
Salt-Mediated Organic Solvent Precipitation for Enhanced Recovery of Peptides Generated by Pepsin Digestion


Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Pepsin Digestion
2.3. Peptide Precipitation
2.4. Peptide Quantitation by LC-UV
2.5. LC-MS/MS Analysis
2.6. Data Analysis
3. Results
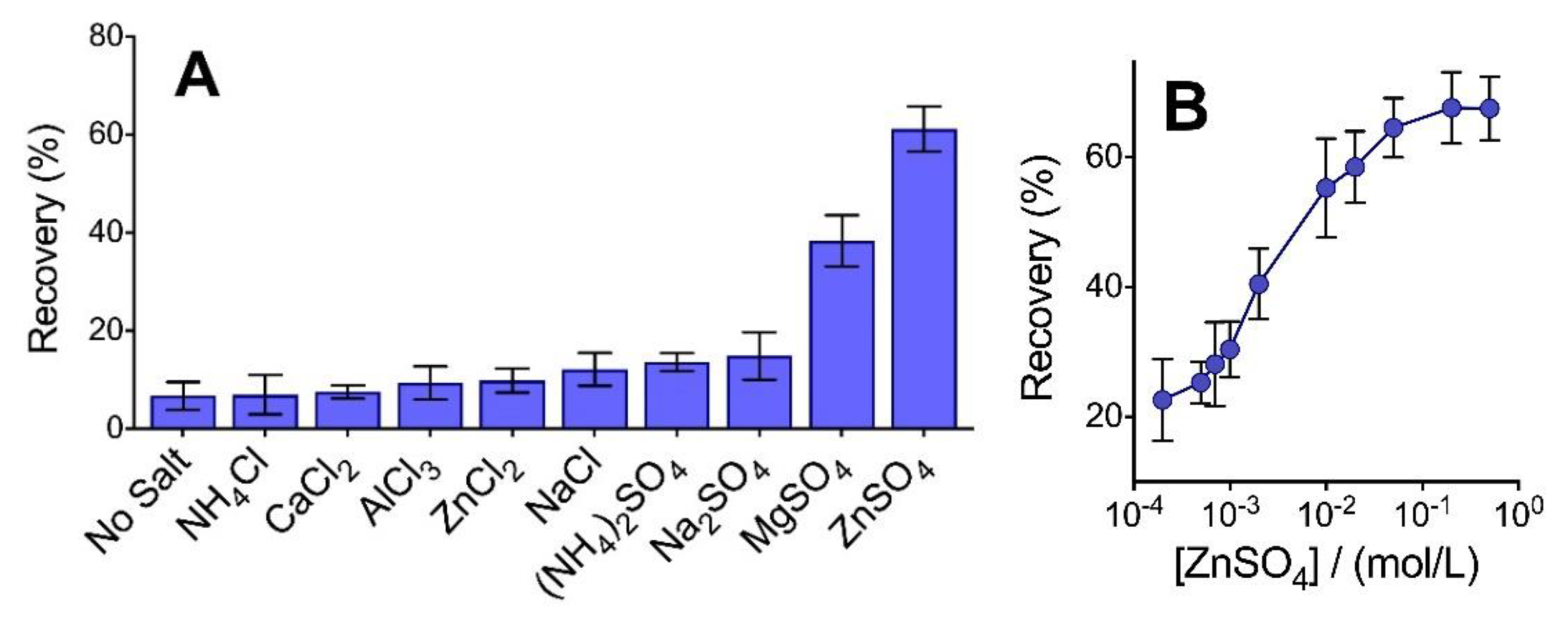
3.1. Salt Controls the Precipitation of Peptic Peptides in Organic Solvents
3.2. Higher Organic Solvent Is Needed to Precipitate Peptides
3.3. Precipitation of Dilute Peptide Solutions
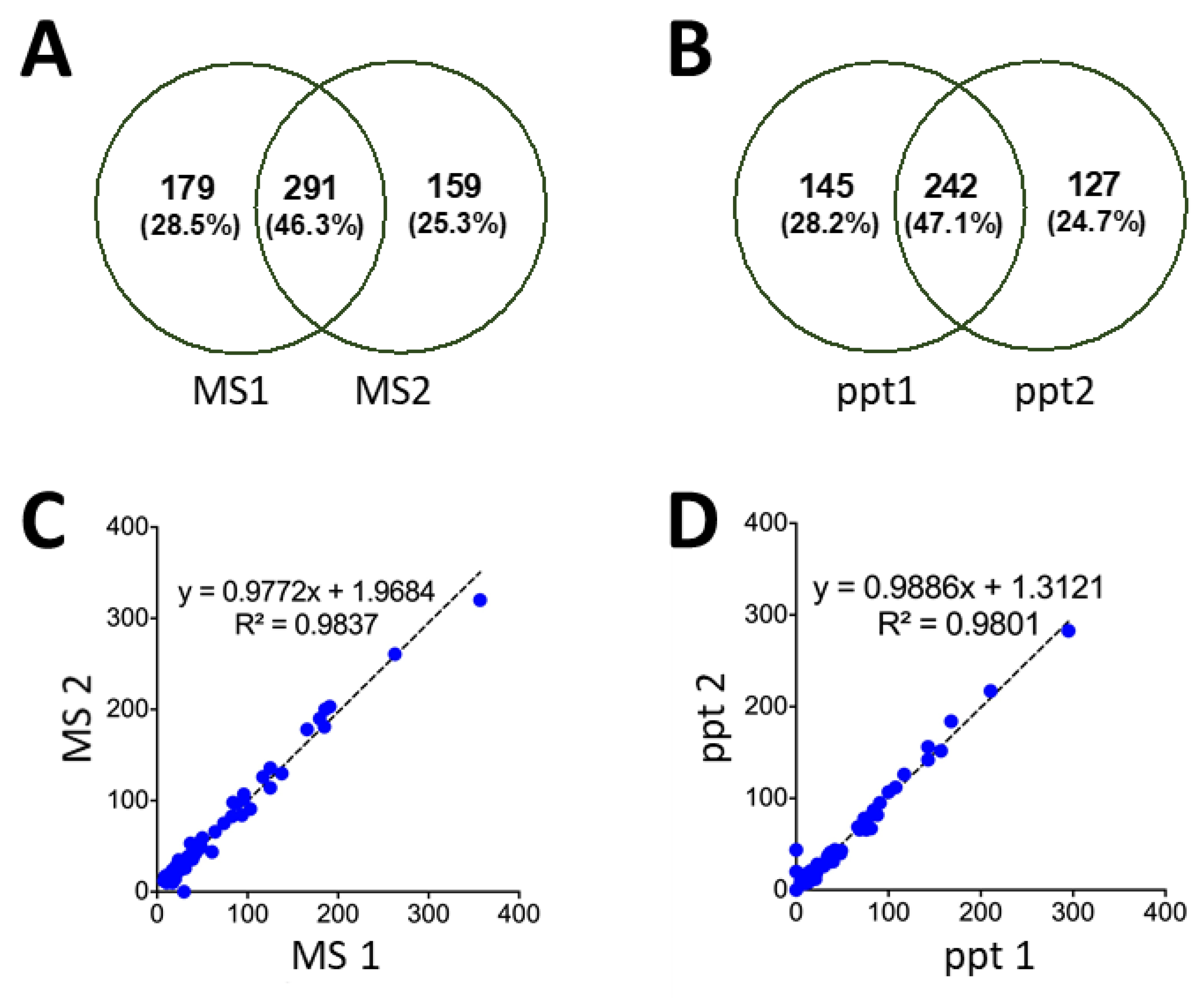
3.4. Mass Spectrometry of Precipitated Peptides
3.5. High Sequence Coverage for Pepsin-Digested Proteins
3.6. Reproducibility of Peptide Precipitation
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Nickerson, J.L.; Doucette, A.A. Rapid and quantitative protein precipitation for proteome analysis by mass sectrometry. J. Proteome Res. 2020, 19, 2035–2042. [Google Scholar] [CrossRef] [PubMed]
- Crowell, A.M.J.; Wall, M.J.; Doucette, A.A. Maximizing recovery of water-soluble proteins through acetone precipitation. Anal. Chim. Acta 2013, 796, 48–54. [Google Scholar] [CrossRef] [PubMed]
- Baghalabadi, V.; Doucette, A.A. Mass spectrometry profiling of low molecular weight proteins and peptides isolated by acetone precipitation. Anal. Chim. Acta 2020, 1138, 38–48. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Budge, S.M.; Ghaly, A.E.; Brooks, M.S.; Dave, D. Extraction, purification and characterization of fish pepsin: A critical review. J. Food Process Technol. 2011, 2, 1000126. [Google Scholar] [CrossRef]
- Jongjareonrak, A.; Benjakul, S.; Visessanguan, W.; Nagai, T.; Tanaka, M. Isolation and characterisation of acid and pepsin-solubilised collagens from the skin of Brownstripe red snapper (Lutjanus vitta). Food Chem. 2005, 93, 475–484. [Google Scholar] [CrossRef]
- Zhang, Y.; Liu, W.; Li, G.; Shi, B.; Miao, Y.; Wu, X. Isolation and partial characterization of pepsin-soluble collagen from the skin of grass carp (Ctenopharyngodon idella). Food Chem. 2007, 103, 906–912. [Google Scholar] [CrossRef]
- Nalinanon, S.; Benjakul, S.; Visessanguan, W.; Kishimura, H. Use of pepsin for collagen extraction from the skin of bigeye snapper (Priacanthus tayenus). Food Chem. 2007, 104, 593–601. [Google Scholar] [CrossRef]
- Nalinanon, S.; Benjakul, S.; Visessanguan, W.; Kishimura, H. Improvement of gelatin extraction from bigeye snapper skin using pepsin-aided process in combination with protease inhibitor. Food Hydrocoll. 2008, 22, 615–622. [Google Scholar] [CrossRef]
- Sun, S.; Mo, W.; Ji, Y.; Liu, S. Preparation and mass spectrometric study of egg yolk antibody (Igy) against rabies virus. Rapid Commun. Mass Spectrom. 2001, 15, 708–712. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.-M.; Bou-Assaf, G.M.; Emmett, M.R.; Marshall, A.G. Fast reversed-phase liquid chromatography to reduce back exchange and increase throughput in H/D exchange monitored by FT-ICR mass spectrometry. J. Am. Soc. Mass Spectrom. 2009, 20, 520–524. [Google Scholar] [CrossRef][Green Version]
- Zhang, H.M.; Kazazic, S.; Schaub, T.M.; Tipton, J.D.; Emmett, M.R.; Marshall, A.G. Enhanced digestion efficiency, peptide ionization efficiency, and sequence resolution for protein hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2008, 80, 9034–9041. [Google Scholar] [CrossRef] [PubMed]
- Endo, Y. Studies on protease produced by thermophilic bacteria. J. Ferment. Technol. 1962, 40, 346–353. [Google Scholar]
- Herriott, R.M. Pepsinogen and pepsin. J. Gen. Physiol. 1962, 45, 57. [Google Scholar] [CrossRef]
- MacWright, R.; Benson, V.; Lovello, K.; van der Rest, M.; Fietzek, P. Isolation and characterization of pepsin-solubilized human basement membrane (type IV) collagen peptides. Biochemistry 1983, 22, 4940–4948. [Google Scholar] [CrossRef]
- Epstein, E.; Munderloh, N. Isolation and characterization of CNBr peptides of human (alpha 1 (III))3 collagen and tissue distribution of (alpha 1 (I))2 alpha 2 and (alpha 1 (III))3 collagens. J. Biol. Chem. 1975, 250, 9304–9312. [Google Scholar] [CrossRef]
- Oliveros, J.C. VENNY. An Interactive Tool for Comparing Lists with Venn’s Diagrams. 2007. Available online: https://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 23 September 2021).
- Stothard, P. The sequence manipulation suite: JavaScript programs for analyzing and formatting protein and DNA sequences. Biotechniques 2000, 28, 1102–1104. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, L.P. IPC–Isoelectric Point Calculator. Biol. Direct 2016, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Hyde, A.M.; Zultanski, S.L.; Waldman, J.H.; Zhong, Y.-L.; Shevlin, M.; Peng, F. General principles and strategies for salting-out informed by the Hofmeister series. Org. Process Res. Dev. 2017, 21, 1355–1370. [Google Scholar] [CrossRef]
- Tucholska, M.; Florentinus, A.; Williams, D.; Marshall, J.G. The endogenous peptides of normal human serum extracted from the acetonitrile-insoluble precipitate using modified aqueous buffer with analysis by LC–ESI–Paul ion trap and Qq-TOF. J. Proteom. 2010, 73, 1254–1269. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, S.; Tang, K.-C.; Raj, M. Amide bond activation of biological molecules. Molecules 2018, 23, 2615. [Google Scholar] [CrossRef] [PubMed]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amino Acid Residues | Average of Normalized 1 Residues to the Length of the Peptides) | p Value of t-Test | |
|---|---|---|---|
| Pellet | Supernatant | Pellet vs. Supernatant | |
| Basic Residues (K + R + H) | 0.12 ± 0.08 | 0.06 ± 0.07 | 1.46 × 10−34 |
| Histidine (H) | 0.02 ± 0.03 | 0.01 ± 0.02 | 7.84 × 10−22 |
| Lysine (K) | 0.06 ± 0.06 | 0.03 ± 0.05 | 5.96 × 10−18 |
| Arginine (R) | 0.04 ± 0.05 | 0.02 ± 0.04 | 7.31 × 10−15 |
| Leucine (L) | 0.10 ± 0.09 | 0.14 ± 0.11 | 2.40 × 10−15 |
| Phenylalanine (F) | 0.04 ± 0.06 | 0.05 ± 0.07 | 5.32 × 10−3 |
| Cysteine (C) | 0.00 ± 0.02 | 0.00 ± 0.01 | 5.69 × 10−3 |
| Aspartic Acid (D) | 0.05 ± 0.06 | 0.05 ± 0.07 | 2.50 × 10−2 |
| Serine (S) | 0.06 ± 0.07 | 0.05 ± 0.07 | 3.34 × 10−2 |
| Tyrosine (Y) | 0.03 ± 0.05 | 0.04 ± 0.07 | 4.30 × 10−2 |
| Isoleucine (I) | 0.06 ± 0.07 | 0.07 ± 0.09 | 4.52 × 10−2 |
| Acidic Residues (D+E) | 0.11 ± 0.09 | 0.11 ± 0.09 | ND |
| Alanine (A) | 0.09 ± 0.09 | 0.09 ± 0.09 | ND |
| Glutamic Acid (E) | 0.06 ± 0.07 | 0.05 ± 0.07 | ND |
| Tryptophane (W) | 0.01 ± 0.03 | 0.01 ± 0.02 | ND |
| Glutamine (Q) | 0.03 ± 0.05 | 0.03 ± 0.05 | ND |
| Glycine (G) | 0.08 ± 0.08 | 0.07 ± 0.08 | ND |
| Proline (P) | 0.06 ± 0.06 | 0.06 ± 0.07 | ND |
| Valine (V) | 0.08 ± 0.08 | 0.08 ± 0.09 | ND |
| Methionine (M) | 0.01 ± 0.03 | 0.01 ± 0.04 | ND |
| Threonine (T) | 0.05 ± 0.06 | 0.05 ± 0.07 | ND |
| Asparagine (N) | 0.04 ± 0.06 | 0.04 ± 0.06 | ND |
| Protein/Enzyme Ratio | Time (min) | # Peptide | # PSMs 1 | % Coverage | Avg Seq Length | Avg MW (u) |
|---|---|---|---|---|---|---|
| 10:1 | 1 | 89 | 274 | 52.6% | 14.3 ± 5.5 | 1695 ± 639 |
| 10 | 133 | 440 | 62.8% | 13.8 ± 5.1 | 1634 ± 607 | |
| Total | 157 | 714 | 64.5% | 14.0 ± 5.3 | 1657 ± 628 | |
| 100:1 | 1 | 83 | 200 | 55.2% | 14.7 ± 5.2 | 1722 ± 623 |
| 10 | 87 | 210 | 49.1% | 14.10 ± 4.7 | 1666 ± 565 | |
| Total | 116 | 410 | 57.9% | 14.6 ± 5.0 | 1714 ± 595 | |
| 1000:1 | 1 | 48 | 111 | 31.8% | 14.4 ± 5.0 | 1677 ± 591 |
| 10 | 53 | 104 | 47.2% | 14.3 ± 5.2 | 1658 ± 625 | |
| Total | 73 | 215 | 523% | 14.8 ± 5.4 | 1710 ± 636 | |
| Aggregated Total | 196 | 1339 | 78.3% | 14.6 ± 5.4 | 1713 ± 629 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baghalabadi, V.; Razmi, H.; Doucette, A. Salt-Mediated Organic Solvent Precipitation for Enhanced Recovery of Peptides Generated by Pepsin Digestion. Proteomes 2021, 9, 44. https://doi.org/10.3390/proteomes9040044
Baghalabadi V, Razmi H, Doucette A. Salt-Mediated Organic Solvent Precipitation for Enhanced Recovery of Peptides Generated by Pepsin Digestion. Proteomes. 2021; 9(4):44. https://doi.org/10.3390/proteomes9040044
Chicago/Turabian StyleBaghalabadi, Venus, Habib Razmi, and Alan Doucette. 2021. "Salt-Mediated Organic Solvent Precipitation for Enhanced Recovery of Peptides Generated by Pepsin Digestion" Proteomes 9, no. 4: 44. https://doi.org/10.3390/proteomes9040044
APA StyleBaghalabadi, V., Razmi, H., & Doucette, A. (2021). Salt-Mediated Organic Solvent Precipitation for Enhanced Recovery of Peptides Generated by Pepsin Digestion. Proteomes, 9(4), 44. https://doi.org/10.3390/proteomes9040044