
Selecting Sample Preparation Workflows for Mass Spectrometry-Based Proteomic and Phosphoproteomic Analysis of Patient Samples with Acute Myeloid Leukemia

 , ,
, , 
Abstract
:1. Introduction
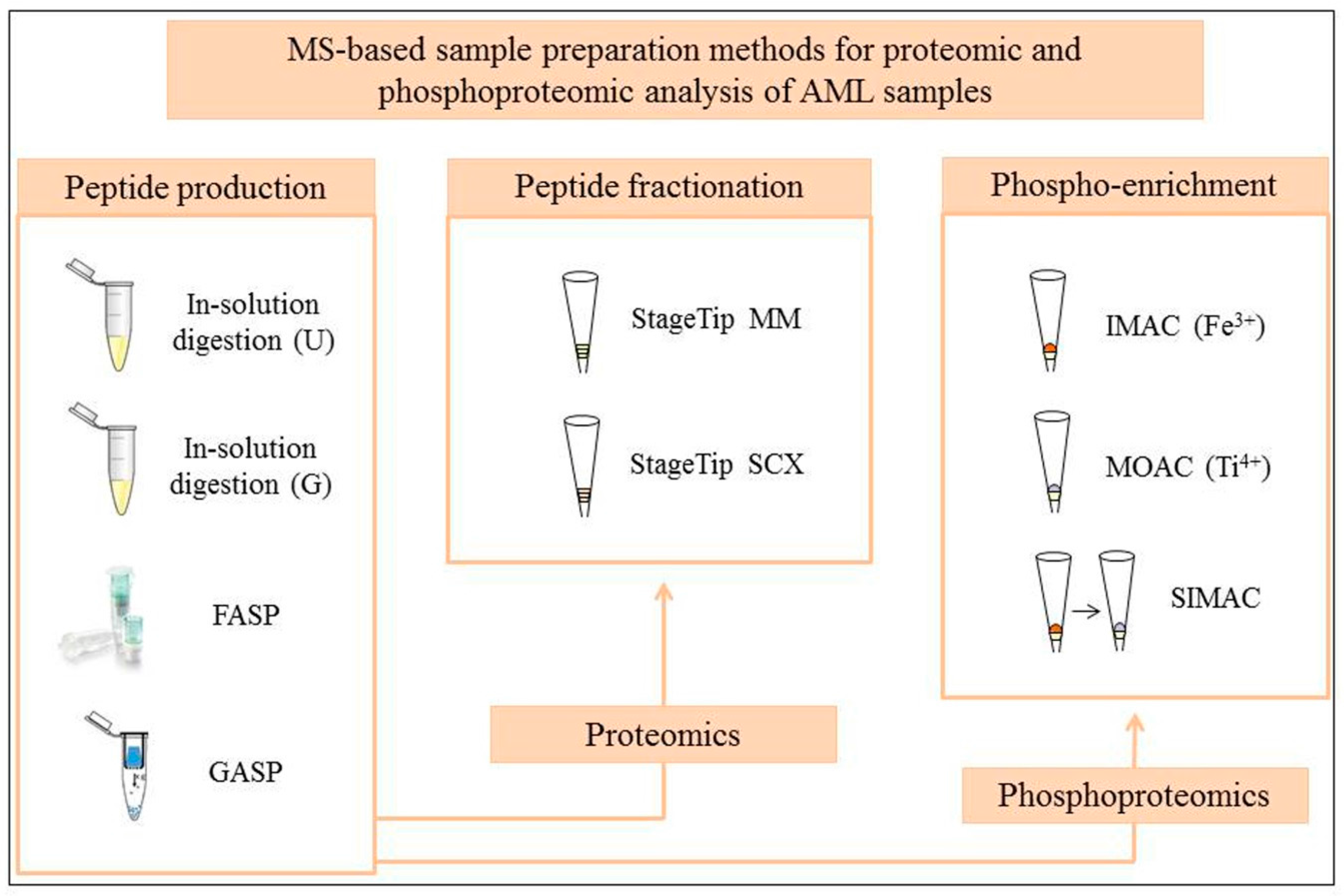
2. Sample Preparation Workflows in Global MS Proteome Studies of AML
3. Testing of Standard and Novel MS-Based Proteomic and Phosphoproteomic Workflows
4. Discussion and Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| 1DE | One-dimensional electrophoresis |
| 2DE | Two-dimensional electrophoresis |
| AAI | Anti-apoptosis index |
| AML | Acute myeloid leukemia |
| CAA | Chloroacetamide |
| CID | Collision-induced dissociation |
| DDA | Data-dependent acquisition |
| ESI | Electrospray ionization |
| FAB | French-American-British |
| FASP | Filter-aided sample preparation |
| GASP | Gel-aided sample preparation |
| IMAC | Immobilized metal affinity chromatography |
| KSEA | Kinase-substrate enrichment analysis |
| LC | Liquid chromatography |
| LDS | Lithium dodecyl sulfate |
| MALDI-TOF | Matrix-assisted laser desorption/ionization time-of-flight |
| MM | Mixed mode |
| MOAC | Metal oxide affinity chromatography |
| MS | Mass spectrometry |
| PTM | Post-translational modifications |
| SCX | Strong cation exchange |
| SDB-RPS | Polystyrenedivinylbenzene reversed phase sulfonate |
| SDS | Sodium dodecyl sulfate |
| SH2 | Src homology 2 |
| SILAC | Stable isotope labelling with amino acids in cell culture |
| SIMAC | Sequential elution from IMAC |
| TCEP | Tris(2-carboxyethyl)phosphine |
| WHO | World Health Organization |
References
- Estey, E.H. Acute myeloid leukemia: 2014 Update on risk-stratification and management. Am. J. Hematol. 2014, 89, 1063–1081. [Google Scholar] [CrossRef] [PubMed]
- Lowenberg, B.; Downing, J.R.; Burnett, A. Acute myeloid leukemia. N. Engl. J. Med. 1999, 341, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Howlader, N.; Noone, A.M.; Krapcho, M.; Miller, D.; Bishop, K.; Altekruse, S.F.; Kosary, C.L.; Yu, M.; Ruhl, J.; Tatalovich, Z.; et al. Leukemia: SEER Cancer Statistics Review 1975–2013. Available online: http://seer.cancer.gov/csr/1975_2013/ (accessed on 18 April 2016).
- Bennett, J.M.; Catovsky, D.; Daniel, M.T.; Flandrin, G.; Galton, D.A.; Gralnick, H.R.; Sultan, C. Proposed revised criteria for the classification of acute myeloid leukemia: A report of the French-American-British Cooperative Group. Ann. Intern. Med. 1985, 103, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Vardiman, J.W.; Thiele, J.; Arber, D.A.; Brunning, R.D.; Borowitz, M.J.; Porwit, A.; Harris, N.L.; Le Beau, M.M.; Hellstrom-Lindberg, E.; Tefferi, A.; et al. The 2008 revision of the world health organization (who) classification of myeloid neoplasms and acute leukemia: Rationale and important changes. Blood 2009, 114, 937–951. [Google Scholar] [CrossRef] [PubMed]
- Godley, L.A. Inherited predisposition to acute myeloid leukemia. Semin. Hematol. 2014, 51, 306–321. [Google Scholar] [CrossRef] [PubMed]
- Dohner, H.; Weisdorf, D.J.; Bloomfield, C.D. Acute myeloid leukemia. N. Engl. J. Med. 2015, 373, 1136–1152. [Google Scholar] [PubMed]
- Roboz, J.; Roboz, G.J. Mass spectrometry in leukemia research and treatment. Expert Rev. Hematol. 2015, 8, 225–235. [Google Scholar] [CrossRef] [PubMed]
- Aasebo, E.; Forthun, R.B.; Berven, F.; Selheim, F.; Hernandez-Valladares, M. Global cell proteome profiling, phospho-signaling and quantitative proteomics for identification of new biomarkers in acute myeloid leukemia patients. Curr. Pharm. Biotechnol. 2016, 17, 52–70. [Google Scholar] [CrossRef] [PubMed]
- Luczak, M.; Kazmierczak, M.; Handschuh, L.; Lewandowski, K.; Komarnicki, M.; Figlerowicz, M. Comparative proteome analysis of acute myeloid leukemia with and without maturation. J. Proteom. 2012, 75, 5734–5748. [Google Scholar] [CrossRef] [PubMed]
- Casado, P.; Rodriguez-Prados, J.C.; Cosulich, S.C.; Guichard, S.; Vanhaesebroeck, B.; Joel, S.; Cutillas, P.R. Kinase-substrate enrichment analysis provides insights into the heterogeneity of signaling pathway activation in leukemia cells. Sci. Signal. 2013, 6, rs6. [Google Scholar] [CrossRef] [PubMed]
- Schaab, C.; Oppermann, F.S.; Klammer, M.; Pfeifer, H.; Tebbe, A.; Oellerich, T.; Krauter, J.; Levis, M.; Perl, A.E.; Daub, H.; et al. Global phosphoproteome analysis of human bone marrow reveals predictive phosphorylation markers for the treatment of acute myeloid leukemia with quizartinib. Leukemia 2014, 28, 716–719. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.L.; Wybenga-Groot, L.E.; Tong, J.; Taylor, P.; Minden, M.D.; Trudel, S.; McGlade, C.J.; Moran, M.F. Tyrosine phosphorylation of the Lyn Src homology 2 (SH2) domain modulates its binding affinity and specificity. Mol. Cell. Proteom. 2015, 14, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Wojtuszkiewicz, A.; Schuurhuis, G.J.; Kessler, F.L.; Piersma, S.R.; Knol, J.C.; Pham, T.V.; Jansen, G.; Musters, R.J.; van Meerloo, J.; Assaraf, Y.G.; et al. Exosomes secreted by apoptosis-resistant acute myeloid leukemia (aml) blasts harbor regulatory network proteins potentially involved in antagonism of apoptosis. Mol. Cell. Proteom. 2016, 15, 1281–1298. [Google Scholar] [CrossRef] [PubMed]
- Aasebo, E.; Mjaavatten, O.; Vaudel, M.; Farag, Y.; Selheim, F.; Berven, F.; Bruserud, O.; Hernandez-Valladares, M. Freezing effects on the acute myeloid leukemia cell proteome and phosphoproteome revealed using optimal quantitative workflows. J. Proteom. 2016, 145, 214–225. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Zougman, A.; Nagaraj, N.; Mann, M. Universal sample preparation method for proteome analysis. Nat. Methods 2009, 6, 359–362. [Google Scholar] [CrossRef] [PubMed]
- Thingholm, T.E.; Larsen, M.R. Phosphopeptide enrichment by immobilized metal affinity chromatography. Methods Mol. Biol. 2016, 1355, 123–133. [Google Scholar] [PubMed]
- Thingholm, T.E.; Larsen, M.R. The use of titanium dioxide for selective enrichment of phosphorylated peptides. Methods Mol. Biol. 2016, 1355, 135–146. [Google Scholar] [PubMed]
- Thingholm, T.E.; Larsen, M.R. Sequential elution from IMAC (SIMAC): An efficient method for enrichment and separation of mono- and multi-phosphorylated peptides. Methods Mol. Biol. 2016, 1355, 147–160. [Google Scholar] [PubMed]
- Montoya, A.; Beltran, L.; Casado, P.; Rodriguez-Prados, J.C.; Cutillas, P.R. Characterization of a TiO2 enrichment method for label-free quantitative phosphoproteomics. Methods 2011, 54, 370–378. [Google Scholar] [CrossRef] [PubMed]
- St-Germain, J.R.; Taylor, P.; Tong, J.; Jin, L.L.; Nikolic, A.; Stewart, I.I.; Ewing, R.M.; Dharsee, M.; Li, Z.; Trudel, S.; et al. Multiple myeloma phosphotyrosine proteomic profile associated with FGFR3 expression, ligand activation, and drug inhibition. Proc. Natl. Acad. Sci. USA 2009, 106, 20127–20132. [Google Scholar] [CrossRef] [PubMed]
- Manza, L.L.; Stamer, S.L.; Ham, A.J.; Codreanu, S.G.; Liebler, D.C. Sample preparation and digestion for proteomic analyses using spin filters. Proteomics 2005, 5, 1742–1745. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Mann, M. Consecutive proteolytic digestion in an enzyme reactor increases depth of proteomic and phosphoproteomic analysis. Anal. Chem. 2012, 84, 2631–2637. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R.; Rakus, D. Multi-enzyme digestion fasp and the ‘total protein approach’-based absolute quantification of the Escherichia coli proteome. J. Proteom. 2014, 109, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Wisniewski, J.R. Quantitative evaluation of filter aided sample preparation (FASP) and multienzyme digestion FASP protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [PubMed]
- Kulak, N.A.; Pichler, G.; Paron, I.; Nagaraj, N.; Mann, M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in Eukaryotic cells. Nat. Methods 2014, 11, 319–324. [Google Scholar] [CrossRef] [PubMed]
- Scheltema, R.A.; Hauschild, J.P.; Lange, O.; Hornburg, D.; Denisov, E.; Damoc, E.; Kuehn, A.; Makarov, A.; Mann, M. The Q exactive HF, a Benchtop mass spectrometer with a pre-filter, high-performance quadrupole and an ultra-high-field Orbitrap analyzer. Mol. Cell. Proteom. 2014, 13, 3698–3708. [Google Scholar] [CrossRef] [PubMed]
- Kelstrup, C.D.; Jersie-Christensen, R.R.; Batth, T.S.; Arrey, T.N.; Kuehn, A.; Kellmann, M.; Olsen, J.V. Rapid and deep proteomes by faster sequencing on a benchtop quadrupole ultra-high-field Orbitrap mass spectrometer. J. Proteome Res. 2014, 13, 6187–6195. [Google Scholar] [CrossRef] [PubMed]
- Fischer, R.; Kessler, B.M. Gel-aided sample preparation (GASP)—A simplified method for gel-assisted proteomic sample generation from protein extracts and intact cells. Proteomics 2015, 15, 1224–1229. [Google Scholar] [CrossRef] [PubMed]
- Aasebo, E.; Vaudel, M.; Mjaavatten, O.; Gausdal, G.; Van der Burgh, A.; Gjertsen, B.T.; Doskeland, S.O.; Bruserud, O.; Berven, F.S.; Selheim, F. Performance of super-silac based quantitative proteomics for comparison of different acute myeloid leukemia (AML) cell lines. Proteomics 2014, 14, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Siepen, J.A.; Keevil, E.J.; Knight, D.; Hubbard, S.J. Prediction of missed cleavage sites in tryptic peptides aids protein identification in proteomics. J. Proteome Res. 2007, 6, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Aasebo, E.; Mjaavatten, O.; Vaudel, M.; Bruserud, O.; Berven, F.; Selheim, F. Reliable FASP-based procedures for optimal quantitative proteomic and phosphoproteomic analysis on samples from acute myeloid leukemia patients. Biol. Proced. Online 2016, 18. [Google Scholar] [CrossRef] [PubMed]
- Fila, J.; Honys, D. Enrichment techniques employed in phosphoproteomics. Amino Acids 2012, 43, 1025–1047. [Google Scholar] [CrossRef] [PubMed]
- Leon, I.R.; Schwammle, V.; Jensen, O.N.; Sprenger, R.R. Quantitative assessment of in-solution digestion efficiency identifies optimal protocols for unbiased protein analysis. Mol. Cell. Proteom. 2013, 12, 2992–3005. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Peng, F.; Li, G.; Fu, Y.; Huang, Y.; Chen, Z.; Chen, Y. Proteomic analysis of stromal proteins in different stages of colorectal cancer establishes Tenascin-C as a stromal biomarker for colorectal cancer metastasis. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Jehmlich, N.; Stegmaier, P.; Golatowski, C.; Salazar, M.G.; Rischke, C.; Henke, M.; Volker, U. Proteome data of whole saliva which are associated with development of oral mucositis in head and neck cancer patients undergoing radiotherapy. Data Brief 2016, 8, 501–505. [Google Scholar] [CrossRef] [PubMed]
- Camisasca, D.R.; da Ros Goncalves, L.; Soares, M.R.; Sandim, V.; Nogueira, F.C.; Garcia, C.H.; Santana, R.; de Oliveira, S.P.; Buexm, L.A.; de Faria, P.A.; et al. A proteomic approach to compare saliva from individuals with and without oral leukoplakia. J. Proteom. 2016. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Cox, J.; Ostasiewicz, P.; Wisniewski, J.R.; Mann, M. Super-silac mix for quantitative proteomics of human tumor tissue. Nat. Methods 2010, 7, 383–385. [Google Scholar] [CrossRef] [PubMed]
- Pozniak, Y.; Balint-Lahat, N.; Rudolph, J.D.; Lindskog, C.; Katzir, R.; Avivi, C.; Ponten, F.; Ruppin, E.; Barshack, I.; Geiger, T. System-wide clinical proteomics of breast cancer reveals global remodeling of tissue homeostasis. Cell Syst. 2016, 2, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Valladares, M.; Aasebø, E.; Vaudel, M.; Berven, F.S.; Selheim, F.; Doskeland, S.Ø.; Bruserud, Ø. Unpublished work. 2016.


{kind=link}
| Method 2 (SILAC Labeling) | Quantified Protein Groups | Quantified Peptides | Missed Cleavages 3 (%) |
|---|---|---|---|
| In-solution digestion, U, SD | 1011 | 8082 | 42 |
| In-solution digestion, U, SD, MM | 1627 | 11,096 | 29 |
| In-solution digestion, U, SD, SCX | 1201 | 7948 | 29 |
| In-solution digestion, G, DD | 1091 | 10,816 | 55 |
| In-solution digestion, G, DD, MM | 2006 | 15,894 | 31 |
| In-solution digestion, G, DD, SCX | 2051 | 16,110 | 35 |
| FASP, SD | 1480 | 12,607 | 15 |
| FASP, SD, MM | 2141 | 16,469 | 11 |
| FASP, SD, SCX | 1500 | 10,414 | 11 |
| Method 2 (SILAC Labeling) | Localized Phosphosites 3 | Mono-Phosphopeptides | Di- and Multi-Phosphopeptides |
|---|---|---|---|
| IMAC, SD | 2708 | 2745 | 607 |
| IMAC, DD | 2792 | 2489 | 843 |
| MOAC, SD | 738 | 945 | 29 |
| MOAC, DD | 897 | 1118 | 46 |
| SIMAC, SD | 1817 | 883 | 954 |
| SIMAC, DD | 1825 | 998 | 883 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hernandez-Valladares, M.; Aasebø, E.; Selheim, F.; Berven, F.S.; Bruserud, Ø. Selecting Sample Preparation Workflows for Mass Spectrometry-Based Proteomic and Phosphoproteomic Analysis of Patient Samples with Acute Myeloid Leukemia. Proteomes 2016, 4, 24. https://doi.org/10.3390/proteomes4030024
Hernandez-Valladares M, Aasebø E, Selheim F, Berven FS, Bruserud Ø. Selecting Sample Preparation Workflows for Mass Spectrometry-Based Proteomic and Phosphoproteomic Analysis of Patient Samples with Acute Myeloid Leukemia. Proteomes. 2016; 4(3):24. https://doi.org/10.3390/proteomes4030024
Chicago/Turabian StyleHernandez-Valladares, Maria, Elise Aasebø, Frode Selheim, Frode S. Berven, and Øystein Bruserud. 2016. "Selecting Sample Preparation Workflows for Mass Spectrometry-Based Proteomic and Phosphoproteomic Analysis of Patient Samples with Acute Myeloid Leukemia" Proteomes 4, no. 3: 24. https://doi.org/10.3390/proteomes4030024
APA StyleHernandez-Valladares, M., Aasebø, E., Selheim, F., Berven, F. S., & Bruserud, Ø. (2016). Selecting Sample Preparation Workflows for Mass Spectrometry-Based Proteomic and Phosphoproteomic Analysis of Patient Samples with Acute Myeloid Leukemia. Proteomes, 4(3), 24. https://doi.org/10.3390/proteomes4030024