Microextraction of Polycyclic Musks from Surface Water with Deep-Eutectic-Solvent-Coated Membrane Followed by Gas-Chromatography–Mass Spectrometry Analysis

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Chemicals and Standard Solutions
2.2. Preparation of DES
2.3. Characterization of the Coated Membranes
2.4. Microextraction Procedure
2.5. Gas Chromatography–Mass Spectrometry Analysis
2.6. Statistical Analyses
3. Results and Discussion
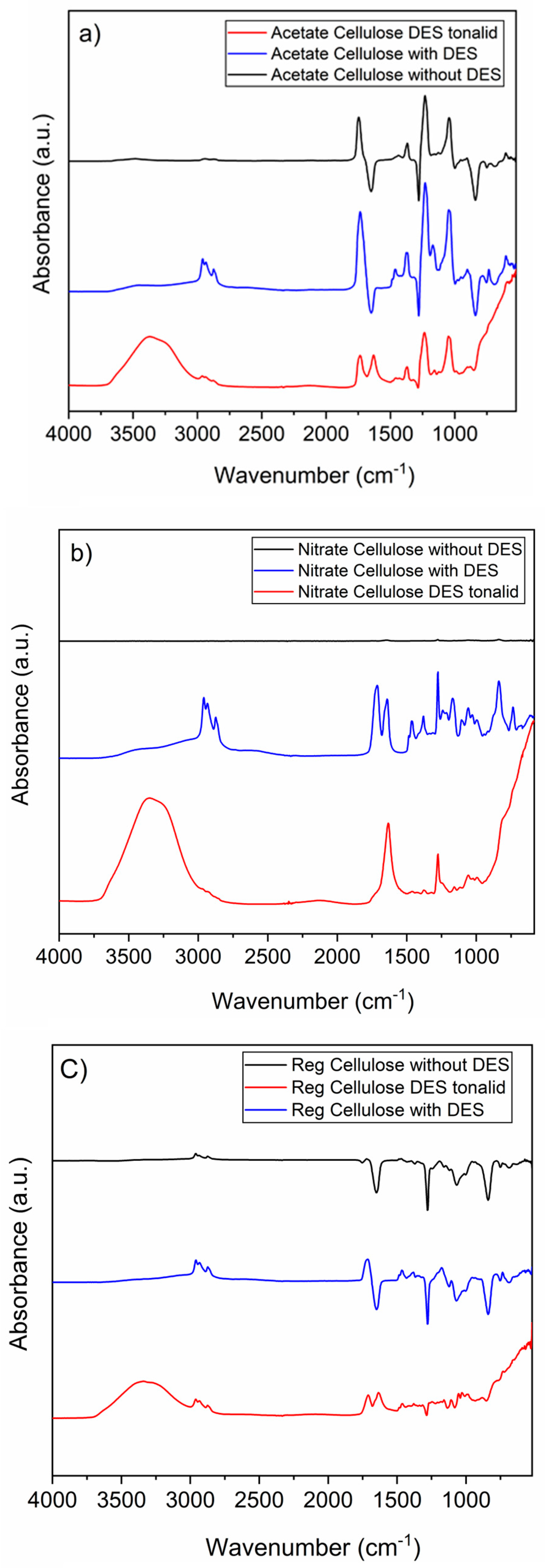
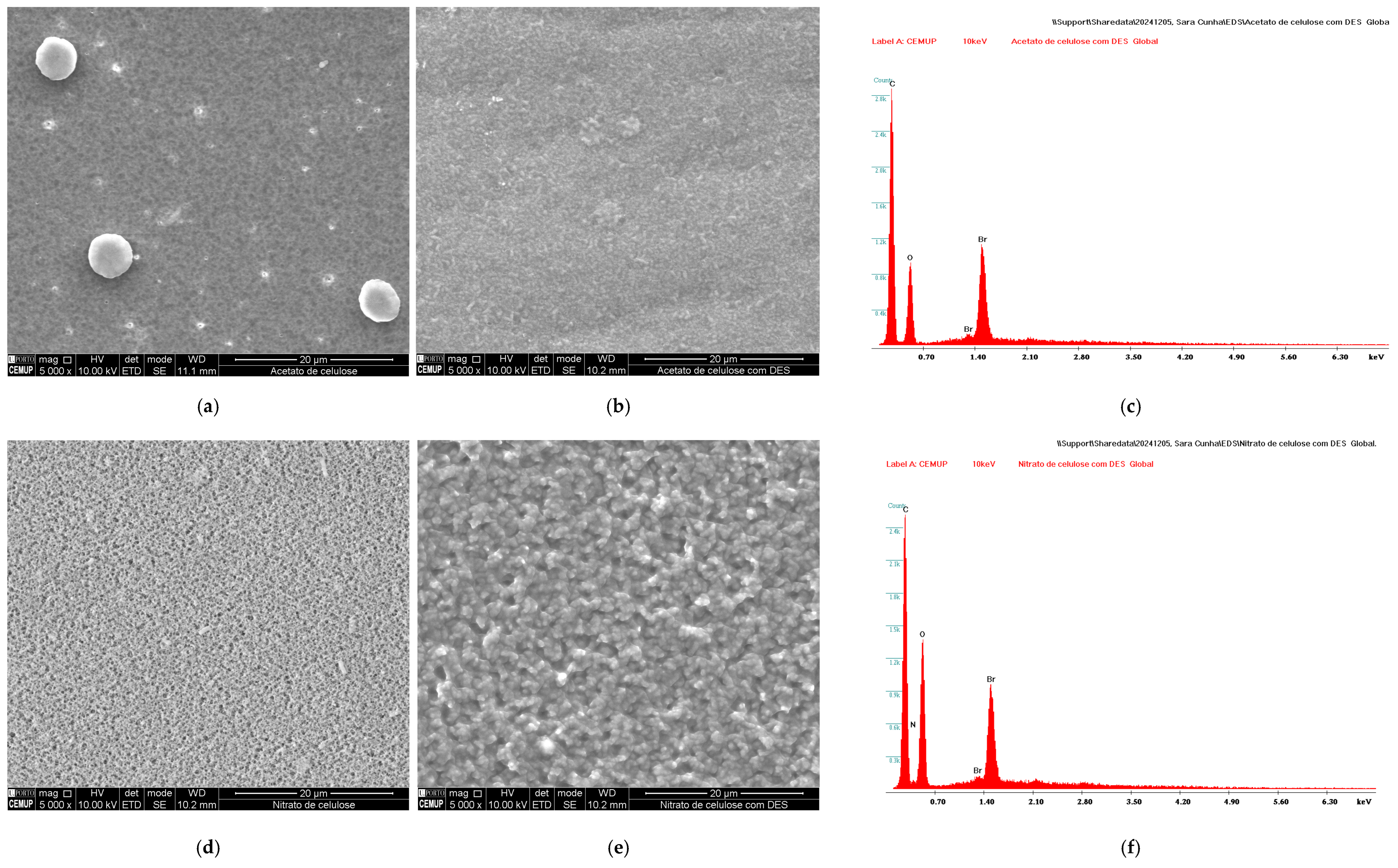
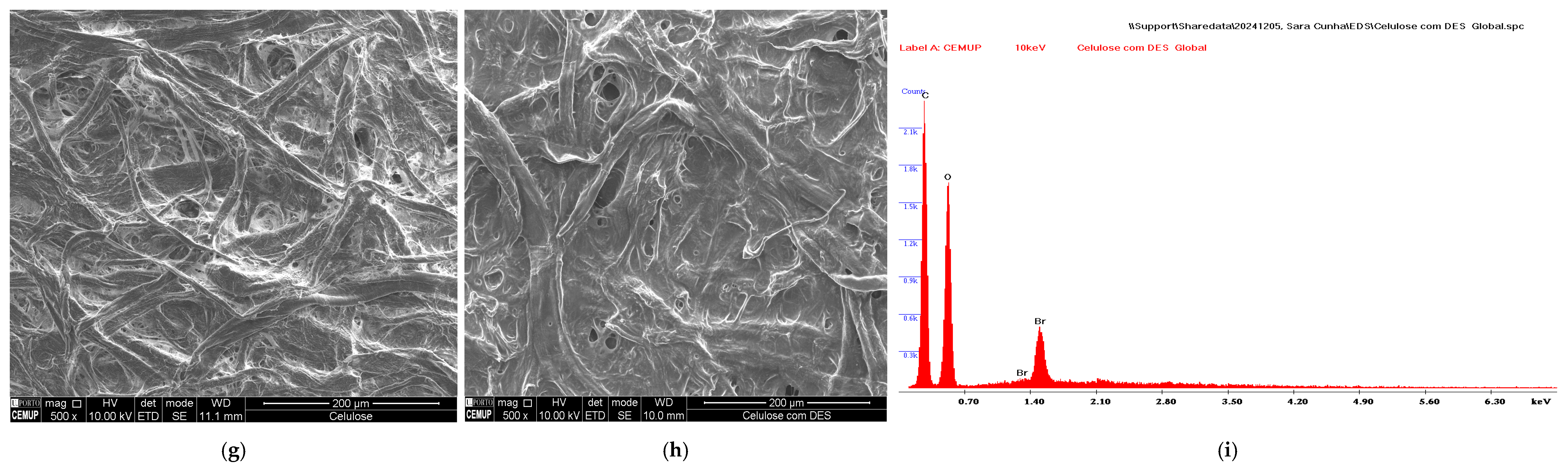
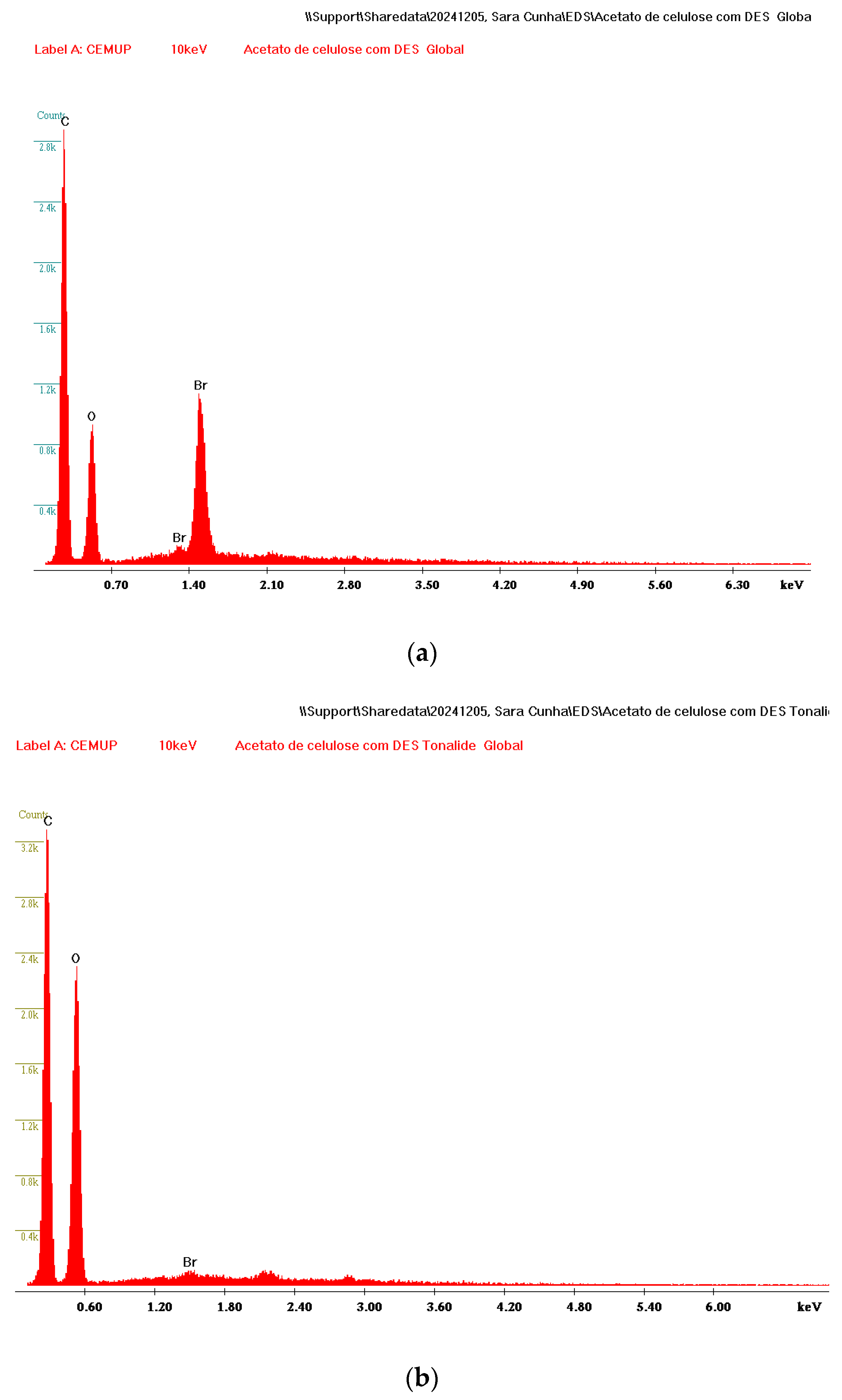
3.1. Characterization of DES-Coated Membrane
3.2. Optimization of DES-Membrane Extraction
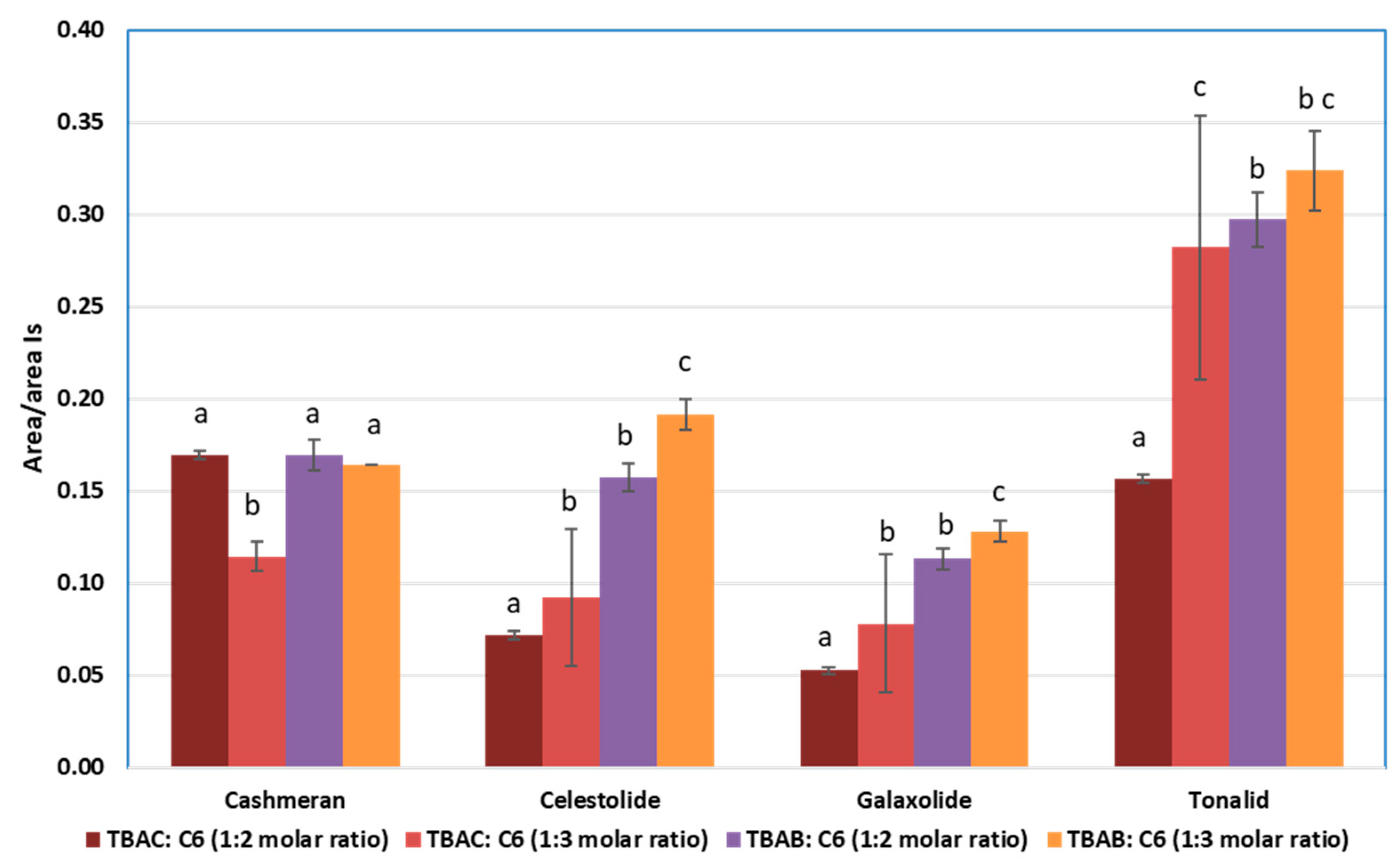
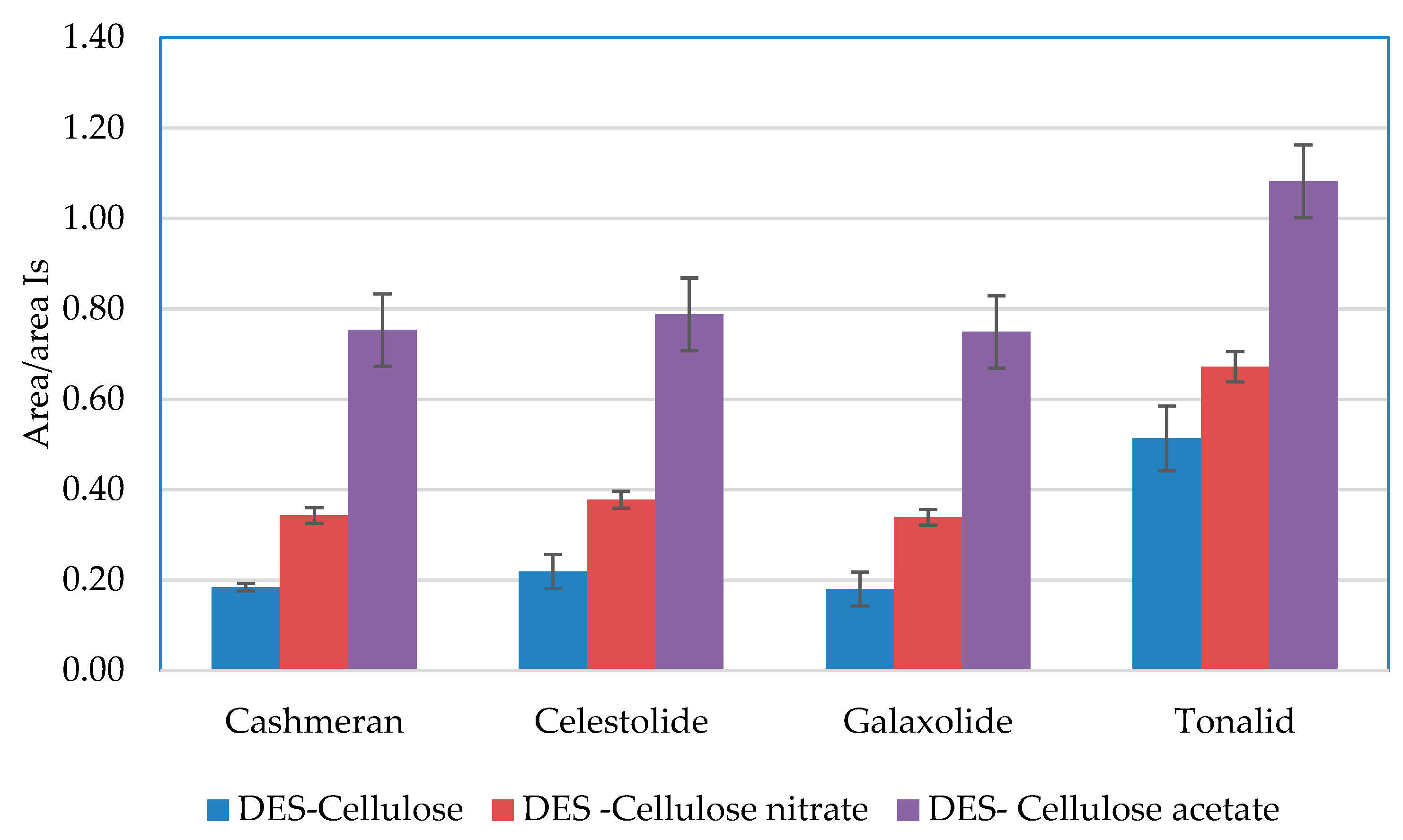
3.2.1. Selection of the DES
3.2.2. Selection of the Membranes
3.2.3. Selection of Dissolving Solvent
3.2.4. Multivariate Approach to Optimize the Volumes of DES and Dissolving Solvent
3.3. In-House Validation
3.4. Real Samples
3.5. Evaluation of the Analytical Method Through Metric Tools
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cunha, S.C.; Menezes-Sousa, D.; Mello, F.M.; Miranda, J.A.T.; Fogaca, F.; Alonso, M.B.; Torres, J.P.M.; Fernandes, J.O. Survey on endocrine-disrupting chemicals in seafood: Occurrence and distribution. Environ. Res. 2022, 210, 112886. [Google Scholar] [CrossRef]
- Aminot, Y.; Munschy, C.; Héas-Moisan, K.; Pollono, C.; Tixier, C. Levels and trends of synthetic musks in marine bivalves from French coastal areas. Chemosphere 2021, 268, 129312. [Google Scholar] [CrossRef] [PubMed]
- Košnář, Z.; Mercl, F.; Chane, A.D.; Pierdonà, L.; Míchal, P.; Tlustoš, P. Occurrence of synthetic polycyclic and nitro musk compounds in sewage sludge from municipal wastewater treatment plants. Sci. Total Environ. 2021, 801, 149777. [Google Scholar] [CrossRef]
- Tasselli, S.; Valenti, E.; Guzzella, L. Polycyclic musk fragrance (PMF) removal, adsorption and biodegradation in a conventional activated sludge wastewater treatment plant in Northern Italy. Environ. Sci. Pollut. Res. 2021, 28, 38054–38064. [Google Scholar] [CrossRef]
- Ren, S.; Tan, F.; Wang, Y.; Zhao, H.; Zhang, Y.; Zhai, M.; Chen, J.; Wang, X. In situ measurement of synthetic musks in wastewaters using diffusive gradients in thin film technique. Water Res. 2020, 185, 116239. [Google Scholar] [CrossRef]
- Juksu, K.; Liu, Y.; Zhao, J.; Yao, L.; Sarin, C.; Sreesai, S.; Klomjek, P.; Traitangwong, A.; Ying, G. Emerging contaminants in aquatic environments and coastal waters affected by urban wastewater discharge in Thailand: An ecological risk perspective. Ecotox. Environ. Safe. 2020, 204, 110952. [Google Scholar] [CrossRef]
- Wang, T.; Zou, H.; Li, D.; Gao, J.; Bu, Q.; Wang, Z. Global distribution and ecological risk assessment of synthetic musks in the environment. Environ. Pollut. 2023, 331, 121893. [Google Scholar] [CrossRef] [PubMed]
- Lee, I.S.; Lee, S.H.; Oh, J.E. Occurrence and fate of synthetic musk compounds in water environment. Water Res. 2010, 44, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Ding, T.; Li, W.; Cai, M.; Jia, X.; Yang, M.; Yang, B.; Li, J. Algal toxicity, accumulation and metabolic pathways of galaxolide. J. Hazard. Mater. 2020, 384, 121360. [Google Scholar] [CrossRef]
- Parolini, M.; Magni, S.; Traversi, I.; Villa, S.; Finizio, A.; Binelli, A. Environmentally relevant concentrations of galaxolide (HHCB) and tonalide (AHTN) induced oxidative and genetic damage in Dreissena polymorpha. J. Hazard. Mater. 2015, 285, 1–10. [Google Scholar] [CrossRef]
- Pedersen, S.; Selck, H.; Salvito, D.; Forbes, V. Effects of the polycyclic musk HHCB on individual-and population-level endpoints in Potamopyrgus antipodarum. Ecotoxicol. Environ. Saf. 2009, 72, 1190–1199. [Google Scholar] [CrossRef]
- Li, Z.; Yin, N.; Liu, Q.; Wang, C.; Wang, T.; Wang, Y.; Qu, G.; Liu, J.; Cai, Y.; Zhou, Q.; et al. Effects of polycyclic musks HHCB and AHTN on steroidogenesis in H295R cells. Chemosphere 2013, 90, 1227–1235. [Google Scholar] [CrossRef] [PubMed]
- Vallecillos, L.; Borrull, F.; Pocurull, E. Recent approaches for the determination of synthetic musk fragrances in environmental samples. TrAC Trends Anal. Chem. 2015, 72, 80–92. [Google Scholar] [CrossRef]
- Cunha, S.C.; Lapa, R.; Fernandes, J.O. Deep eutectic solvents for microextraction of endocrine (Cunha et al., 2022)disruptor contaminants from water and food: Review. TrAC Trends Anal. Chem. 2024, 171, 117495. [Google Scholar] [CrossRef]
- Abbott, A.P.; Capper, G.; Davies, D.L.; Rasheed, R.K.; Tambyrajah, V. Novel solvent properties of choline chloride/urea mixtures. Chem. Commun. 2003, 1, 70–71. [Google Scholar] [CrossRef]
- Li, T.; Song, Y.; Xu, J.; Fan, J. A hydrophobic deep eutectic solvent mediated sol-gel coating of solid phase microextraction fiber for determination of toluene, ethylbenzene and o-xylene in water coupled with GC-FID. Talanta 2019, 195, 298–305. [Google Scholar] [CrossRef]
- Werner, J.; Zgoła-Grześkowiak, A.; Grześkowiak, T. Development of novel thin-film solid-phase microextraction materials based on deep eutectic solvents for preconcentration of trace amounts of parabens in surface waters. J. Sep. Sci. 2022, 45, 1374–1384. [Google Scholar] [CrossRef]
- Quintanilla, I.; Fontàs, C.; Anticó, E. Deep eutectic solvents incorporated in a polymeric film for organophosphorus pesticide microextraction from water samples. Anal. Chim. Acta 2024, 1318, 342940. [Google Scholar] [CrossRef]
- Ríos-Gómez, J.; García-Valverde, M.T.; López-Lorente, Á.I.; Toledo-Neira, C.; Lucena, R. Soledad Cárdenas Polymeric ionic liquid immobilized onto paper as sorptive phase in microextraction. Anal. Chim. Acta 2020, 1094, 47–56. [Google Scholar] [CrossRef]
- Mansour, F.R.; Omer, K.M.; Płotka-Wasylka, J. A total scoring system and software for complex modified GAPI (ComplexMoGAPI) application in the assessment of method greenness. Green Anal. Chem. 2024, 10, 100126. [Google Scholar] [CrossRef]
- Pena-Pereira, F.; Wojnowski, W.; Tobiszewski, M. AGREE—Analytical greenness metric approach and software. Anal. Chem. 2020, 92, 10076–10082. [Google Scholar] [CrossRef] [PubMed]
- Manousi, N.; Wojnowski, W.; Plotka-Wasylka, J.; Samanidou, V. Blue applicability grade index (BAGI) and software: A new tool for the evaluation of method practicability. Green Chem. 2023, 25, 7598–7604. [Google Scholar] [CrossRef]
- Shishov, A.; Makoś-Chełstowska, P.; Bulatov, A.; Andruch, V. Deep Eutectic Solvents or Eutectic Mixtures? Characterization of Tetrabutylammonium Bromide and Nonanoic Acid Mixtures. J. Phys. Chem. B 2022, 126, 3889–3896. [Google Scholar] [CrossRef]
- Rodrigues, M.I.; Iemma, A.F. Experimental Design and Process Optimization, 1st ed.; CRC Press: Boca Raton, FL, USA, 2014. [Google Scholar]
- European Commission. Analytical Quality Control and Method Validation Procedures for Pesticide Residues Analysis in Food and Feed. SANTE/11312/2021. 2022. Available online: https://www.eurl-pesticides.eu/docs/public/tmplt_article.asp?CntID=727 (accessed on 1 December 2024).
- Shafie, M.H.; Yusof, R.; Gan, C.Y. Synthesis of citric acid monohydrate-choline chloride based deep eutectic solvents (DES) and characterization of their physicochemical properties. J. Mol. Liq. 2019, 288, 111081. [Google Scholar] [CrossRef]
- Tang, N.; Liu, L.; Yin, C.; Zhu, G.; Huang, Q.; Dong, J.; Yang, X.; Wang, S. Environmentally benign hydrophobic deep eutectic solvents for palladium (II) extraction from hydrochloric acid solution. J. Taiwan Inst. Chem. Eng. 2021, 121, 92–100. [Google Scholar] [CrossRef]
- Hansen, B.B.; Spittle, S.; Chen, B.; Poe, D.; Zhang, Y.; Klein, M.J.; Horton, A.; Adhikari, L.; Zelovich, T.; Doherty, W.B.; et al. Deep Eutectic Solvents: A Review of Fundamentals and Applications. Am. Chem. Soc. 2021, 121, 1232–1285. [Google Scholar] [CrossRef]
- Alhadid, A.; Mokrushina, L.; Minceva, M. Formation of glassy phases and polymorphism in deep eutectic solvents. J. Mol. Liq. 2020, 314, 113667. [Google Scholar] [CrossRef]
- Drebushchak, V.A. Thermophysical theory of DSC melting peak. J. Therm. Anal. Calorim. 2012, 545–553. [Google Scholar] [CrossRef]
- Menczel, J.D.; Kohl, W.S. Differential scanning calorimetry (DSC) in fiber research. In The Textile Institute Book Series, Thermal Analysis of Textiles and Fibers; Jaffe, M., Menczel, J.D., Eds.; Woodhead Publishing: Cambridge, UK, 2020; pp. 17–69. [Google Scholar] [CrossRef]
- Fatahi, H.; Claverie, J.; Poncet, S.; Fatahi, H.; Claverie, J.; Poncet, S. Thermal Characterization of Phase Change Materials by Differential Scanning Calorimetry: A Review. Appl. Sci. 2022, 12, 12019. [Google Scholar] [CrossRef]
- Schawe, J.E.K.; Pogatscher, S.; Löffler, J.F. Thermodynamics of polymorphism in a bulk metallic glass: Heat capacity measurements by fast differential scanning calorimetry. Thermochim. Acta 2020, 685, 178518. [Google Scholar] [CrossRef]
- Morrison, H.G.; Sun, C.C.; Neervannan, S. Characterization of thermal behavior of deep eutectic solvents and their potential as drug solubilization vehicles. Int. J. Pharm. 2009, 378, 136–139. [Google Scholar] [CrossRef] [PubMed]
- Pavia, D.L.; Lampman, G.M.; Kriz, G.S.; Vyvyan, J.R. Introdução à Espectroscopia, 4th ed.; Cengage Learning: Singapore, 2010. [Google Scholar]
- Delgado-Mellado, N.; Larriba, M.; Navarro, P.; Rigual, V.; Ayuso, M.; García, J.; Rodríguez, F. Thermal stability of choline chloride deep eutectic solvents by TGA/FTIR-ATR analysis. J. Mol. Liq. 2018, 260, 37–43. [Google Scholar] [CrossRef]
- Ribeiro, B.D.; Florindo, C.; Iff, L.C.; Coelho, M.A.; Marrucho, I.M. Menthol-based Eutectic Mixtures: Hydrophobic Low Viscosity Solvents. ACS Sustain. Chem. Eng. 2015, 3, 2469–2477. [Google Scholar] [CrossRef]
- Lange, C.; Kuch, B.; Metzger, J.W. Occurrence and fate of synthetic musk fragrances in a small German river. J. Hazard. Mater. 2015, 282, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Siyan, L.; Fang, Z.; Ruifen, J.; Gangfeng, O. Preparation and evaluation of amino modified graphene solid-phase microextraction fiber and its application to the determination of synthetic musks in water samples. J. Chromat. A 2016, 1429, 1–7. [Google Scholar]
- Vimalkumar, K.; Nikhil, N.P.; Arun, E.; Babu-Rajendran, M.R. Synthetic musks in surface water and fish from the rivers in India: Seasonal distribution and toxicological risk assessment. J. Hazard. Mater. 2021, 414, 125558. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trial | X1 (DES Volume, µL) | X2 (DES Time, Min) | X3 (Extraction Time, Min) | X4 (Dissolving Solvent Volume, µL) | Recovery (%) |
|---|---|---|---|---|---|
| 1 | +1 (100) | −1 (15) | −1 (5) | +1 (1000) | 16.8 |
| 2 | +1 (100) | +1 (60) | −1 (5) | −1 (250) | 117.7 |
| 3 | +1 (100) | +1 (60) | +1 (60) | −1 (250) | 92.9 |
| 4 | −1 (25) | +1 (60) | +1 (60) | +1 (1000) | 38.2 |
| 5 | +1 (100) | −1 (15) | +1 (60) | +1 (1000) | 19.6 |
| 6 | −1 (25) | +1 (60) | −1 (5) | +1 (1000) | 14.5 |
| 7 | −1 (25) | −1 (15) | +1 (60) | −1 (250) | 49.7 |
| 8 | −1 (25) | −1 (15) | −1 (5) | −1 (250) | 49.9 |
| CP 1 | 0 (62.5) | 0 (37.5) | 0 (32.5) | 0 (625) | 17.1 |
| CP 2 | 0 (62.5) | 0 (37.5) | 0 (32.5) | 0 (625) | 11.6 |
| CP 3 | 0 (62.5) | 0 (37.5) | 0 (32.5) | 0 (625) | 22.5 |
| Variable (Range) | Effect (%) | Standard Error | t (5) | p-Value |
|---|---|---|---|---|
| DES volume (25–100 µL) | 23.64 | 8.05 | 2.94 | 0.0323 * |
| DES time (15–60 min) | 31.81 | 8.05 | 3.95 | 0.0108 * |
| Extraction time (5–60 min) | 0.37 | 8.05 | 0.05 | 0.9649 |
| Dissolving solvent volume (250–1000 µL) | −55.28 | 8.05 | −6.87 | 0.0009 * |
| Analytes | Linear Range a (µg/L) | Slope ± SD (n = 3) | Intercept ± SD (n = 3) | R2 b | LOD c (µg/L) | LOQ d (µg/L) |
|---|---|---|---|---|---|---|
| Cashmeran | 2.5–100 | 0.1214 ± 0.0014 | 0.6486 ± 0.07166 | 0.9997 | 0.15 | 0.5 |
| Celestolide | 2.5–100 | 0.2004 ± 0.0053 | 1.07332 ± 0.2778 | 0.9982 | 0.06 | 0.2 |
| Galaxolide | 2.5–100 | 0.1020 ± 0.0034 | 0.6025 ± 0.1772 | 0.9972 | 0.15 | 0.5 |
| Tonalid | 2.5–100 | 0.1511 ± 0.0056 | 0.7375 ± 0.2905 | 0.9966 | 0.06 | 0.2 |
| Analytes | 5 µg/L % Recovery (RSD) | 25 µg/L % Recovery (RSD) | 75 µg/L % Recovery (RSD) |
|---|---|---|---|
| Cashmeran | 71 (10) | 85 (6) | 97 (4) |
| Celestolide | 74 (4) | 96 (7) | 99 (3) |
| Galaxolide | 83 (13) | 109 (14) | 107 (4) |
| Tonalid | 89 (8) | 104 (8) | 106 (6) |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Terlizzi, F.; Fernandes, J.O.; Gonzalez, T.; Petrarca, M.H.; Silva, L.d.S.; Lamarca, R.S.; Gonzalez, M.H.; Lapa, R.; Cunha, S.C. Microextraction of Polycyclic Musks from Surface Water with Deep-Eutectic-Solvent-Coated Membrane Followed by Gas-Chromatography–Mass Spectrometry Analysis. Chemosensors 2025, 13, 88. https://doi.org/10.3390/chemosensors13030088
Terlizzi F, Fernandes JO, Gonzalez T, Petrarca MH, Silva LdS, Lamarca RS, Gonzalez MH, Lapa R, Cunha SC. Microextraction of Polycyclic Musks from Surface Water with Deep-Eutectic-Solvent-Coated Membrane Followed by Gas-Chromatography–Mass Spectrometry Analysis. Chemosensors. 2025; 13(3):88. https://doi.org/10.3390/chemosensors13030088
Chicago/Turabian StyleTerlizzi, Fabiana, José O. Fernandes, Tamara Gonzalez, Mateus H. Petrarca, Leandro dos S. Silva, Rafaela S. Lamarca, Mario H. Gonzalez, Rui Lapa, and Sara C. Cunha. 2025. "Microextraction of Polycyclic Musks from Surface Water with Deep-Eutectic-Solvent-Coated Membrane Followed by Gas-Chromatography–Mass Spectrometry Analysis" Chemosensors 13, no. 3: 88. https://doi.org/10.3390/chemosensors13030088
APA StyleTerlizzi, F., Fernandes, J. O., Gonzalez, T., Petrarca, M. H., Silva, L. d. S., Lamarca, R. S., Gonzalez, M. H., Lapa, R., & Cunha, S. C. (2025). Microextraction of Polycyclic Musks from Surface Water with Deep-Eutectic-Solvent-Coated Membrane Followed by Gas-Chromatography–Mass Spectrometry Analysis. Chemosensors, 13(3), 88. https://doi.org/10.3390/chemosensors13030088