
Comparative “Golgi” Proteome Study of Lolium multiflorum and Populus trichocarpa


 ,
,
Abstract
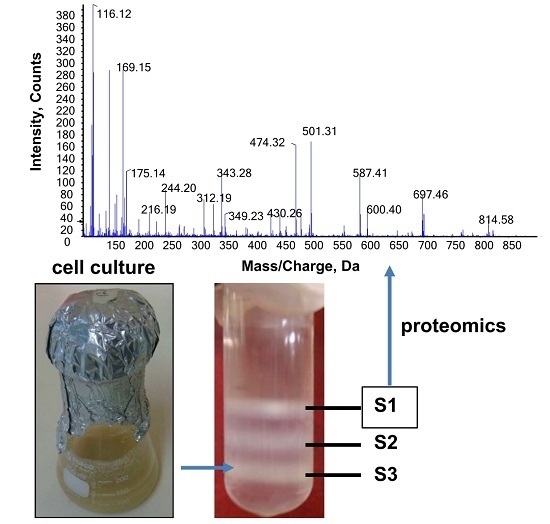
:
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Tissue Extraction
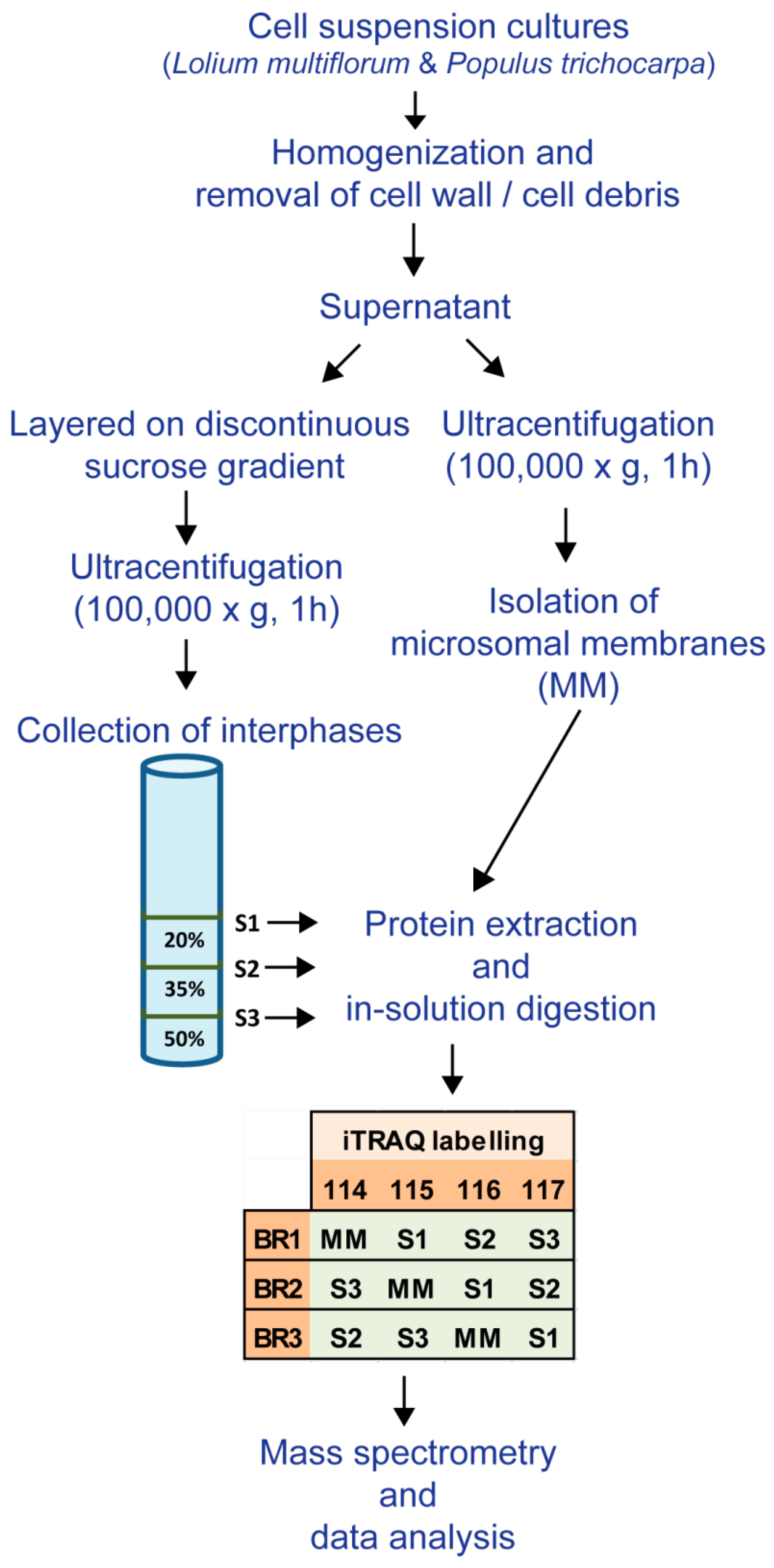
2.3. Microsomal Membrane Preparation
2.4. Membrane Fractionation
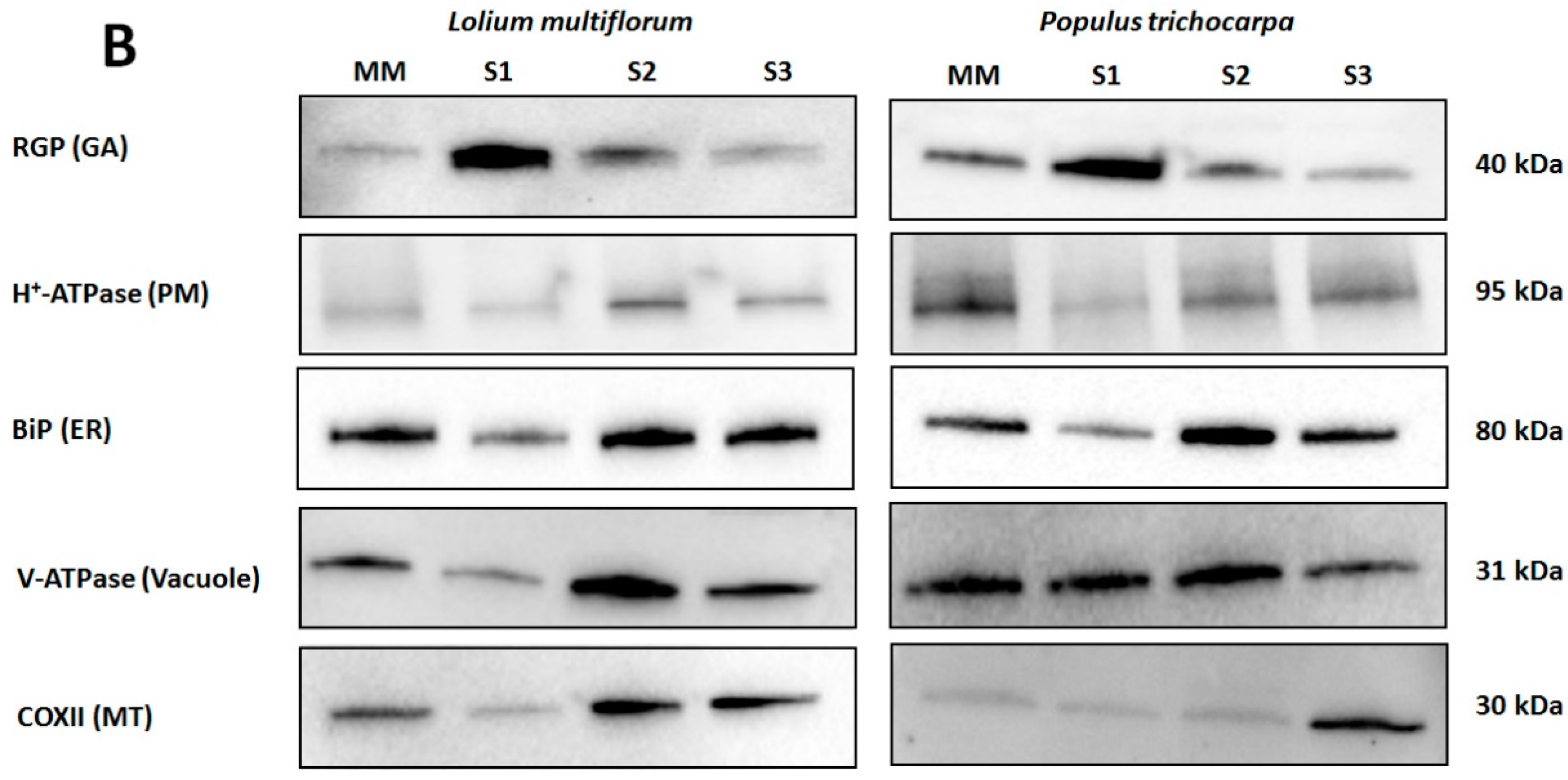
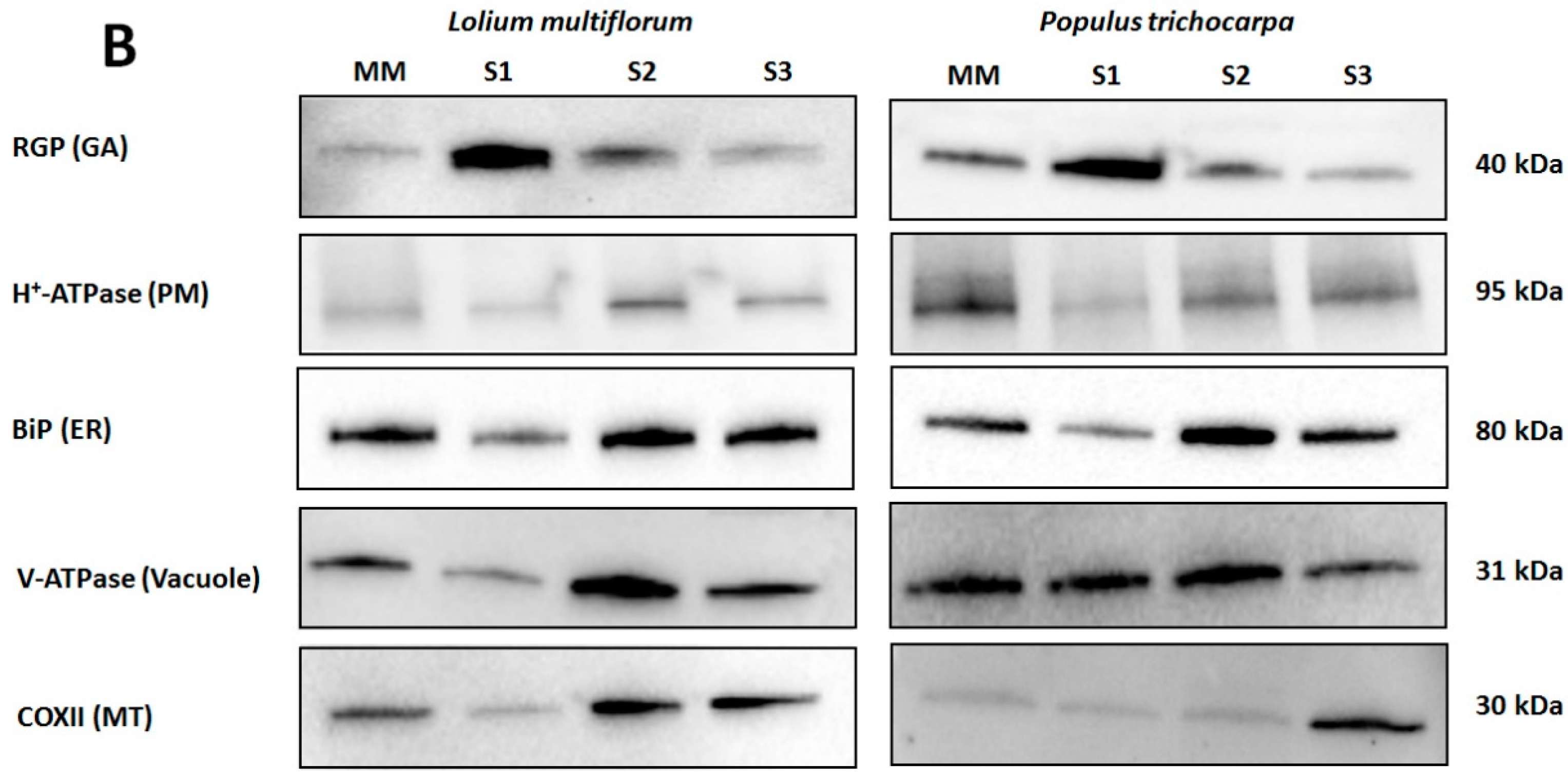
2.5. Enzyme Marker Assays
2.6. SDS-PAGE and Western Blot Analysis
2.7. Protein Digestion and iTRAQ Labelling
2.8. Non-labelled Protein Hydrolysis
2.9. Peptide Fractionation and Mass Spectrometry
2.10. Protein Identification
2.11. Relative Quantification
3. Results
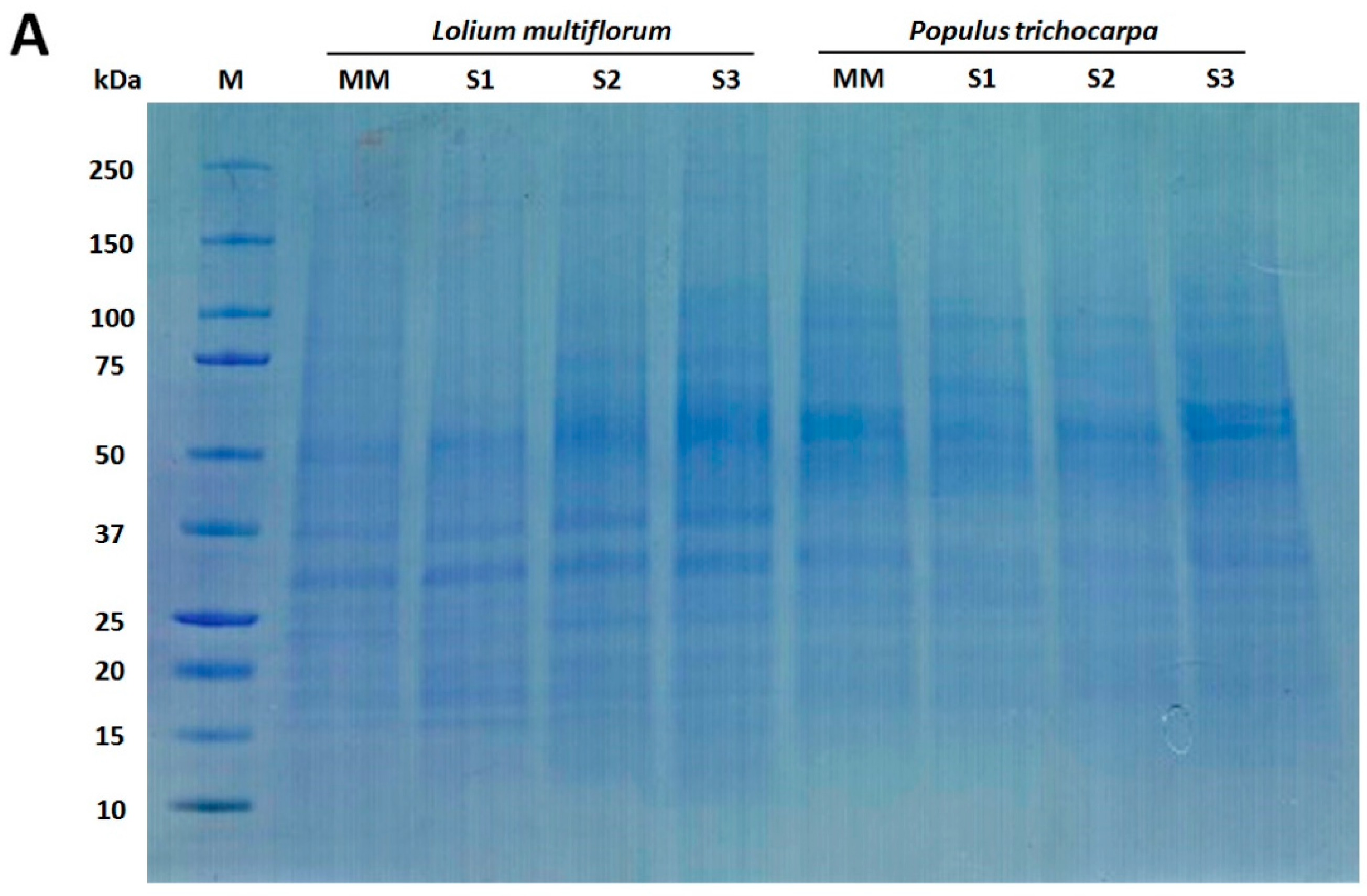
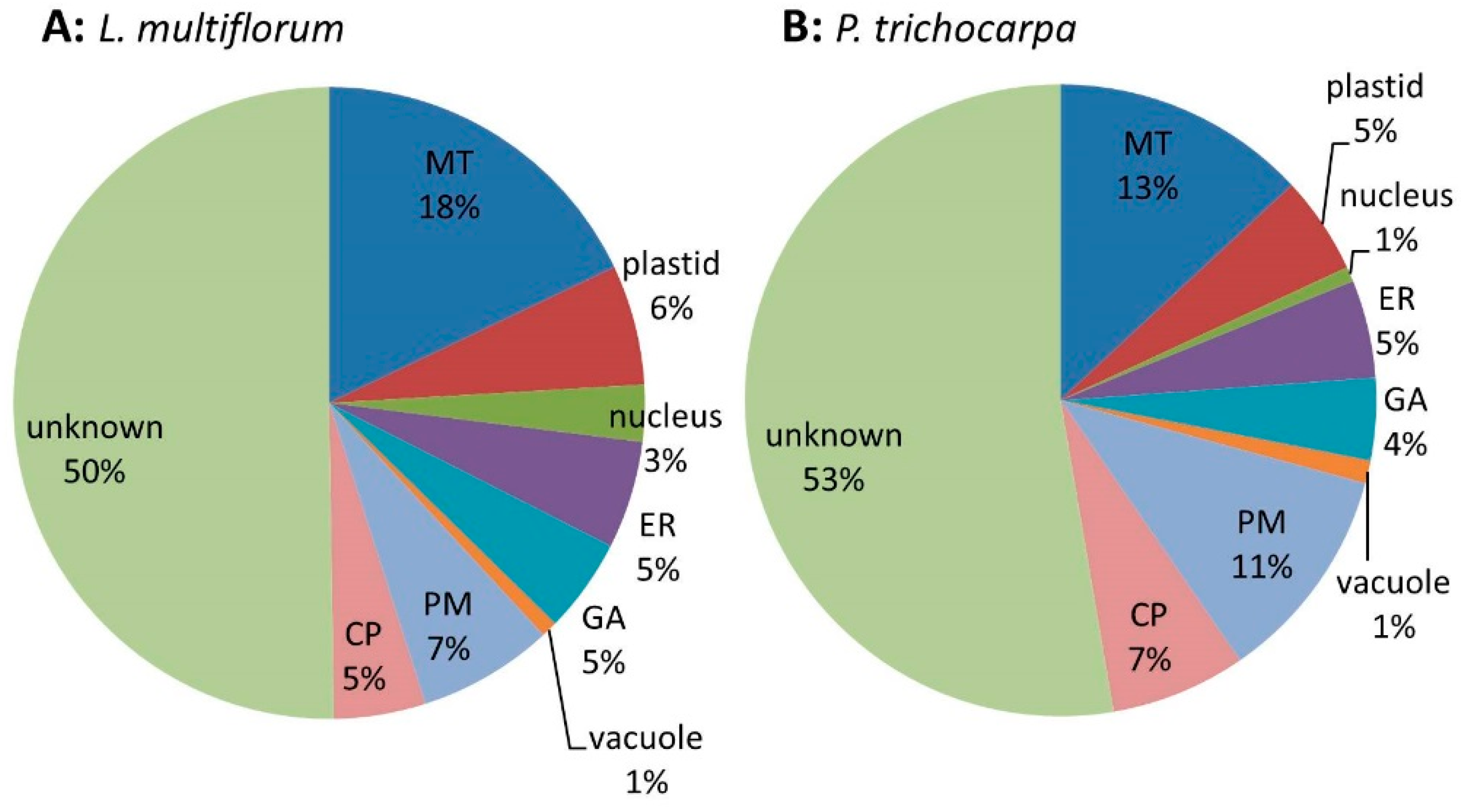
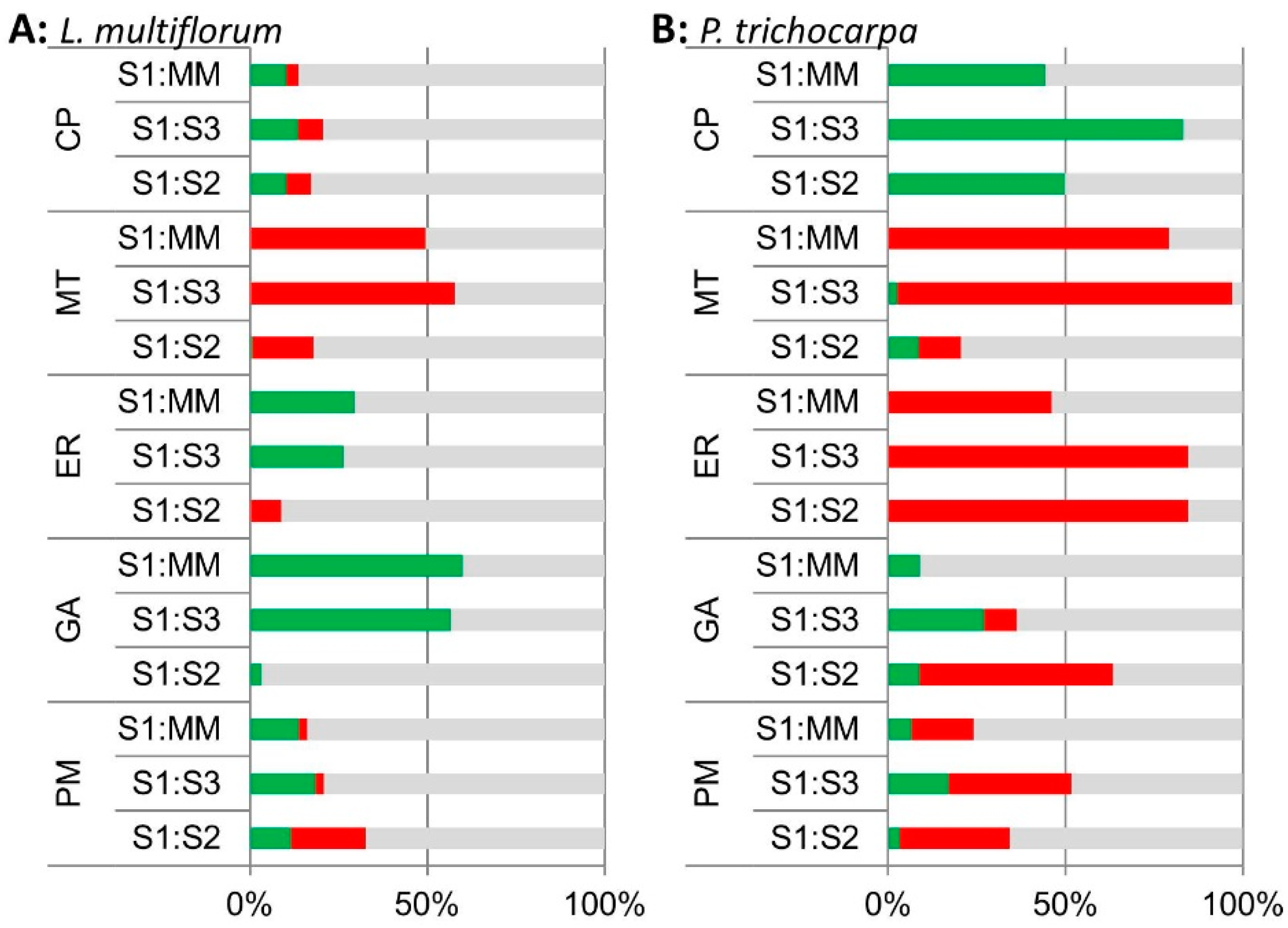
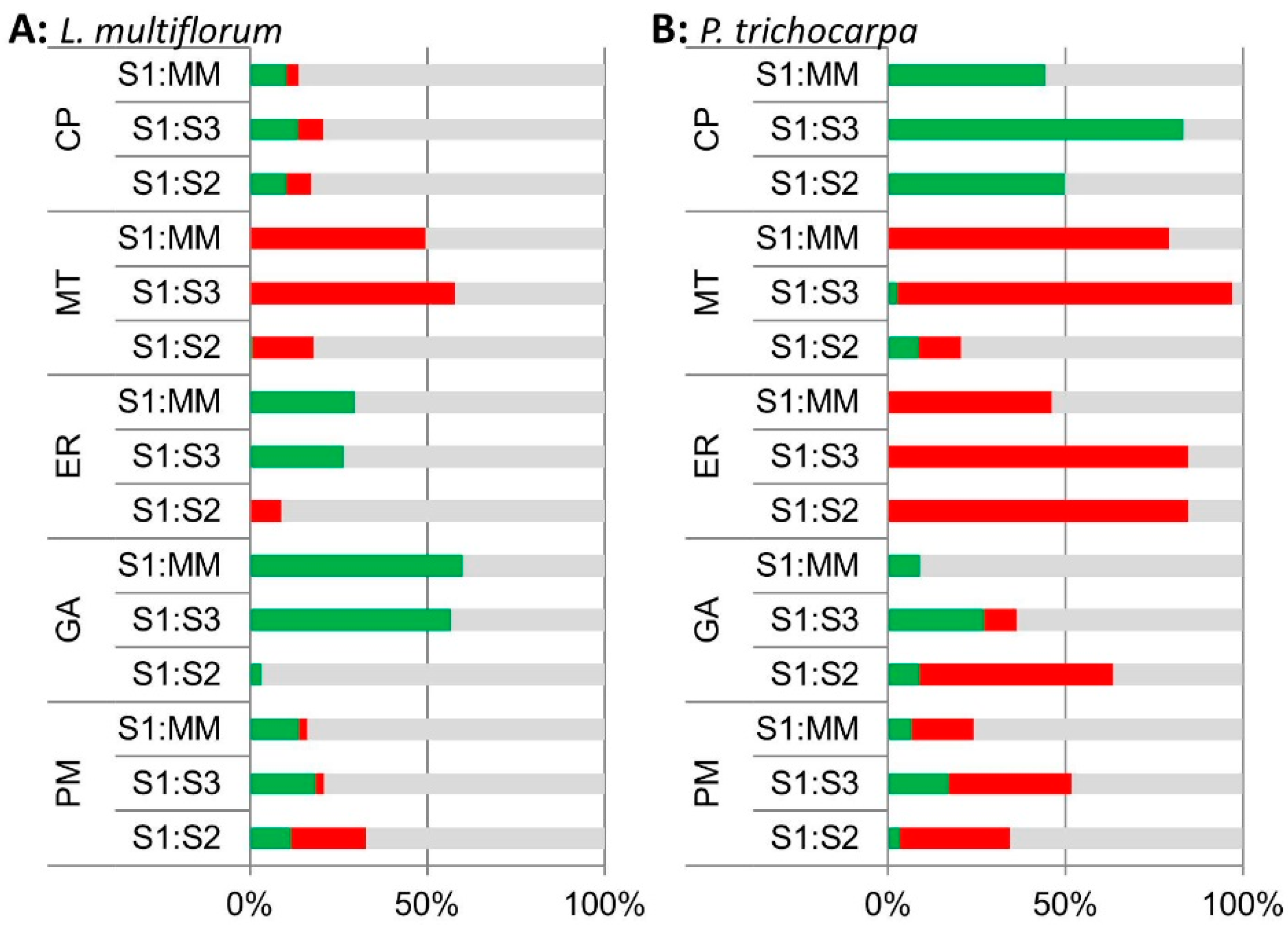
3.1. Enrichment of Microsomal Membranes
3.2. Mass Spectrometric Analyses
3.2.1. Protein Profiling of the Non-labelled GA-Enriched Fractions from L. multiflorum and P. trichocarpa
3.2.2. iTRAQ Analysis of the L. multiflorum and P. trichocarpa GA-enriched Fractions
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ATTPS | trehalose phosphatase/synthase |
| BiP | luminal-binding protein |
| COG | oligomeric Golgi complex |
| COX | cytochrome-c oxidase |
| CP | cytoplasmic |
| CSL | cellulose synthase-like |
| DTT | dithiothreitol |
| EMP70 | endomembrane 70 protein |
| ER | endoplasmic reticulum |
| FFE | Free Flow Electrophoresis |
| GA | Golgi apparatus |
| GC6 | golgin candidate 6 |
| GSL | glucan synthase-like |
| GT | glycosyltransferase |
| IDPase | inosine diphosphatase |
| iTRAQ | Isobaric tags for relative and absolute quantitation |
| LOPIT | Localization of Organelle Proteins by Isotope Tagging |
| MLG | β-1,3;1,4-glucans |
| MM | microsomal membrane |
| MMTS | methylmethane thiosulphate |
| MT | mitochondria |
| PM | plasma membrane |
| RGP | reversibly glycosylated protein |
| SAM | S-adenosyl-l-methionine-dependent methyltransferase |
| SCCs | suspension cell cultures |
| SNARE | N-ethylmaleimide-sensitive factor attachment protein receptors |
| TCEP | tris-(2-carboxyethyl) phosphine |
| TGN | trans-Golgi network |
| TRAPP | transport protein particle |
| UGE | UDP-d-glucose/UDP-d-galactose 4 epimerase |
| UGP | UDP-d-glucose pyrophosphorylase |
| VAMP | vesicle-associated membrane proteins |
| VSR | vacuolar sorting receptors |
References
- Van de Meene, A.M.; Doblin, M.S.; Bacic, A. The plant secretory pathway seen through the lens of the cell wall. Protoplasma 2016. [Google Scholar] [CrossRef] [PubMed]
- Parsons, H.T.; Drakakaki, G.; Heazlewood, J.L. Proteomic dissection of the Arabidopsis golgi and trans-golgi network. Front. Plant Sci. 2013, 3, 298. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.G.; Brandizzi, F.; Hawes, C.; Nakano, A. Vesicles versus tubes: Is endoplasmic reticulum-golgi transport in plants fundamentally different from other eukaryotes? Plant Physiol. 2015, 168, 393–406. [Google Scholar] [CrossRef] [PubMed]
- Gibeaut, D.M.; Carpita, N.C. Separation of membranes by flotation centrifugation for in vitro synthesis of plant cell wall polysaccharides. Protoplasma 1990, 156, 82–93. [Google Scholar] [CrossRef]
- Van der Woude, W.J.; Lembi, C.A.; Morré, D.J.; Kindinger, J.I.; Ordin, L. Β-glucan synthetases of plasma membrane and golgi apparatus from onion stem. Plant Physiol. 1974, 54, 333–340. [Google Scholar] [CrossRef] [PubMed]
- Dunkley, T.P.J.; Hester, S.; Shadforth, I.P.; Runions, J.; Weimar, T.; Hanton, S.L.; Griffin, J.L.; Bessant, C.; Brandizzi, F.; Hawes, C.; et al. Mapping the Arabidopsis organelle proteome. Proc. Natl. Acad. Sci. USA 2006, 103, 6518–6523. [Google Scholar] [CrossRef] [PubMed]
- Nikolovski, N.; Rubtsov, D.; Segura, M.P.; Miles, G.P.; Stevens, T.J.; Dunkley, T.P.J.; Munro, S.; Lilley, K.S.; Dupree, P. Putative glycosyltransferases and other plant golgi apparatus proteins are revealed by lopit proteomics. Plant Physiol. 2012, 160, 1037–1051. [Google Scholar] [CrossRef] [PubMed]
- Nikolovski, N.; Shliaha, P.V.; Gatto, L.; Dupree, P.; Lilley, K.S. Label-free protein quantification for plant golgi protein localization and abundance. Plant Physiol. 2014, 166, 1033–1043. [Google Scholar] [CrossRef] [PubMed]
- Parsons, H.T.; Christiansen, K.; Knierim, B.; Carroll, A.; Ito, J.; Batth, T.S.; Smith-Moritz, A.M.; Morrison, S.; McInerney, P.; Hadi, M.Z.; et al. Isolation and proteomic characterization of the Arabidopsis golgi defines functional and novel components involved in plant cell wall biosynthesis. Plant Physiol. 2012, 159, 12–26. [Google Scholar] [CrossRef] [PubMed]
- Heard, W.; Sklenář, J.; Tomé, D.F.A.; Robatzek, S.; Jones, A.M.E. Identification of regulatory and cargo proteins of endosomal and secretory pathways in Arabidopsis thaliana by proteomic dissection. Mol. Cell. Proteomics 2015, 14, 1796–1813. [Google Scholar] [CrossRef] [PubMed]
- Asakura, T.; Hirose, S.; Katamine, H.; Kitajima, A.; Hori, H.; Sato, M.H.; Fujiwara, M.; Shimamoto, K.; Mitsui, T. Isolation and proteomic analysis of rice golgi membranes: Cis-golgi membranes labeled with GFP-SYP31. Plant Biotechnol. 2006, 23, 475–485. [Google Scholar] [CrossRef]
- Tanaka, N.; Fujita, M.; Handa, H.; Murayama, S.; Uemura, M.; Kawamura, Y.; Mitsui, T.; Mikami, S.; Tozawa, Y.; Yoshinaga, T.; et al. Proteomics of the rice cell: Systematic identification of the protein populations in subcellular compartments. Mol. Genet. Genomics 2004, 271, 566–576. [Google Scholar] [CrossRef] [PubMed]
- Parsons, H.T.; Weinberg, C.S.; Macdonald, L.J.; Adams, P.D.; Petzold, C.J.; Strabala, T.J.; Wagner, A.; Heazlewood, J.L. Golgi enrichment and proteomic analysis of developing Pinus radiata xylem by free-flow electrophoresis. PLoS ONE 2013, 8, e84669. [Google Scholar] [CrossRef] [PubMed]
- Mast, S.; Peng, L.; Jordan, T.W.; Flint, H.; Phillips, L.; Donaldson, L.; Strabala, T.J.; Wagner, A. Proteomic analysis of membrane preparations from developing Pinus radiata compression wood. Tree Physiol. 2010, 30, 1456–1468. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.M.; Stone, B.A. Chemical composition of the cell walls of lolium multiflorum endosperm. Phytochemistry 1973, 12, 1361–1367. [Google Scholar] [CrossRef]
- Smith, M.M.; Stone, B.A. Studies on lolium multiflorum endosperm in tissue-culture. Aust. J. Biol. Sci. 1973, 26, 123–133. [Google Scholar]
- Wilson, S.M.; Ho, Y.Y.; Lampugnani, E.R.; van de Meene, A.M.L.; Bain, M.P.; Bacic, A.; Doblin, M.S. Determining the subcellular location of synthesis and assembly of the cell wall polysaccharide (1,3; 1,4)-β-d-glucan in grasses. Plant Cell 2015, 27, 754–771. [Google Scholar] [CrossRef] [PubMed]
- Briskin, D.P.; Leonard, R.T.; Hodges, T.K. Isolation of plasma membrane: Membrane markers and general principles. Methods Enzymol. 1987, 148, 542–558. [Google Scholar]
- Natera, S.H.A.; Ford, K.L.; Cassin, A.M.; Patterson, J.H.; Newbigin, E.J.; Bacic, A. Analysis of the oryza sativa plasma membrane proteome using combined protein and peptide fractionation approaches in conjunction with mass spectrometry. J. Proteome Res. 2008, 7, 1159–1187. [Google Scholar] [CrossRef] [PubMed]
- Ford, K.L.; Cassin, A.; Bacic, A. Quantitative proteomic analysis of wheat cultivars with differing drought stress tolerance. Front. Plant Sci. 2011, 2, 44. [Google Scholar] [CrossRef] [PubMed]
- Byrne, S.L.; Nagy, I.; Pfeifer, M.; Armstead, I.; Swain, S.; Studer, B.; Mayer, K.; Campbell, J.D.; Czaban, A.; Hentrup, S.; et al. A synteny-based draft genome sequence of the forage grass lolium perenne. Plant J. 2015, 84, 816–826. [Google Scholar] [CrossRef] [PubMed]
- Tuskan, G.A.; Difazio, S.; Jansson, S.; Bohlmann, J.; Grigoriev, I.; Hellsten, U.; Putnam, N.; Ralph, S.; Rombauts, S.; Salamov, A.; et al. The genome of black cottonwood, Populus trichocarpa (torr. & gray). Science 2006, 313, 1596–1604. [Google Scholar] [PubMed]
- Berthold, M.R.; Cebron, N.; Dill, F.; Gabriel, T.R.; Kötter, T.; Meinl, T.; Ohl, P.; Sieb, C.; Thiel, K.; Wiswedel, B. Knime: The konstanz information miner. In Data Analysis, Machine Learning and Applications; Preisach, C., Burkhardt, H., Schmidt-Thieme, L., Decker, R., Eds.; Springer Berlin Heidelberg: Berlin, Heidelberg, Germany, 2008; pp. 319–326. [Google Scholar]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis information resource (tair): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef] [PubMed]
- Consortium, T.U. Uniprot: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef] [PubMed]
- Tanz, S.K.; Castleden, I.; Hooper, C.M.; Vacher, M.; Small, I.; Millar, H.A. Suba3: A database for integrating experimentation and prediction to define the subcellular location of proteins in Arabidopsis. Nucleic Acids Res. 2013, 41, D1185–D1191. [Google Scholar] [CrossRef] [PubMed]
- Lombard, V.; Golaconda Ramulu, H.; Drula, E.; Coutinho, P.M.; Henrissat, B. The carbohydrate-active enzymes database (cazy) in 2013. Nucleic Acids Res. 2014, 42, D490–D495. [Google Scholar] [CrossRef] [PubMed]
- Petersen, T.N.; Brunak, S.; von Heijne, G.; Nielsen, H. Signalp 4.0: Discriminating signal peptides from transmembrane regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Drakakaki, G.; van de Ven, W.; Pan, S.; Miao, Y.; Wang, J.; Keinath, N.F.; Weatherly, B.; Jiang, L.; Schumacher, K.; Hicks, G.; et al. Isolation and proteomic analysis of the syp61 compartment reveal its role in exocytic trafficking in Arabidopsis. Cell Res. 2012, 22, 413–424. [Google Scholar] [CrossRef] [PubMed]
- Burton, R.A.; Wilson, S.M.; Hrmova, M.; Harvey, A.J.; Shirley, N.J.; Medhurst, A.; Stone, B.A.; Newbigin, E.J.; Bacic, A.; Fincher, G.B. Cellulose synthase-like cslf genes mediate the synthesis of cell wall (1,3;1,4)-β-d-glucans. Science 2006, 311, 1940–1942. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Szumlanski, A.L.; Gu, F.; Guo, F.; Nielsen, E. A role for CSLD3 during cell-wall synthesis in apical plasma membranes of tip-growing root-hair cells. Nat. Cell Biol. 2011, 13, 973–980. [Google Scholar] [CrossRef] [PubMed]
- Yin, L.; Verhertbruggen, Y.; Oikawa, A.; Manisseri, C.; Knierim, B.; Prak, L.; Jensen, J.K.; Knox, J.P.; Auer, M.; Willats, W.G.; et al. The cooperative activities of CSLD2, CSLD3, and CSLD5 are required for normal Arabidopsis development. Mol. Plant 2011, 4, 1024–1037. [Google Scholar] [CrossRef] [PubMed]
- Ellinger, D.; Voigt, C.A. Callose biosynthesis in Arabidopsis with a focus on pathogen response: What we have learned within the last decade. Ann. Bot. 2014, 114, 1349–1358. [Google Scholar] [CrossRef] [PubMed]
- Zeng, W.; Jiang, N.; Nadella, R.; Killen, T.L.; Nadella, V.; Faik, A. A glucurono(arabino)xylan synthase complex from wheat contains members of the GT43, GT47, and GT75 families and functions cooperatively. Plant Physiol. 2010, 154, 78–97. [Google Scholar] [CrossRef] [PubMed]
- Anders, N.; Wilkinson, M.D.; Lovegrove, A.; Freeman, J.; Tryfona, T.; Pellny, T.K.; Weimar, T.; Mortimer, J.C.; Stott, K.; Baker, J.M.; et al. Glycosyl transferases in family 61 mediate arabinofuranosyl transfer onto xylan in grasses. Proc. Natl. Acad. Sci. USA 2012, 109, 989–993. [Google Scholar] [CrossRef] [PubMed]
- Dumont, M.; Lehner, A.; Bouton, S.; Kiefer-Meyer, M.C.; Voxeur, A.; Pelloux, J.; Lerouge, P.; Mollet, J.C. The cell wall pectic polymer rhamnogalacturonan-II is required for proper pollen tube elongation: Implications of a putative sialyltransferase-like protein. Ann. Bot. 2014, 114, 1177–1188. [Google Scholar] [CrossRef] [PubMed]
- Lerouxel, O.; Mouille, G.; Andeme-Onzighi, C.; Bruyant, M.P.; Seveno, M.; Loutelier-Bourhis, C.; Driouich, A.; Hofte, H.; Lerouge, P. Mutants in defective glycosylation, an Arabidopsis homolog of an oligosaccharyltransferase complex subunit, show protein underglycosylation and defects in cell differentiation and growth. Plant J. 2005, 42, 455–468. [Google Scholar] [CrossRef] [PubMed]
- Lunn, J.E.; Delorge, I.; Figueroa, C.M.; van Dijck, P.; Stitt, M. Trehalose metabolism in plants. Plant J. 2014, 79, 544–567. [Google Scholar] [CrossRef] [PubMed]
- Woo, C.H.; Gao, C.; Yu, P.; Tu, L.; Meng, Z.; Banfield, D.K.; Yao, X.; Jiang, L. Conserved function of the lysine-based KXD/E motif in golgi retention for endomembrane proteins among different organisms. Mol. Biol. Cell 2015, 26, 4280–4293. [Google Scholar] [CrossRef] [PubMed]
- Dell, A.; Galadari, A.; Sastre, F.; Hitchen, P. Similarities and differences in the glycosylation mechanisms in prokaryotes and eukaryotes. Int. J. Microbiol. 2010, 2010, 148178. [Google Scholar] [CrossRef] [PubMed]
- Ow, S.Y.; Salim, M.; Noirel, J.; Evans, C.; Rehman, I.; Wright, P.C. Itraq underestimation in simple and complex mixtures: “The good, the bad and the ugly”. J. Proteome Res. 2009, 8, 5347–5355. [Google Scholar] [CrossRef] [PubMed]
- Philippe, S.; Saulnier, L.; Guillon, F. Arabinoxylan and (1→3),(1→4)-β-glucan deposition in cell walls during wheat endosperm development. Planta 2006, 224, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.F.; Staehelin, L.A. Functional compartmentation of the golgi apparatus of plant cells: Immunocytochemical analysis of high-pressure frozen- and freeze-substituted sycamore maple suspension culture cells. Plant Physiol. 1992, 99, 1070–1083. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Marker Activity | Membrane | MM | S1 | S2 | S3 |
|---|---|---|---|---|---|
| IDPase (nmol/min/mg) | GA | 1.04 (0.02) | 1.70 (0.17) | 1.22 (0.18) | 0.47 (0.01) |
| Cyto.-c reductase (nmol/min/mg) | ER | 280 (10) | 200 (20) | 240 (10) | 340 (20) |
| Nitrate sensitive ATPase (nmol/min/mg) | Vacuole | 66.24 (3.42) | 39.25 (3.85) | 137.26 (32.63) | 178.51 (0.01) |
| Vanadate sensitive ATPase (nmol/min/mg) | PM | 389.95 (13.51) | 188.65 (13.87) | 448.43 (2.32) | 681.63 (46.81) |
| Cyto.-c oxidase (nmol/min/mg) | MT | 2140 (1100) | 210 (50) | 720 (210) | 2560 (590) |
| Protein ID | TAIR10 ID | TAIR10 Description | Previous GA Proteomes | Function |
|---|---|---|---|---|
| LP_11479 | AT3G19820.1 | DWARF1 | [10,29] | Cell elongation |
| p6229 | AT4G12700.1 | Unknown, contains a O-FucT-like domain | Non-classified glycosyltransferase | |
| LP_2216 | AT3G62830.1 | UDP-glucuronic acid decarboxylase | [7,8,9,10] | Nucleotide sugar biosynthesis |
| p2877 | AT1G30620.1 | UDP-arabinose 4-epimerase 1 | [9] | Nucleotide sugar biosynthesis |
| p20671 | AT2G30500.1 | kinase interacting family protein | Signaling | |
| p4201 | AT3G04080.1 | apyrase 1 | [7,8,9,10] | Transport |
| p3712 | AT3G57330.1 | autoinhibited Ca2+-ATPase 11 | [9] | Transport |
| p45403 | AT1G04120.1 | multidrug resistance-associated protein 5 | Transport | |
| p42314 | AT2G25520.1 | putative sugar phosphate/phosphate translocator | [7,10] | Transport |
| p2354 | AT4G35300.1 | tonoplast monosaccharide transporter2 | [9] | Transport |
| LP_6010 | AT3G52850.1 | Vacuolar Sorting Receptor-1 (VSR-1) | [8,9,10] | Transport |
| p4069 | AT2G14740.1 | vaculolar sorting receptor 3 | [7,8,9] | Transport |
| p3235 | AT4G39080.1 | vacuolar proton ATPase A3 | [9,10,29] | Transport |
| p3662 | AT4G14240.1 | CBS domain-containing protein with a DUF21 | Unknown | |
| p5363 | AT5G10840.1 | Endomembrane protein 70 protein | [6,7,8,9,10,29] | Unknown |
| p5735 | AT5G35160.2 | Endomembrane protein 70 protein | [9] | Unknown |
| LP_598 | AT2G24170.1 | Endomembrane protein 70 protein | [9] | Unknown |
| p820 | AT3G13772.1 | transmembrane nine 7 | [6,7,8,9,10] | Unknown |
| p7705 | AT5G51570.1 | SPFH/Band 7/PHB domain-containing protein | [9,10] | Unknown |
| p2170 | AT5G16250.1 | unknown protein | Unknown | |
| p9698 | AT4G23790.1 | unknown protein | Unknown | |
| p50050 | predicted protein | Unknown |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ford, K.L.; Chin, T.; Srivastava, V.; Zeng, W.; Doblin, M.S.; Bulone, V.; Bacic, A. Comparative “Golgi” Proteome Study of Lolium multiflorum and Populus trichocarpa. Proteomes 2016, 4, 23. https://doi.org/10.3390/proteomes4030023
Ford KL, Chin T, Srivastava V, Zeng W, Doblin MS, Bulone V, Bacic A. Comparative “Golgi” Proteome Study of Lolium multiflorum and Populus trichocarpa. Proteomes. 2016; 4(3):23. https://doi.org/10.3390/proteomes4030023
Chicago/Turabian StyleFord, Kristina L., Tony Chin, Vaibhav Srivastava, Wei Zeng, Monika S. Doblin, Vincent Bulone, and Antony Bacic. 2016. "Comparative “Golgi” Proteome Study of Lolium multiflorum and Populus trichocarpa" Proteomes 4, no. 3: 23. https://doi.org/10.3390/proteomes4030023
APA StyleFord, K. L., Chin, T., Srivastava, V., Zeng, W., Doblin, M. S., Bulone, V., & Bacic, A. (2016). Comparative “Golgi” Proteome Study of Lolium multiflorum and Populus trichocarpa. Proteomes, 4(3), 23. https://doi.org/10.3390/proteomes4030023