Raman Spectroscopy as an Assay to Disentangle Zinc Oxide Carbon Nanotube Composites for Optimized Uric Acid Detection



Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Nanocomposite Synthesis
2.3. Characterization
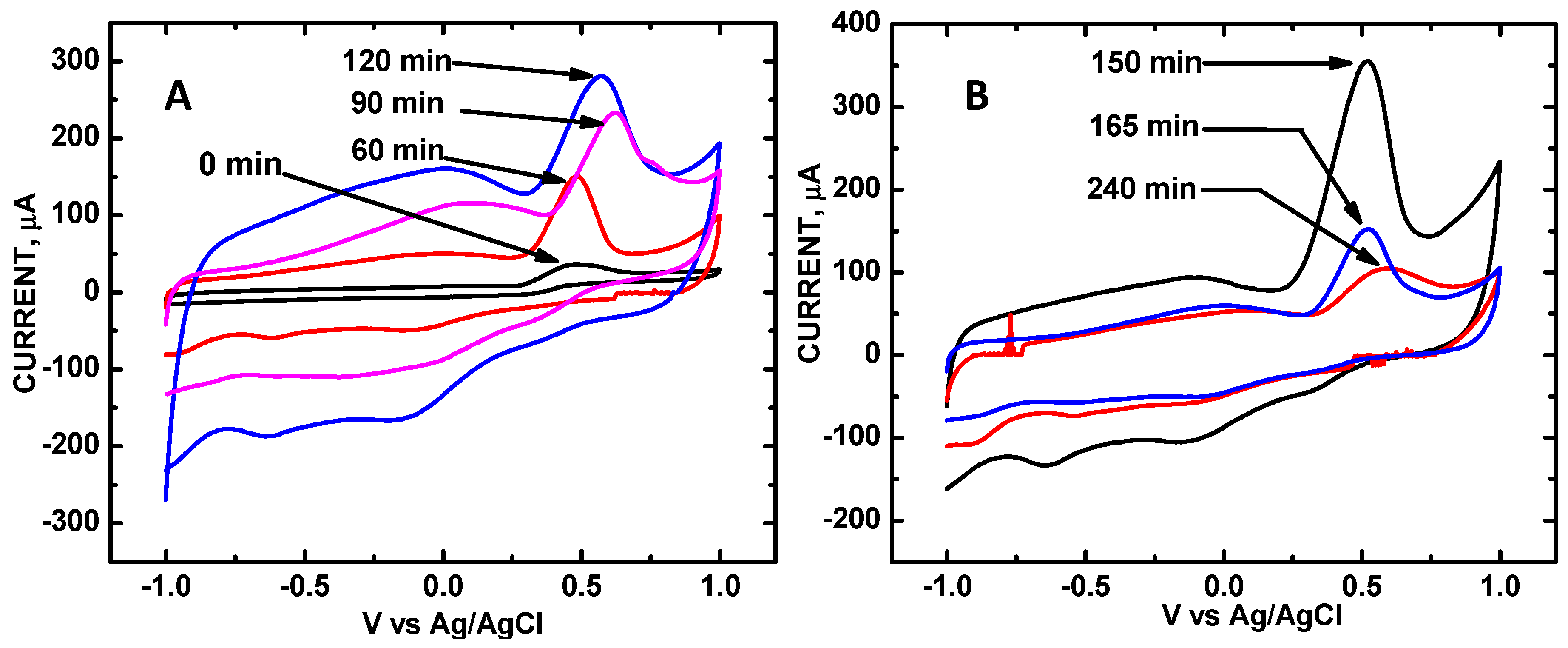
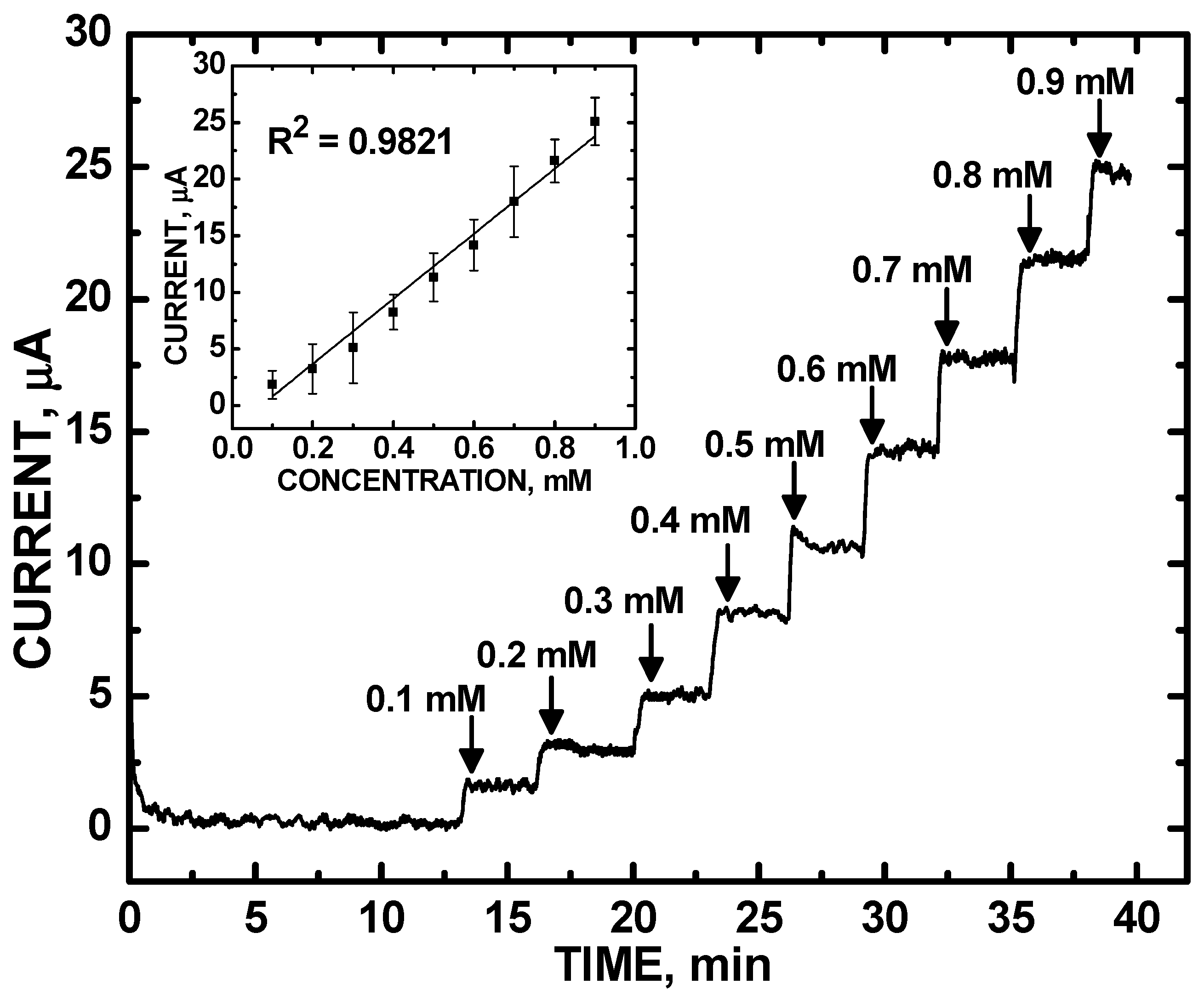
3. Results and Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Perlstein, T.S.; Gumieniak, O.; Williams, G.H.; Sparrow, D.; Vokonas, P.S.; Gaziano, M.; Weiss, S.T.; Litonjua, A.A. Uric acid and the development of hypertension: The normative aging study. Hypertension 2006, 48, 1031–1036. [Google Scholar] [CrossRef] [PubMed]
- Usama, A.A.; El Din, S.; Salem, M.M.; Abdulazim, D.O. Uric acid in the pathogenesis of metabolic, renal, and cardiovascular diseases: A review. J. Adv. Res. 2017, 8, 537–548. [Google Scholar]
- Sakhaee, K. Epidemiology and clinical pathophysiology of uric acid kidney stones. J. Nephrol. 2014, 27, 241–245. [Google Scholar] [CrossRef] [PubMed]
- Kelley, W.N.; Rosenbloom, F.M.; Henderson, J.F.; Seegmiller, J.E. A specific enzyme defect in gout associated with overproduction of uric acid. Proc. Natl. Acad. Sci. USA 1967, 57, 1735–1739. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.-C.; Do, J.-S.; Liu, C.-C. An amperometric uric acid biosensor based on modified Ir-C electrode. Biosens. Bioelectron. 2006, 22, 482–488. [Google Scholar] [CrossRef] [PubMed]
- Perez-Ruiz, F.; Dalbeth, N.; Bardin, T. A review of uric acid, crystal deposition disease, and gout. Adv. Ther. 2015, 32, 31–41. [Google Scholar] [CrossRef] [PubMed]
- El Ridi, R.; Tallima, H. Physiological functions and pathogenic potential of uric acid: A review. J. Adv. Res. 2017, 8, 487–493. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Doherty, M.; Bardin, T.; Pascual, E.; Barskova, V.; Conaghan, P.; Gerster, J.; Jacobs, J.; Leeb, B.; Lioté, F.; et al. EULAR evidence based recommendations for gout. Part II: Management. Report of a task force of the EULAR standing committee for international clinical studies including therapeutics (ESCISIT). Ann. Rheum. Dis. 2006, 65, 1312–1324. [Google Scholar] [CrossRef] [PubMed]
- Khanna, D.; Fitzgerald, J.D.; Khanna, P.P.; Bae, S.; Singh, M.K.; Neogi, T.; Pillinger, M.H.; Merill, J.; Lee, S.; Prakash, S.; et al. 2012 American College of Rheumatology Guidelines for Management of Gout. Part 1: Systematic Nonpharmacologic and Pharmacologic Therapeutic Approaches to Hyperuricemia. Arthritis Care Res. 2012, 64, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Stack, A.G.; Hanley, A.; Casserly, L.F.; Cronin, C.J.; Abdalla, A.A.; Kernan, T.J.; Murthy, B.V.R.; Hegarty, A.; Hannigan, A.; Nguyen, H.T. Independent and conjoint associations of gout and hyperuricemia with total and cardiovascular mortality. J. Med. 2013, 106, 647–658. [Google Scholar]
- Choi, H.K.; Curhan, G. Independent impact of gout on mortality and risk for coronary heart disease. Circulation 2007, 107, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, E.; Svendsen, K.; Neaton, J.D. Long-term cardiovascular mortality among middle-aged men with gout. Arch. Intern. Med. 2008, 168, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.M.; Henderson, G.N.; Ouyang, X.; Frye, R.F.; Sautin, Y.Y.; Feig, D.I.; Johnson, R.J. A sensitive and specific liquid chromatography-tandem mass spectrometry method for the determination of intracellular and extracellular uric acid. J. Chromatogr. B 2009, 877, 2032–2038. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. Carbon-nanotube based electrochemical biosensors: A review. Electroanalysis 2005, 17, 7–14. [Google Scholar] [CrossRef]
- Erden, P.E.; Kiliç, E. A review of enzymatic uric acid biosensors based on amperometric detection. Talanta 2013, 107, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Wayu, M.B.; Spidle, R.T.; Devkota, T.; Deb, A.K.; Delong, R.K.; Ghosh, K.C.; Wanekaya, A.K.; Chusuei, C.C. Morphology of hydrothermally synthesized ZnO nanoparticles tethered to carbon nanotubes affects electrocatalytic activity for H2O2 detection. Electrochim. Acta 2013, 97, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Conway, G.E.; Lamberston, R.H.; Schwarzmann, M.A.; Pannell, M.J.; Kerins, H.W.; Rubenstien, K.J.; Dattelbaum, J.D.; Leopold, M.C. Layer-by-Layer desgin and optimization of xerogel-based amperometric first generation biosensors of uric acid. J. Electroanal. Chem. 2016, 775, 135–145. [Google Scholar] [CrossRef]
- Wayu, M.B.; DiPasquale, L.T.; Schwarzmann, M.A.; Gillspie, S.D.; Leopold, M.C. Electropolymerization of ß-cyclodextrin onto multi-walled carbon nanotube composite films for enhanced selective detection of uric acid. J. Electroanal. Chem. 2016, 783, 192–200. [Google Scholar] [CrossRef]
- Wayu, M.B.; Pannell, M.J.; Leopold, M.C. Layered xerogel films incorporating monolayer-protected cluster networks on platinum-black-modified electrodes for enhanced sensitivity in first-generation uric acid biosensing. ChemElectroChem 2016, 3, 1245–1252. [Google Scholar] [CrossRef]
- Wayu, M.B.; Schwarzmann, M.A.; Gillespie, S.D.; Leopold, M.C. Enzyme-free uric acid electrochemical sensors using ß-cyclodextrin-modified carboxylic acid-functionalized carbon nanotubes. J. Mater. Sci. 2017, 52, 6050–6062. [Google Scholar] [CrossRef]
- Donniah, S.K.; Periakarruppan, P.; Vellaichamy, B. New electrochemical sensor based on a silver-doped iron oxide nanocomposite coupled with polyaniline and its sensing application for picomolar-level detection of uric acid in human blood and urine samples. J. Phys. Chem. B 2018, 122, 3037–3046. [Google Scholar]
- Wu, C.; Qiao, X.; Chen, J.; Wang, H.; Tan, F.; Li, S. A novel chemical route to prepare ZnO nanoparticles. Mater. Lett. 2006, 60, 1828–1832. [Google Scholar] [CrossRef]
- Bai, H.P.; Lu, X.X.; Yang, Y.H. Hydrogen peroxide biosensor based on electrodeposition of zinc oxide nanoflowers onto carbon nanotubes film electrode. Chin. Chem. Lett. 2008, 19, 314–318. [Google Scholar] [CrossRef]
- Baig, N.; Rana, A.; Kawde, A.-N. Modified electrodes for selective detection of biomolecules. Electroanalysis 2018, 30, 2551–2574. [Google Scholar] [CrossRef]
- Xing, Y. Synthesis and electrochemical characterization of uniformly-dispersed high loading Pt nanoparticles on sonochemically-treated carbon nanotubes. J. Phys. Chem. B 2004, 108, 19255–19259. [Google Scholar] [CrossRef]
- Xing, Y.; Li, L.; Chusuei, C.C.; Hull, R.V. Sonochemical oxidation of multiwalled carbon nanotubes. Langmuir 2005, 21, 4185–4190. [Google Scholar] [CrossRef] [PubMed]
- Hull, R.V.; Li, L.; Xing, Y.; Chusuei, C.C. Pt nanoparticle binding on functionalized multiwalled carbon nanotubes. Chem. Mater. 2006, 18, 1780–1788. [Google Scholar] [CrossRef]
- Deb, A.K.; Das, S.C.; Saha, A.; Wayu, M.B.; Marksberry, M.H.; Baltz, R.J.; Chusuei, C.C. Ascorbic acid, acetaminophen, and hydrogen peroxide detection using a dendrimer-encapsulated Pt nanoparticle carbon nanotube composite. J. Appl. Electrochem. 2016, 46, 289–298. [Google Scholar] [CrossRef]
- Fang, B.; Zhang, C.; Zhang, G. A novel hydrazine electrochemical sensor based on a carbon nanotube-wired ZnO nanoflower-modified electrode. Electrochim. Acta 2009, 55, 178–182. [Google Scholar] [CrossRef]
- Park, J.; Regalbuto, J.R. A Simple, Accurate Determination of Oxide PZC and the Strong Buffering Effect of Oxide Surfaces at Incipient Wetness. J. Colloid Interface Sci. 1995, 175, 239–252. [Google Scholar] [CrossRef]
- McPhail, M.R.; Sells, J.A.; He, Z.; Chusuei, C.C. Charging Nanowalls: Adjusting the Carbon Nanotube Isoelectric Point via Surface Chemical Functionalization. J. Phys. Chem. C 2009, 113, 14102–14109. [Google Scholar] [CrossRef]
- Chusuei, C.C.; Wayu, M. Characterizing functionalized carbon nanotubes for improved fabrication in aqueous solution environments. In Electronic Properties of Carbon Nanotubes/Book 5; Marulanda, J.M., Ed.; InTech: Rijeka, Croatia, 2011; pp. 55–66. [Google Scholar]
- Deb, A.K.; Chusuei, C.C. Aqueous solution surface chemistry of carbon nanotubes. In Physical and Chemical Properties of Carbon Nanotubes; Suzuki, S., Ed.; InTech: Rijeka, Croatia, 2013; pp. 263–283. [Google Scholar]
- Chusuei, C.C.; Wu, C.-H.; Mallavarapu, S.; Stephen Hou, F.Y.; Hsu, C.-M.; Winiarz, J.G.; Aronstam, R.S.; Huang, Y.-W. Cytotoxicity in the age of nano: The role of fourth period transition metal oxide nanoparticle physicochemical properties. Chem.-Biol. Interact. 2013, 206, 319–326. [Google Scholar] [CrossRef] [PubMed]
- Chusuei, C.C.; Wu, C.-H.; Mallavarapu, S.; Stephen Hou, F.Y.; Hsu, C.-M.; Aronstam, R.S.; Huang, Y.-W. Physicochemical structure effects on metal oxide nanoparticulate cytotoxicity. In Recent Progress in Surface and Colloid Chemistry with Biological Applications; Wang, C., Hauserman, B., Eds.; American Chemical Society: Washington, DC, USA, 2015; Volume 1215, pp. 137–155. [Google Scholar]
- Weiler, M.; Sattel, S.; Jung, K.; Ehrhardt, H.; Veerasamy, V.; Robertson, J. Highly tetrahedral, diamond-like amorphous hydrogenated carbon prepared from a plasma beam source. Appl. Phys. Lett. 1994, 64, 2797–2799. [Google Scholar] [CrossRef]
- McCafferty, E.; Wightman, J.P. Determination of the concentration of surface hydroxyl groups on metal oxide films by a quantitative XPS method. Surf. Interface Anal. 1998, 26, 549–564. [Google Scholar] [CrossRef]
- Haber, J.; Stoch, J.; Ungier, L. X-ray photoelectron spectra of oxygen in oxides of cobalt, nickel, iron, and zinc. J. Electron Spectrosc. Relat. Phenom. 1976, 9, 459–467. [Google Scholar] [CrossRef]
- Kanik, F.E.; Kolb, M.; Timur, S.; Bahadir, M.; Toppare, L. An amperometric acetylcholine biosensor based on a conducting polymer. Int. J. Biol. Macromol. 2013, 59, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Langer, D.W.; Vesely, C.J. Electronic core levels of zinc chalcogenides. Phys. Rev. B 1970, 2, 4885–4892. [Google Scholar] [CrossRef]
- Strohmeier, B.R.; Hercules, D.M. Surface spectroscopic characterization of the interaction between zinc ions and γ-alumina. J. Catal. 1984, 86, 266–279. [Google Scholar] [CrossRef]
- Wayu, M.B.; King, J.E.; Johnson, J.A.; Chusuei, C.C. A zinc oxide carbon nanotubed based sensor for in situ monitoring of hydrogen peroxide in swimming pools. Electroanalysis 2015, 27, 2552–2558. [Google Scholar] [CrossRef]
- Zanello, P. Inorganic Electrochemistry: Theory, Practice and Application; Royal Society of Chemistry: London, UK, 2003. [Google Scholar]
- Dresselhaus, M.S.; Dresselhaus, G.; Avouris, P. Carbon Nanotubes: Synthesis, Structure, Properties and Applications; Springer: New York, NY, USA, 2001. [Google Scholar]
- Lehman, J.H.; Terrones, M.; Mansfield, E.; Hurst, K.E.; Meunir, V. Evaluating the characteristics of multiwall carbon nanotubes. Carbon 2011, 49, 2581–2602. [Google Scholar] [CrossRef]
- Kim, J.H.; Hwang, J.Y.; Hwang, H.R.; Kim, H.S.; Lee, J.H.; Seo, J.W.; Shin, U.S.; Lee, S.H. Simple and cost-effective method of highly conductive and elastic carbon nanotube/polydimethylsiloxane composite for wearable electronics. Sci. Rep. 2018, 8, 1375. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, H.; Togashi, R.; Hakoda, M.; Terai, C.; Kashiwazaki, S.; Dan, T.; Kamatani, N. Optimum range of serum urate concentrations to minimize risk of gouty attacks during anti-hyperuricemic treatment. Adv. Exp. Med. Biol. 1998, 431, 13–18. [Google Scholar] [PubMed]
- Kuo, C.C.; Weaver, V.; Fadrowski, J.J.; Lin, Y.S.; Guallar, E.; Navas-Acien, A. Arsenic exposure, hyperuricemia, and gout in US adults. Environ. Int. 2015, 76, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Sposito, G. On points of zero charge. Environ. Sci. Technol. 1998, 32, 2815–2819. [Google Scholar] [CrossRef]
- Lide, D.R. (Ed.) CRC Handbook of Chemistry and Physics, 71st ed; CRC Press: Boca Raton, FL, USA, 1990. [Google Scholar]
- Szakács, Z.; Noszál, B. Determination of dissociation constants of folic acid, methotrexate, and other photolabile pterdines by pressure-assisted capillary electrophoresis. Electrophoresis 2006, 27, 3399–3409. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Lang, Q.; Zeng, L.; Li, T.; Wei, M.; Liu, A. Substituent effect on the oxidation peak potentials of phenol derivatives at ordered mesoporous carbons modified electrode and its application in determination of acidity coefficients (pKa). Electrochim. Acta 2014, 115, 283–289. [Google Scholar] [CrossRef]
- Perrin, D.D. Ionization Constants of Inorganic Acids and Bases in Aqueous Solution, 2nd ed.; Pergamon: Oxford, UK, 1982. [Google Scholar]
- Ngai, K.S.; Tan, W.T.; Zainal, Z.; Zawawi, R.M.; Zidan, M. Voltammetry detection of ascorbic acid at glassy carbon electrode modified by single-walled carbon nanotube/zinc oxide. Int. J. Electrochem. Sci. 2013, 8, 10557–10567. [Google Scholar]
- Raoof, J.B.; Teymoori, N.; Khalilzadeh, M.A.; Ojani, R. A high sensitive electrochemical nanosensor for simultaneous determination of glutathione, NADH and folic acid. Mater. Sci. Eng. C 2015, 47, 77–84. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sonication Time, min | 0 | 60 | 90 | 120 | 150 | 165 | 180 | 240 |
|---|---|---|---|---|---|---|---|---|
| V | 0.500 | 0.499 | 0.623 | 0.571 | 0.524 | 0.520 | 0.534 | 0.524 |
| μA relative area | 36 0 | 149 1.00 | 233 1.56 | 281 1.87 | 355 2.38 | 153 1.07 | 133 0.892 | 106 0.698 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, S.C.; Pandey, R.R.; Devkota, T.; Chusuei, C.C. Raman Spectroscopy as an Assay to Disentangle Zinc Oxide Carbon Nanotube Composites for Optimized Uric Acid Detection. Chemosensors 2018, 6, 65. https://doi.org/10.3390/chemosensors6040065
Das SC, Pandey RR, Devkota T, Chusuei CC. Raman Spectroscopy as an Assay to Disentangle Zinc Oxide Carbon Nanotube Composites for Optimized Uric Acid Detection. Chemosensors. 2018; 6(4):65. https://doi.org/10.3390/chemosensors6040065
Chicago/Turabian StyleDas, Shawtik C., Raja R. Pandey, Tuphan Devkota, and Charles C. Chusuei. 2018. "Raman Spectroscopy as an Assay to Disentangle Zinc Oxide Carbon Nanotube Composites for Optimized Uric Acid Detection" Chemosensors 6, no. 4: 65. https://doi.org/10.3390/chemosensors6040065
APA StyleDas, S. C., Pandey, R. R., Devkota, T., & Chusuei, C. C. (2018). Raman Spectroscopy as an Assay to Disentangle Zinc Oxide Carbon Nanotube Composites for Optimized Uric Acid Detection. Chemosensors, 6(4), 65. https://doi.org/10.3390/chemosensors6040065