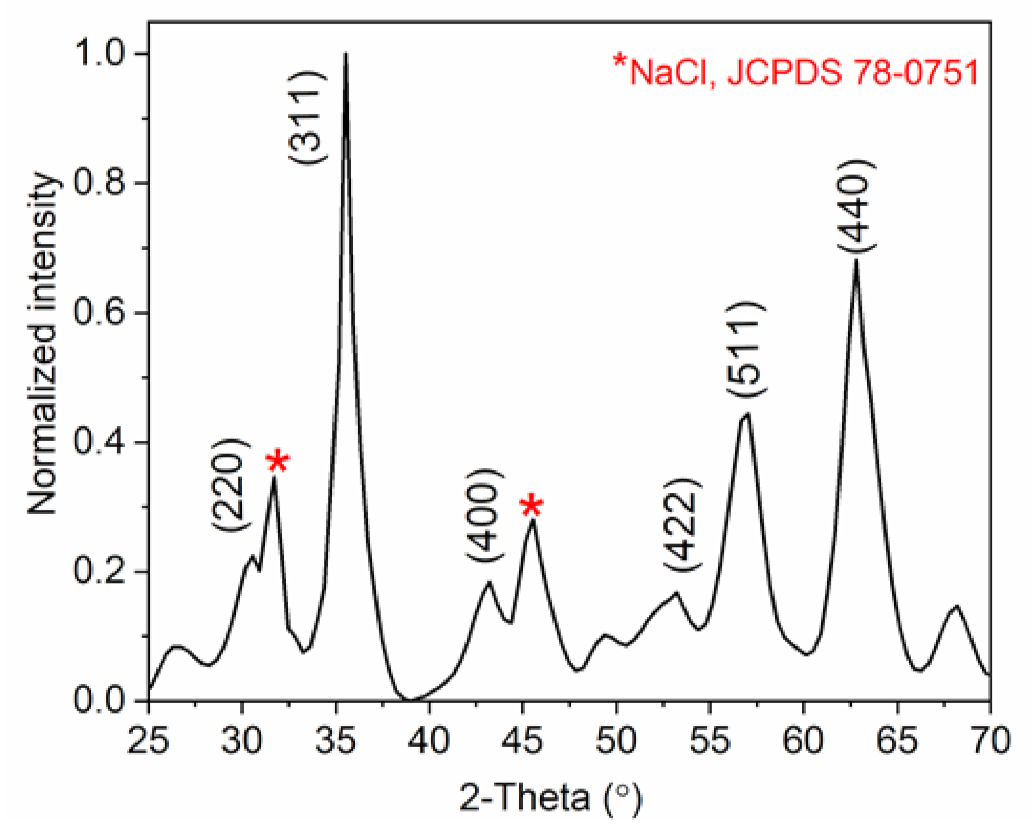
2.3.1. Structural Analysis Using XRD
2.3.2. Morphology Measurements of MNPs@SiO2@Lys
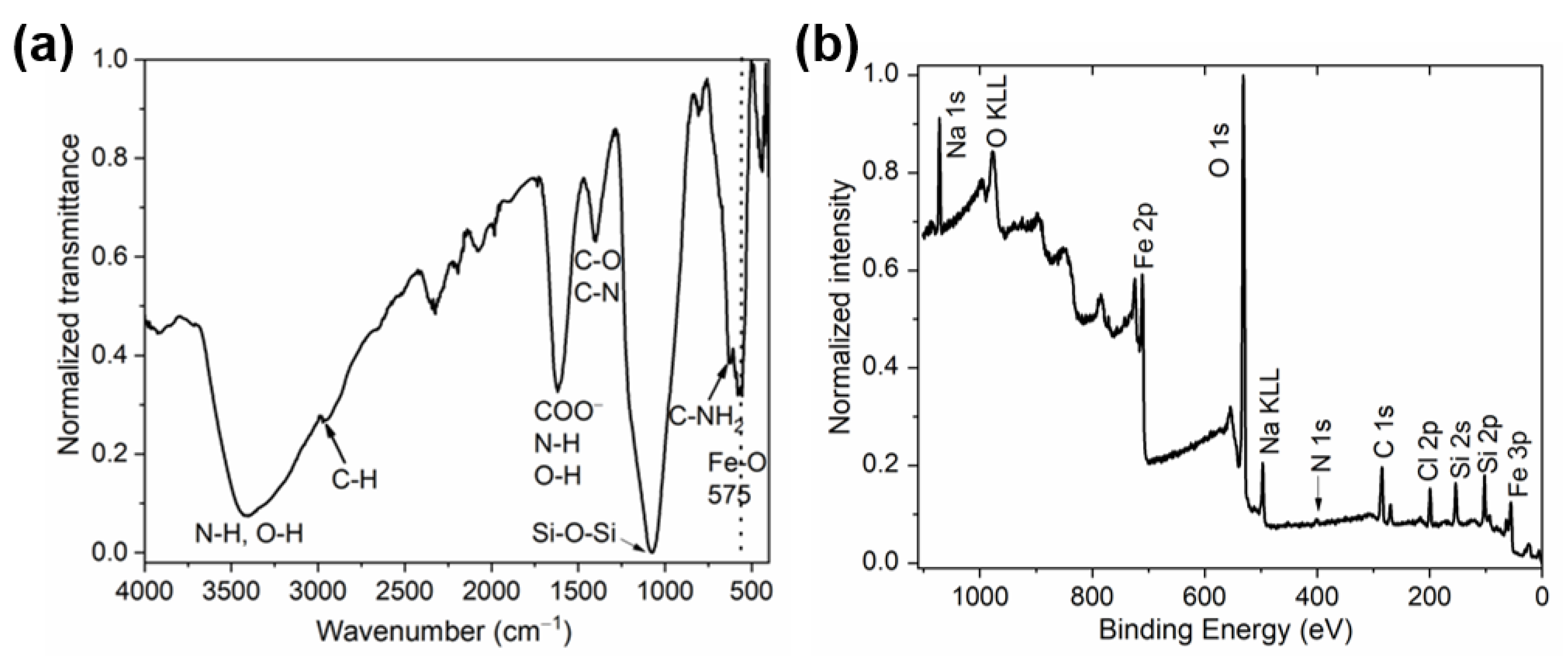
2.3.3. Surface Analysis Using ATR-FTIR Spectroscopy and XPS Analysis
2.3.4. Goniometry Measurements
2.4. Electrochemical Measurements
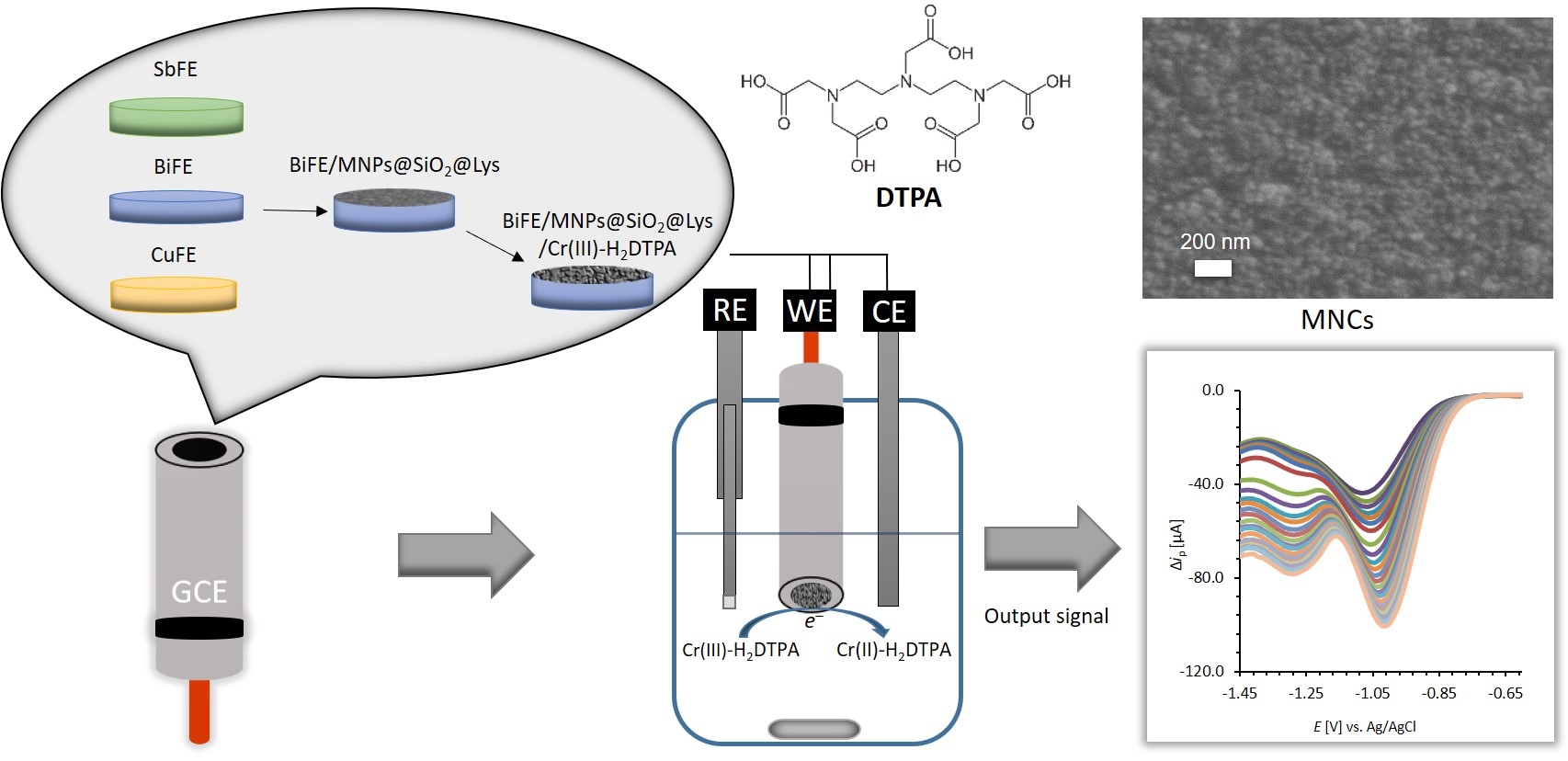
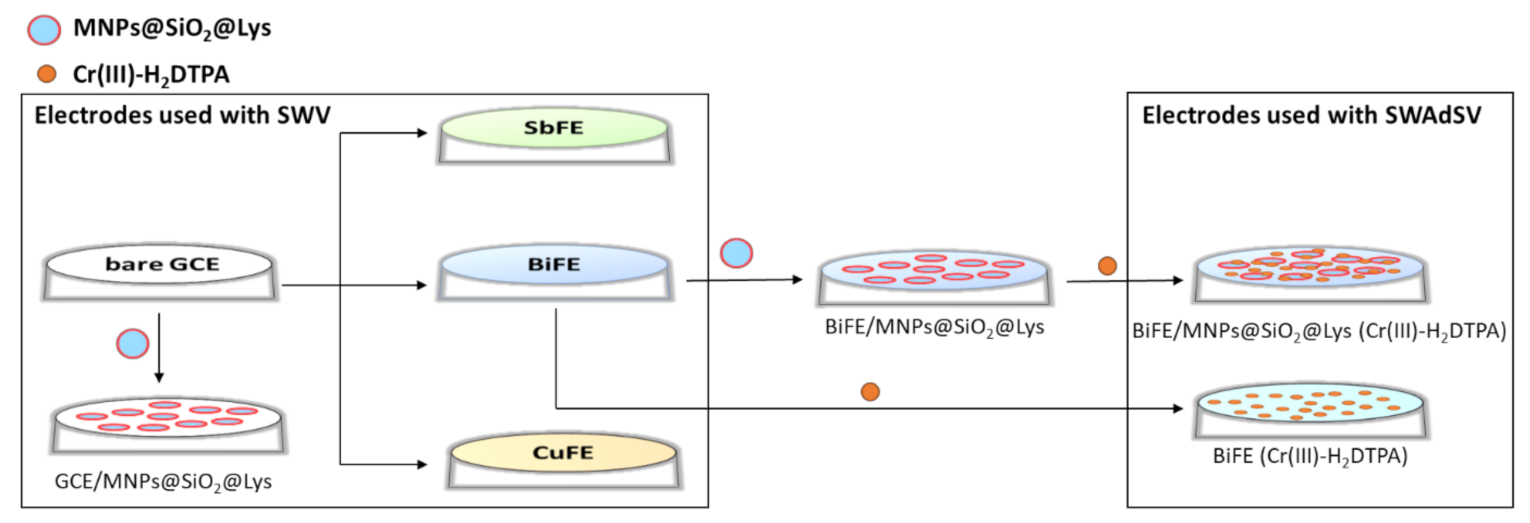
2.4.1. Preparation of the Ex-Situ Electrodes
2.4.2. Cathodic SWV Measurements of Cr(VI) Using Ex Situ Electrodes
2.4.3. SWAdSV Measurements Using DTPA as a Complexing Agent
2.4.4. Modification of the GCE or Ex-Situ BiFE with MNPs@SiO2@Lys
3. Results and Discussion
3.1. Analytical Performance of Different Electrodes
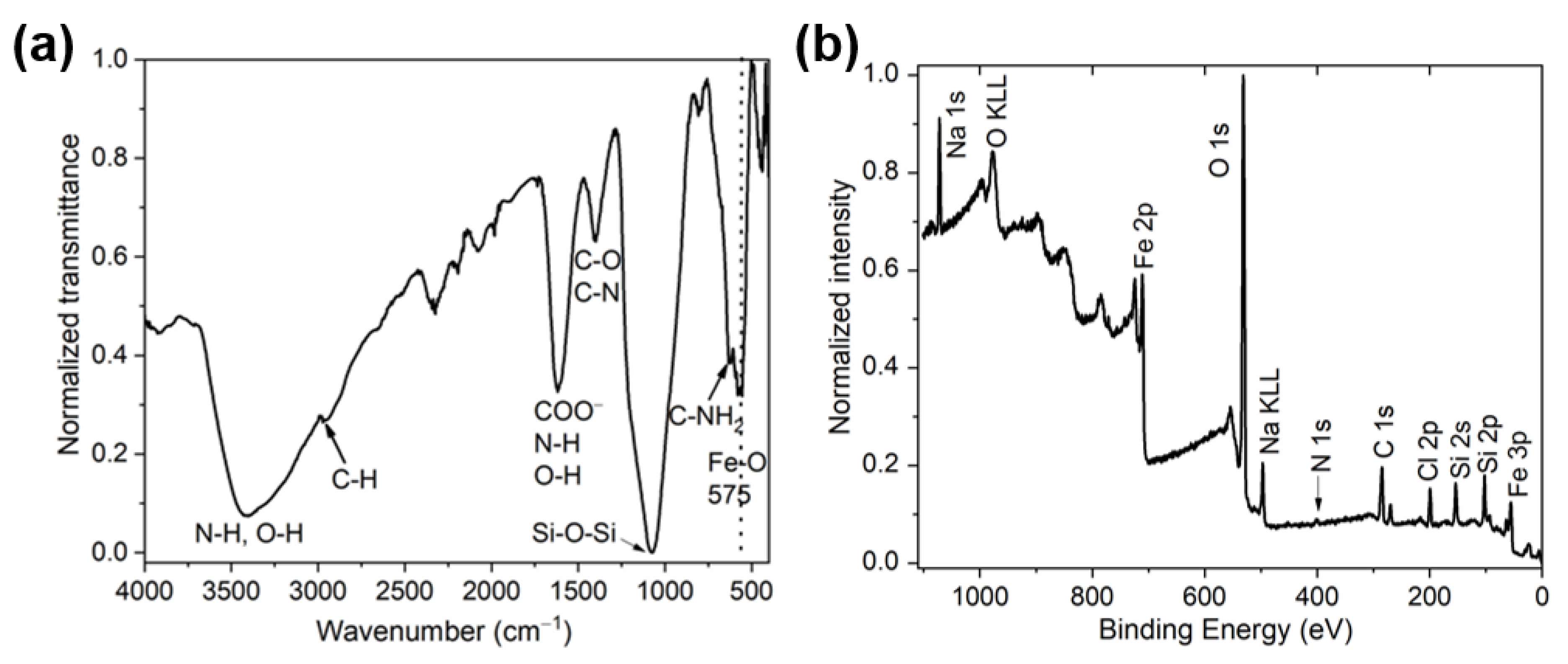
3.2. Characterization of the MNCs
3.3. The Hydrophilicity of the Surface of the Bare GCE and Differently Modified GCEs
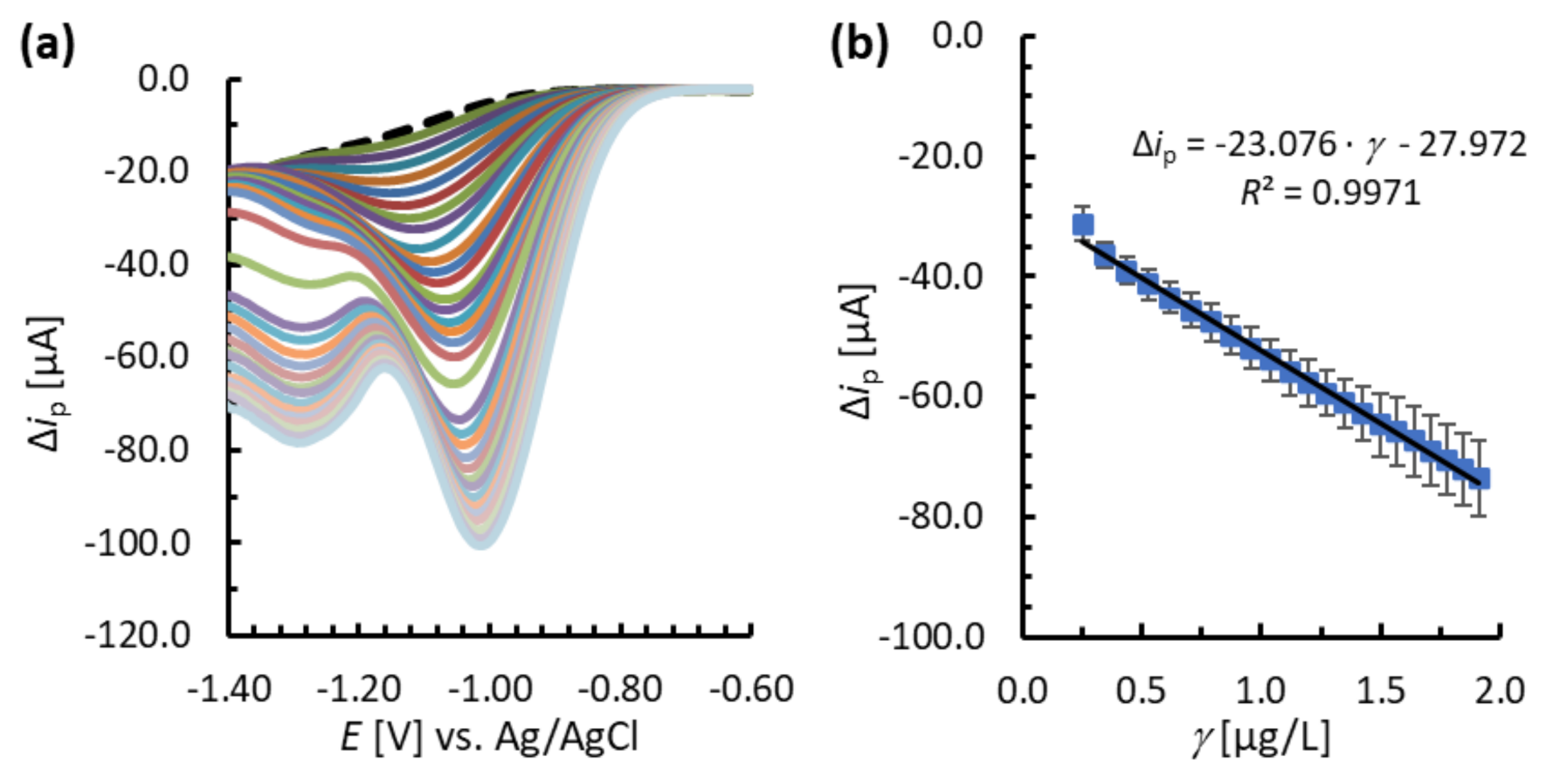
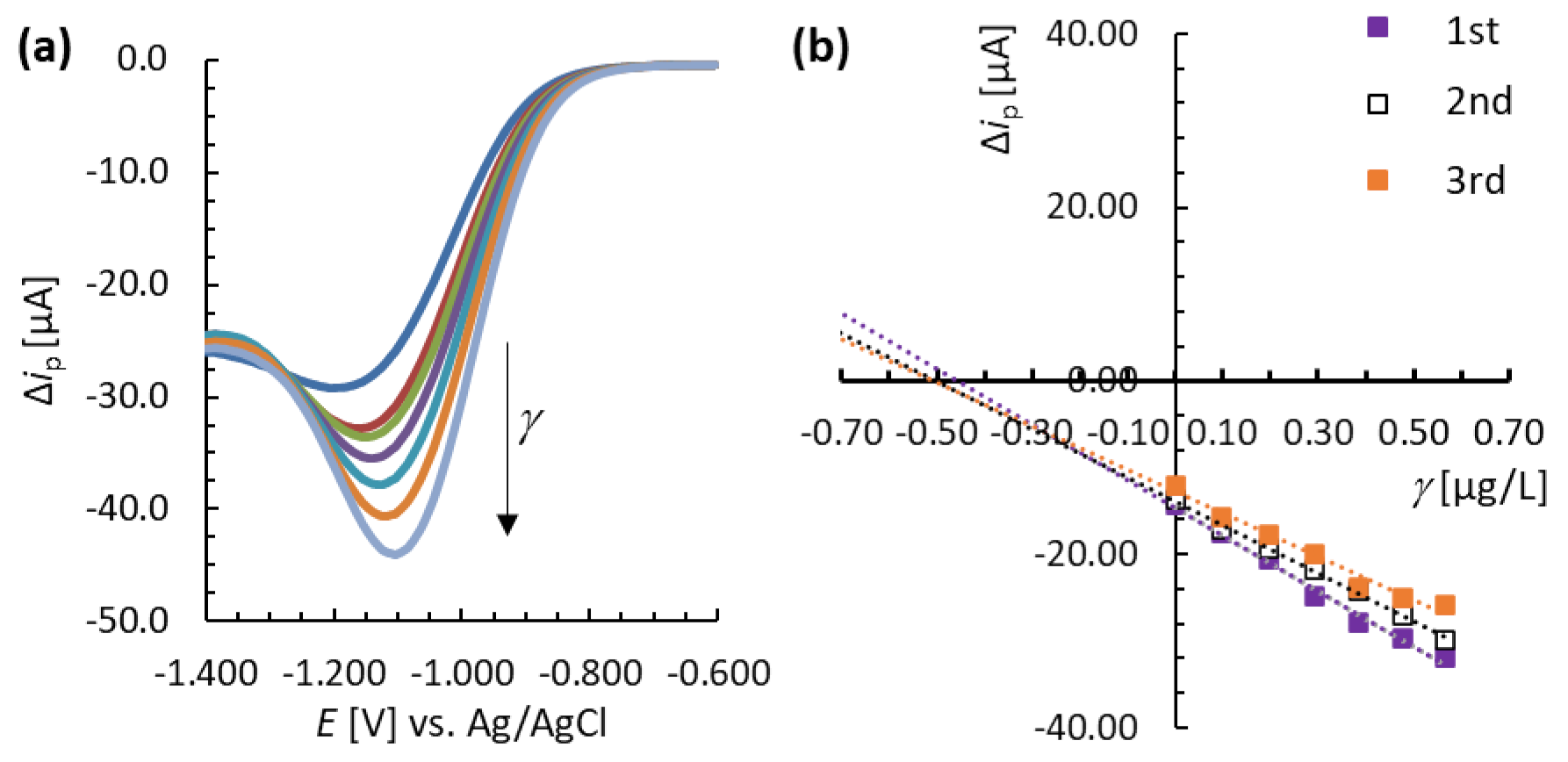
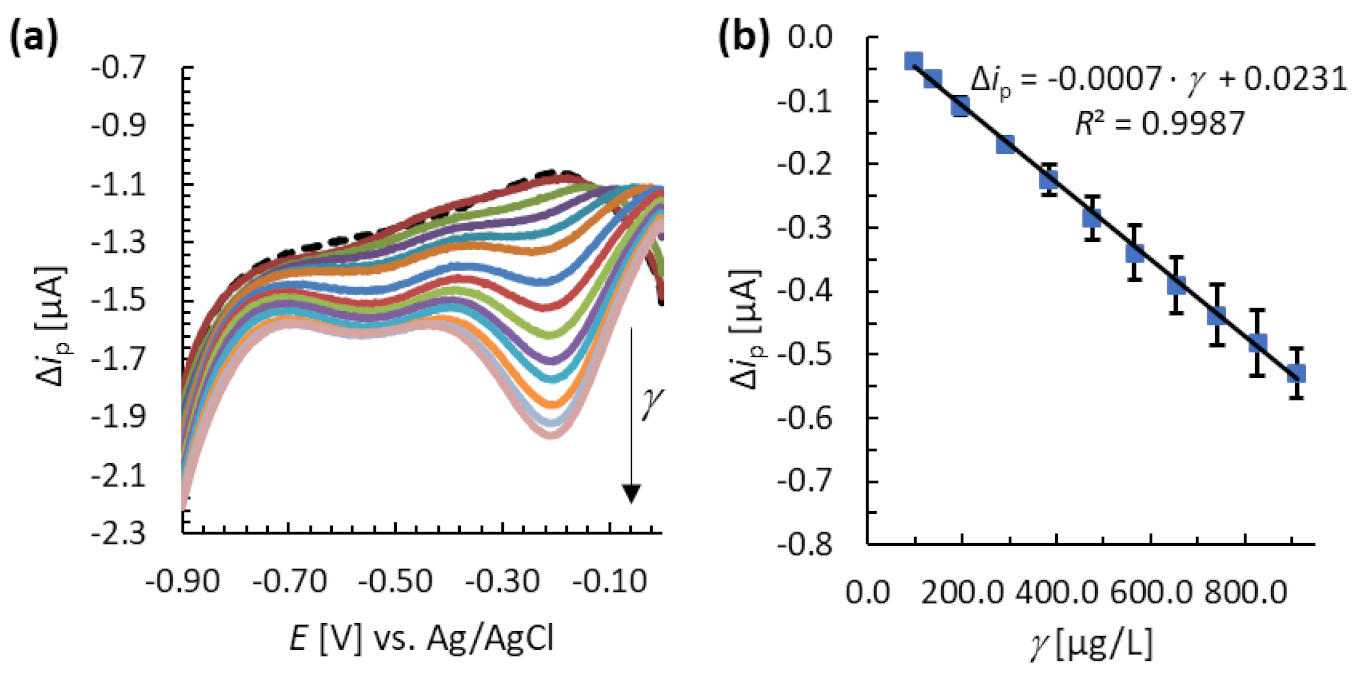
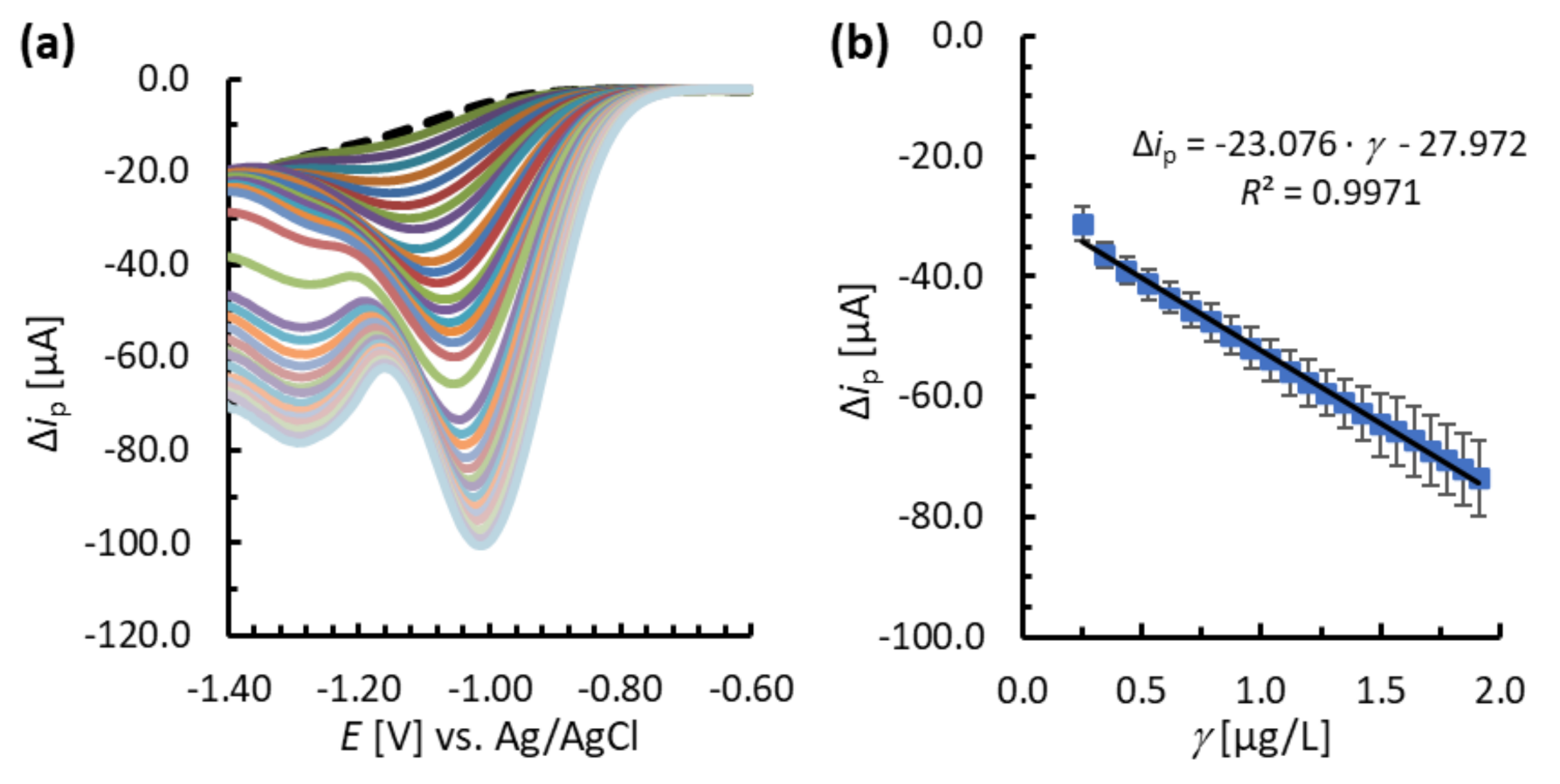
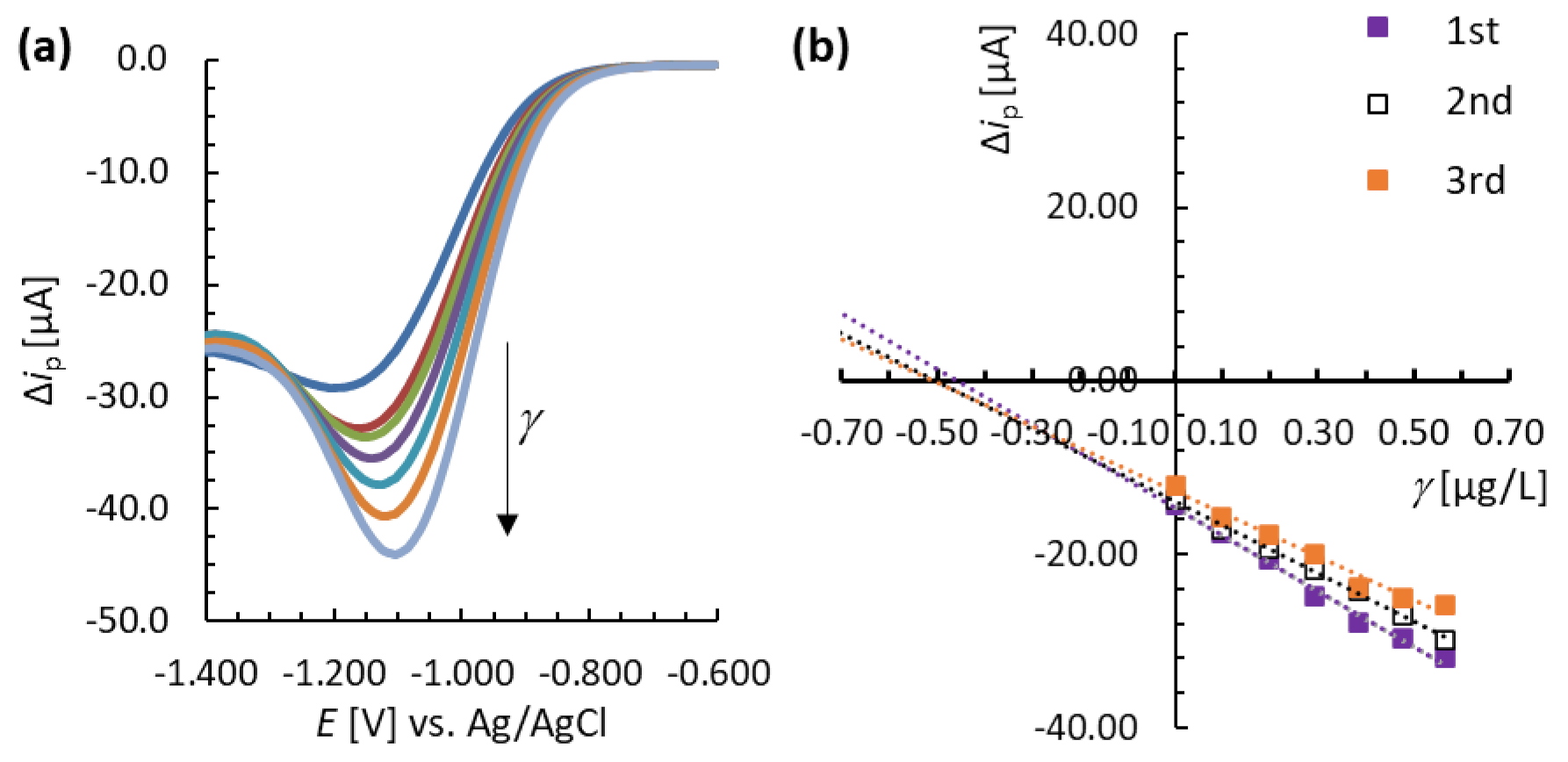
3.4. Analytical Performance of the MNC-Modified Electrodes
3.5. Analysis of a Real Sample
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Flora, S. Threshold mechanisms and site specificity in chromium(VI) carcinogenesis. Carcinogenesis 2000, 21, 533–541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliveira, H. Chromium as an Environmental Pollutant: Insights on Induced Plant Toxicity. J. Bot. 2012, 2012, 375843. [Google Scholar] [CrossRef]
- World Health Organization. Chemical Hazards in Drinking-Water: Chromium; WHO Press: Geneva, Switzerland, 2004. [Google Scholar]
- Almeida, J.C.; Cardoso, C.E.D.; Tavares, D.S.; Freitas, R.; Trindade, T.; Vale, C.; Pereira, E. Chromium removal from contaminated waters using nanomaterials—A review. TrAC Trends Anal. Chem. 2019, 118, 277–291. [Google Scholar] [CrossRef]
- Bobrowski, A.; Królicka, A.; Zarębski, J. Characteristics of Voltammetric Determination and Speciation of Chromium—A Review. Electroanalysis 2009, 21, 1449–1458. [Google Scholar] [CrossRef]
- Gómez, V.; Callao, M.P. Chromium determination and speciation since 2000. TrAC Trends Anal. Chem. 2006, 25, 1006–1015. [Google Scholar] [CrossRef]
- Karosi, R.; Andruch, V.; Posta, J.; Balogh, J. Separation of chromium (VI) using complexation and its determination with GFAAS. Microchem. J. 2006, 82, 61–65. [Google Scholar] [CrossRef]
- Zhao, W.R.; Kang, T.F.; Lu, L.P.; Shen, F.X.; Cheng, S.Y. A novel electrochemical sensor based on gold nanoparticles and molecularly imprinted polymer with binary functional monomers for sensitive detection of bisphenol A. J. Electroanal. Chem. 2017, 786, 102–111. [Google Scholar] [CrossRef]
- Tyszczuk-Rotko, K.; Madejska, K.; Domańska, K. Ultrasensitive hexavalent chromium determination at bismuth film electrode prepared with mediator. Talanta 2018, 182, 62–68. [Google Scholar] [CrossRef]
- Ariño, C.; Serrano, N.; Díaz-Cruz, J.M.; Esteban, M. Voltammetric determination of metal ions beyond mercury electrodes. A review. Anal. Chim. Acta 2017, 990, 11–53. [Google Scholar] [CrossRef] [Green Version]
- Domínguez, O.; Arcos, M.J. Speciation of Chromium by Adsorptive Stripping Voltammetry Using Pyrocatechol Violet. Electroanalysis 2000, 12, 449–458. [Google Scholar] [CrossRef]
- Domínguez, O.; Julia Arcos, M. Simultaneous determination of chromium(VI) and chromium(III) at trace levels by adsorptive stripping voltammetry. Anal. Chim. Acta 2002, 470, 241–252. [Google Scholar] [CrossRef]
- Domínguez, O.; Asunción Alonso, M.; Arcos, M.J. Application of an Optimization Procedure in Adsorptive Stripping Voltammetry for the Determination of Chromium with Ammonium Pyrrolidine Dithiocarbamate. Electroanalysis 2002, 14, 1083–1089. [Google Scholar] [CrossRef]
- Grabarczyk, M. Speciation Analysis of Chromium by Adsorptive Stripping Voltammetry in Tap and River Water Samples. Electroanalysis 2008, 20, 2217–2222. [Google Scholar] [CrossRef]
- Domínguez, O.; Sanllorente, S.; Arcos, M.J. Application of an Optimization Procedure of Adsorptive Stripping Voltammetry for the Determination of Chromium in Wine. Electroanalysis 1999, 11, 1273–1279. [Google Scholar] [CrossRef]
- Lin, L.; Lawrence, N.S.; Thongngamdee, S.; Wang, J.; Lin, Y. Catalytic adsorptive stripping determination of trace chromium (VI) at the bismuth film electrode. Talanta 2005, 65, 144–148. [Google Scholar] [CrossRef]
- Torrance, K.; Gatford, C. Determination of soluble chromium in simulated PWR coolant by differential-pulse adsorptive stripping voltammetry. Talanta 1987, 34, 939–944. [Google Scholar] [CrossRef]
- Boussemart, M.; van den Berg, C.M.G.; Ghaddaf, M. The determination of the chromium speciation in sea water using catalytic cathodic stripping voltammetry. Anal. Chim. Acta 1992, 262, 103–115. [Google Scholar] [CrossRef]
- Li, Y.; Xue, H. Determination of Cr(III) and Cr(VI) species in natural waters by catalytic cathodic stripping voltammetry. Anal. Chim. Acta 2001, 448, 121–134. [Google Scholar] [CrossRef]
- Sander, S.; Navrátil, T.; Novotný, L. Study of the Complexation, Adsorption and Electrode Reaction Mechanisms of Chromium(VI) and (III) with DTPA Under Adsorptive Stripping Voltammetric Conditions. Electroanalysis 2003, 15, 1513–1521. [Google Scholar] [CrossRef]
- Grabarczyk, M.; Kaczmarek, L.; Korolczuk, M. Determination of Cr(VI) in the Presence of Complexing Agents and Humic Substances by Catalytic Stripping Voltammetry. Electroanalysis 2007, 19, 1183–1188. [Google Scholar] [CrossRef]
- Bobrowski, A.; Baś, B.; Dominik, J.; Niewiara, E.; Szalińska, E.; Vignati, D.; Zarębski, J. Chromium speciation study in polluted waters using catalytic adsorptive stripping voltammetry and tangential flow filtration. Talanta 2004, 63, 1003–1012. [Google Scholar] [CrossRef]
- Dobney, A.M.; Greenway, G.M. On-line determination of chromium by adsorptive cathodic stripping voltammetry. Analyst 1994, 119, 293–297. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Lu, J.; Tian, B.; MacDonald, D.; Olsen, K. Flow probe for in situ electrochemical monitoring of trace chromium. Analyst 1999, 124, 349–352. [Google Scholar] [CrossRef]
- Yong, L.; Armstrong, K.C.; Dansby-Sparks, R.N.; Carrington, N.A.; Chambers, J.Q.; Xue, Z.-L. Quantitative analysis of trace chromium in blood samples. Combination of the advanced oxidation process with catalytic adsorptive stripping voltammetry. Anal. Chem. 2006, 78, 7582–7587. [Google Scholar] [CrossRef] [Green Version]
- Brett, C.M.A.; Filipe, O.M.S.; Susanna Neves, C. Determination of Chromium(VI) by Batch Injection Analysis and Adsorptive Stripping Voltammetry. Anal. Lett. 2003, 36, 955–969. [Google Scholar] [CrossRef]
- Chatzitheodorou, E.; Economou, A.; Voulgaropoulos, A. Trace Determination of Chromium by Square-Wave Adsorptive Stripping Voltammetry on Bismuth Film Electrodes. Electroanalysis 2004, 16, 1745–1754. [Google Scholar] [CrossRef]
- Gao, Z.; Siow, K.S. Catalytic-adsorptive stripping voltammetric determination of chromium in environmental materials. Electroanalysis 1996, 8, 602–606. [Google Scholar] [CrossRef]
- Vukomanovic, D.V.; Vanloon, G.W.; Nakatsu, K.; Zoutman, D.E. Determination of Chromium (VI) and (III) by Adsorptive Stripping Voltammetry with Pyrocatechol Violet. Microchem. J. 1997, 57, 86–95. [Google Scholar] [CrossRef]
- Minamata Convention on Mercury. Available online: http://www.mercuryconvention.org/ (accessed on 8 March 2021).
- Jovanovski, V.; Hočevar, S.B.; Ogorevc, B. Bismuth electrodes in contemporary electroanalysis. Curr. Opin. Electrochem. 2017, 3, 114–122. [Google Scholar] [CrossRef]
- Švancara, I.; Prior, C.; Hočevar, S.B.; Wang, J. A Decade with Bismuth-Based Electrodes in Electroanalysis. Electroanalysis 2010, 22, 1405–1420. [Google Scholar] [CrossRef]
- Wang, J. Stripping Analysis at Bismuth Electrodes: A Review. Electroanalysis 2005, 17, 1341–1346. [Google Scholar] [CrossRef]
- Jovanovski, V.; Hrastnik, N.I.; Hočevar, S.B. Copper film electrode for anodic stripping voltammetric determination of trace mercury and lead. Electrochem. Commun. 2015, 57, 1–4. [Google Scholar] [CrossRef]
- Bobrowski, A.; Putek, M.; Zarębski, J. Antimony Film Electrode Prepared In Situ in Hydrogen Potassium Tartrate in Anodic Stripping Voltammetric Trace Detection of Cd(II), Pb(II), Zn(II), Tl(I), In(III) and Cu(II). Electroanalysis 2012, 24, 1071–1078. [Google Scholar] [CrossRef]
- Bobrowski, A.; Maczuga, M.; Królicka, A.; Konstanteli, E.; Sakellaropoulou, C.; Economou, A. Determination of Copper(II) Through Anodic Stripping Voltammetry in Tartrate Buffer Using an Antimony Film Screen-printed Carbon Electrode. Anal. Lett. 2017, 50, 2920–2936. [Google Scholar] [CrossRef]
- Maczuga, M.; Economou, A.; Bobrowski, A.; Prodromidis, M.I. Novel screen-printed antimony and tin voltammetric sensors for anodic stripping detection of Pb(II) and Cd(II). Electrochim. Acta 2013, 114, 758–765. [Google Scholar] [CrossRef]
- Zhu, W.W.; Li, N.B.; Luo, H.Q. Simultaneous determination of chromium(III) and cadmium(II) by differential pulse anodic stripping voltammetry on a stannum film electrode. Talanta 2007, 72, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Li, B.L.; Wu, Z.L.; Xiong, C.H.; Luo, H.Q.; Li, N.B. Anodic stripping voltammetric measurement of trace cadmium at tin-coated carbon paste electrode. Talanta 2012, 88, 707–710. [Google Scholar] [CrossRef]
- Xu, S.; Wang, X.; Zhou, C. A micro electrochemical sensor based on bismuth-modified mesoporous carbon for hexavalent chromium detection. In Proceedings of the 2015 IEEE Sensors, Busan, Korea, 1–4 November 2015; pp. 1–4. [Google Scholar]
- Li, M.; He, R.; Wang, S.; Feng, C.; Wu, H.; Mei, H. Visible light driven photoelectrochemical sensor for chromium(VI) by using BiOI microspheres decorated with metallic bismuth. Microchim. Acta 2019, 186, 345. [Google Scholar] [CrossRef] [PubMed]
- Santhosh, C.; Saranya, M.; Ramachandran, R.; Felix, S.; Velmurugan, V.; Nirmala Grace, A. Graphene/Gold Nanocomposites-Based Thin Films as an Enhanced Sensing Platform for Voltammetric Detection of Cr(VI) Ions. J. Nanotechnol. 2014, 2014, 304526. [Google Scholar] [CrossRef] [Green Version]
- Sari, T.K.; Takahashi, F.; Jin, J.; Zein, R.; Munaf, E. Electrochemical Determination of Chromium(VI) in River Water with Gold Nanoparticles—Graphene Nanocomposites Modified Electrodes. Anal. Sci. 2018, 34, 155–160. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, R.; Zhang, W.; Zhou, S.; Xue, Z.-L.; Xu, L.; Gu, Y.; Miao, Y. Improved Bi film wrapped single walled carbon nanotubes for ultrasensitive electrochemical detection of trace Cr(VI). Electrochim. Acta 2013, 113, 686–693. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salimi, A.; Pourbahram, B.; Mansouri-Majd, S.; Hallaj, R. Manganese oxide nanoflakes/multi-walled carbon nanotubes/chitosan nanocomposite modified glassy carbon electrode as a novel electrochemical sensor for chromium (III) detection. Electrochim. Acta 2015, 156, 207–215. [Google Scholar] [CrossRef]
- Rosolina, S.M.; Bragg, S.A.; Ouyang, R.; Chambers, J.Q.; Xue, Z.-L. Highly sensitive detection of hexavalent chromium utilizing a sol-gel/carbon nanotube modified electrode. J. Electroanal. Chem. 2016, 781, 120–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, R.; Bragg, S.A.; Chambers, J.Q.; Xue, Z.-L. Flower-like self-assembly of gold nanoparticles for highly sensitive electrochemical detection of chromium(VI). Anal. Chim. Acta 2012, 722, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Tu, J.; Gan, Y.; Liang, T.; Wan, H.; Wang, P. A miniaturized electrochemical system for high sensitive determination of chromium(VI) by screen-printed carbon electrode with gold nanoparticles modification. Sens. Actuators B Chem. 2018, 272, 582–588. [Google Scholar] [CrossRef]
- Sari, T.K.; Jin, J.; Zein, R.; Munaf, E. Anodic Stripping Voltammetry for the Determination of Trace Cr(VI) with Graphite/Styrene-Acrylonitrile Copolymer Composite Electrodes. Anal. Sci. 2017, 33, 801–806. [Google Scholar] [CrossRef] [Green Version]
- Plohl, O.; Simonič, M.; Kolar, K.; Gyergyek, S.; Fras Zemljič, L. Magnetic nanostructures functionalized with a derived lysine coating applied to simultaneously remove heavy metal pollutants from environmental systems. Sci. Technol. Adv. Mater. 2021, 22, 55–71. [Google Scholar] [CrossRef]
- Hilali, N.; Mohammadi, H.; Amine, A.; Zine, N.; Errachid, A. Recent Advances in Electrochemical Monitoring of Chromium. Sensors 2020, 20, 5153. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, P.; Wen, F.; Yuan, B.; Wang, H. Fe3O4 nanospheres on MoS2 nanoflake: Electrocatalysis and detection of Cr(VI) and nitrite. J. Electroanal. Chem. 2016, 761, 14–20. [Google Scholar] [CrossRef]
- Sanchayanukun, P.; Muncharoen, S. Chitosan coated magnetite nanoparticle as a working electrode for determination of Cr(VI) using square wave adsorptive cathodic stripping voltammetry. Talanta 2020, 217, 121027. [Google Scholar] [CrossRef]
- Fu, L.; Liu, Z.; Ge, J.; Guo, M.; Zhang, H.; Chen, F.; Su, W.; Yu, A. (001) plan manipulation of α-Fe2O3 nanostructures for enhanced electrochemical Cr(VI) sensing. J. Electroanal. Chem. 2019, 841, 142–147. [Google Scholar] [CrossRef]
- Massart, D.L.; Vandeginste, B.G.M.; Buydens, L.M.; Jong, S.D.; Lewi, P.J.; Smeyers-Verbeke, J. Handbook of Chemometrics and Qualimetrics: Part A; Elsevier: Amsterdam, The Netherlands, 1997. [Google Scholar]
- Jorge, E.O.; Rocha, M.M.; Fonseca, I.T.E.; Neto, M.M.M. Studies on the stripping voltammetric determination and speciation of chromium at a rotating-disc bismuth film electrode. Talanta 2010, 81, 556–564. [Google Scholar] [CrossRef] [PubMed]
- Guideline, I.H.T. Validation of Analytical Procedures. Available online: https://onlinelibrary.wiley.com/doi/10.1002/9781118971147.ch5 (accessed on 22 July 2021).
- Van Loco, J.; Elskens, M.; Croux, C.; Beernaert, H. Linearity of calibration curves: Use and misuse of the correlation coefficient. Accredit. Qual. Assur. 2002, 7, 281–285. [Google Scholar] [CrossRef]
- Rojas-Romo, C.; Aliaga, M.E.; Arancibia, V. Determination of molybdenum(VI) via adsorptive stripping voltammetry using an ex‒situ bismuth screen‒printed carbon electrode. Microchem. J. 2020, 154, 104589. [Google Scholar] [CrossRef]
- Pérez-Ràfols, C.; Serrano, N.; Díaz-Cruz, J.M.; Ariño, C.; Esteban, M. New approaches to antimony film screen-printed electrodes using carbon-based nanomaterials substrates. Anal. Chim. Acta 2016, 916, 17–23. [Google Scholar] [CrossRef] [Green Version]
- Gajdár, J.; Barek, J.; Fischer, J. Antimony film electrodes for voltammetric determination of pesticide trifluralin. J. Electroanal. Chem. 2016, 778, 1–6. [Google Scholar] [CrossRef]
- Laboratory and Scientific Section United Nations Office on Drugs and Crime, V. Guidance for the Validation of Analytical Methodology and Calibration of Equipment Used for Testing of Illicit Drugs in Seized Materials and Biological Specimens, A Commitment to Quality nd Continuous Improvement; United Nations: New York, NY, USA, 2009. [Google Scholar]
- Cornell, R.M.; Schwertmann, U. The Iron Oxides: Structure, Reactions, Occurrences and Uses, 2nd ed.; WILEY-VCH: Weinheim, Germany, 2003. [Google Scholar] [CrossRef]
- Quy, D.V.; Hieu, N.M.; Tra, P.T.; Nam, N.H.; Hai, N.H.; Thai Son, N.; Nghia, P.T.; Anh, N.T.V.; Hong, T.T.; Luong, N.H. Synthesis of Silica-Coated Magnetic Nanoparticles and Application in the Detection of Pathogenic Viruses. J. Nanomater. 2013, 2013, 603940. [Google Scholar] [CrossRef] [Green Version]
- Larkin, P. IR and Raman Spectroscopy-Principles and Spectral Interpretation; Elsevier: Oxford, UK, 2011. [Google Scholar] [CrossRef]
- Kitadai, N.; Yokoyama, T.; Nakashima, S. ATR-IR spectroscopic study of L-lysine adsorption on amorphous silica. J. Colloid Interface Sci. 2009, 329, 31–37. [Google Scholar] [CrossRef]
- Saturno, J.; Valera, D.; Carrero, H.; Fernández, L. Electroanalytical detection of Pb, Cd and traces of Cr at micro/nano-structured bismuth film electrodes. Sens. Actuators B Chem. 2011, 159, 92–96. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode Designation | LOD [µg/L], S/N ≥ 3 | LOQ [µg/L], S/N ≥ 10 | Linear Concentration Range [µg/L] | Slope of the Calibration Curve [µA/(µg/L)] | RSD (%) (n = 6) ** | Average Recovery (%) (n = 6) ** |
|---|---|---|---|---|---|---|
| SWV techniques | ||||||
| bare GCE | 39.8 | 79.4 | 79.4–548.2 | –0.0009 | 20.1 | 95.0 (at 200.0 µg/L) |
| ex situ SbFE | 99.0 | 138.1 | 138.1–825.7 | –0.0006 | 12.9 | 113.1 (at LOQ) |
| ex situ CuFE | 99.0 | 138.1 | 138.1–825.7 | –0.0005 | 0.8 | 118.6 (at LOQ) |
| ex situ BiFE | 59.6 | 99.0 | 99.0–909.1 | –0.0007 | 17.7 | 102.1 (at LOQ) |
| GCE/MNPs@SiO2@Lys | 39.8 | 59.6 | 59.6–384.6 | –0.0007 | 15.3 | 103.3 (at LOQ) |
| SWAdSV techniques (see Section 3.4) | ||||||
| BiFE | * | 0.1 | 0.2–0.5 | –37.572 | 7.1 | 65.5 (at 0.5 µg/L) |
| BiFE/MNPs@SiO2@Lys | * | 0.1 | 0.2–2.0 | –23.076 | 9.0 | 98.5 (at 0.5 µg/L) |
| Electrode Used | Linear Concentration Range [µg/L] | Applied for Real Sample Analysis | References |
|---|---|---|---|
| BiFE | 0.3–2.6 | River water | [16] |
| Rotating-disc BiFE | 0.1–0.5 | River water | [56] |
| Micro/nanoparticle BiFE | 0.1–0.5 | / | [67] |
| ex situ BiFE | 0.1–0.5 | / | [67] |
| Bi/SWNTs/GCE | 0–1.3 (reported LOD = 0.036 nM) | River water | [44] |
| BiFE/MNPs@SiO2@Lys | 0.2–2.0 | Tap water | herein |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hojnik, N.; Plohl, O.; Finšgar, M. A Magnetic Nanocomposite Modifier for Improved Ultrasensitive Detection of Hexavalent Chromium in Water Samples. Chemosensors 2021, 9, 189. https://doi.org/10.3390/chemosensors9080189
Hojnik N, Plohl O, Finšgar M. A Magnetic Nanocomposite Modifier for Improved Ultrasensitive Detection of Hexavalent Chromium in Water Samples. Chemosensors. 2021; 9(8):189. https://doi.org/10.3390/chemosensors9080189
Chicago/Turabian StyleHojnik, Nuša, Olivija Plohl, and Matjaž Finšgar. 2021. "A Magnetic Nanocomposite Modifier for Improved Ultrasensitive Detection of Hexavalent Chromium in Water Samples" Chemosensors 9, no. 8: 189. https://doi.org/10.3390/chemosensors9080189
APA StyleHojnik, N., Plohl, O., & Finšgar, M. (2021). A Magnetic Nanocomposite Modifier for Improved Ultrasensitive Detection of Hexavalent Chromium in Water Samples. Chemosensors, 9(8), 189. https://doi.org/10.3390/chemosensors9080189