
A Staphylococcus aureus Proteome Overview: Shared and Specific Proteins and Protein Complexes from Representative Strains of All Three Clades




Abstract
:1. Introduction
2. Materials and Methods
2.1. Genome-Based Comparisons
2.2. Modeling Complexes
2.3. Structure Annotation with AnDOM
2.4. Phylogenetic Analysis
3. Results
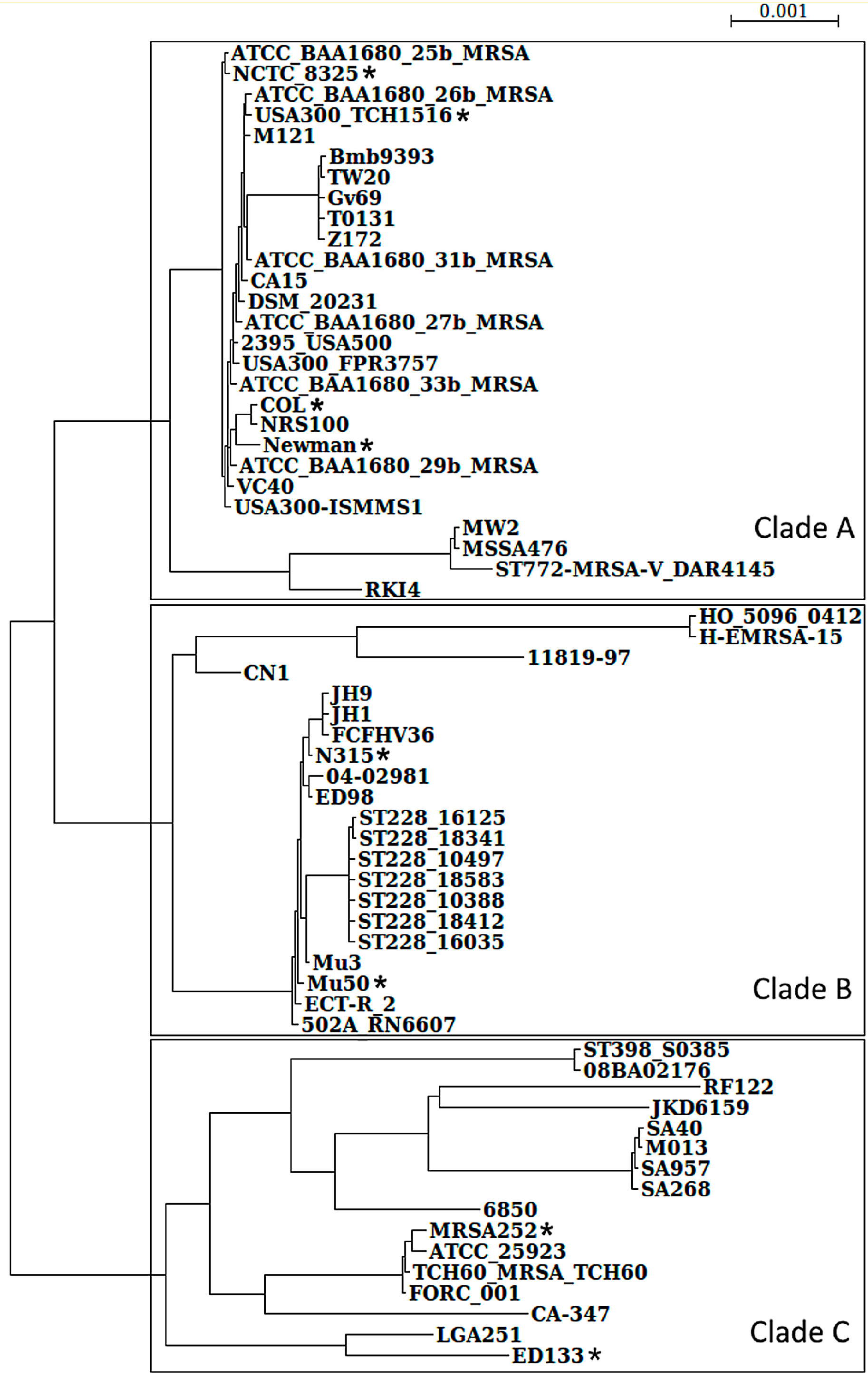
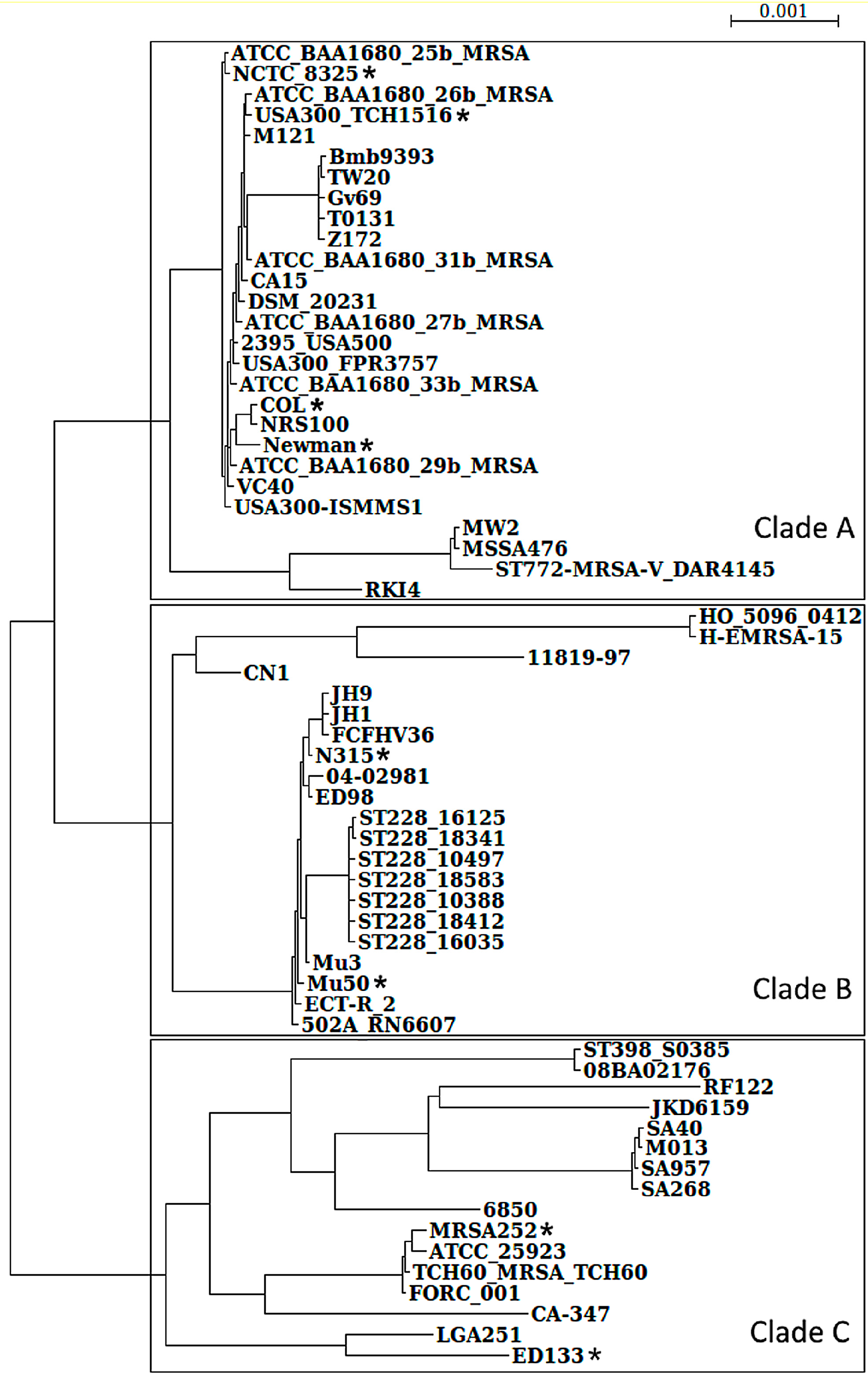
3.1. S. aureus Strains Form Three Clades
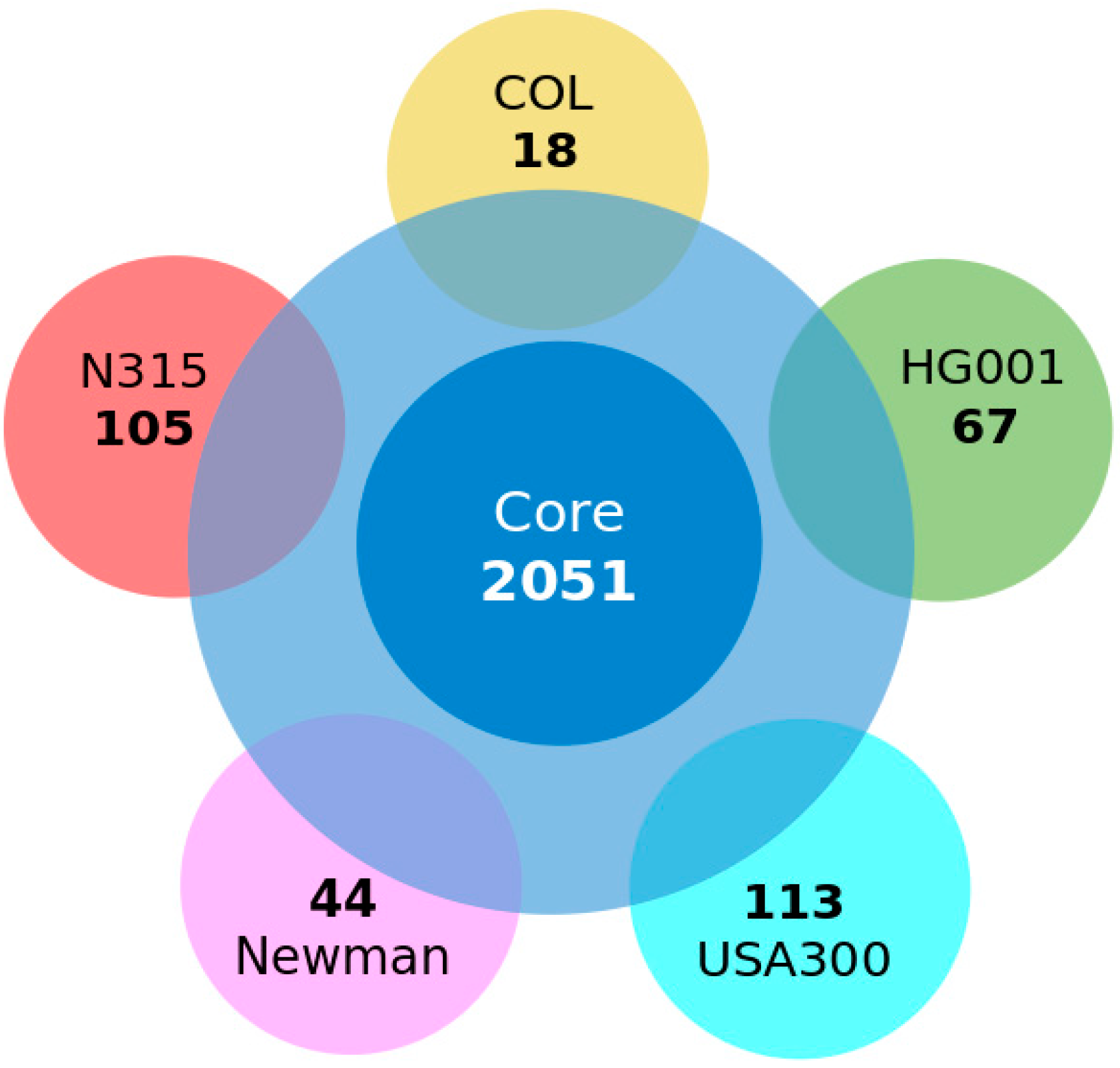
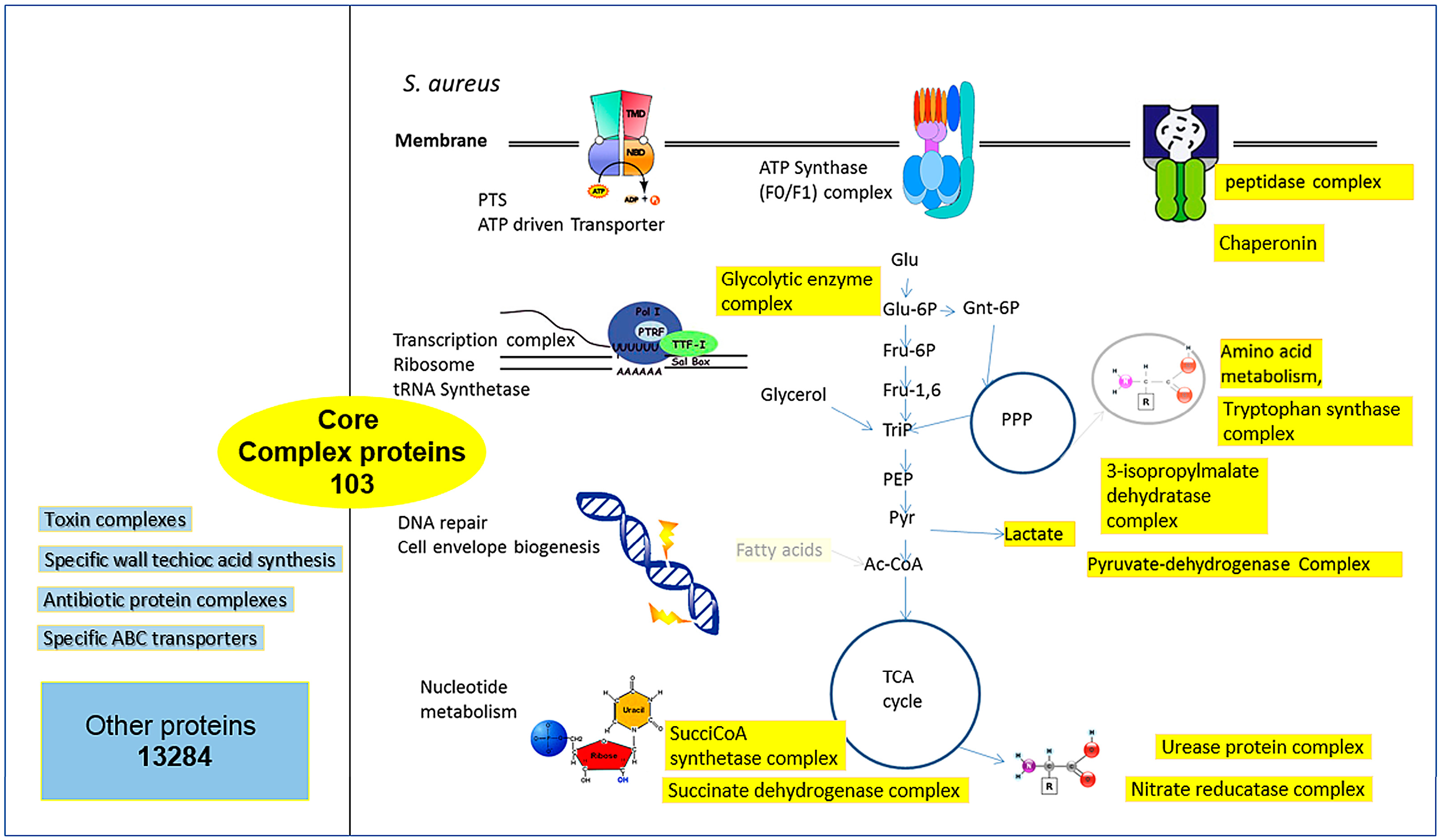
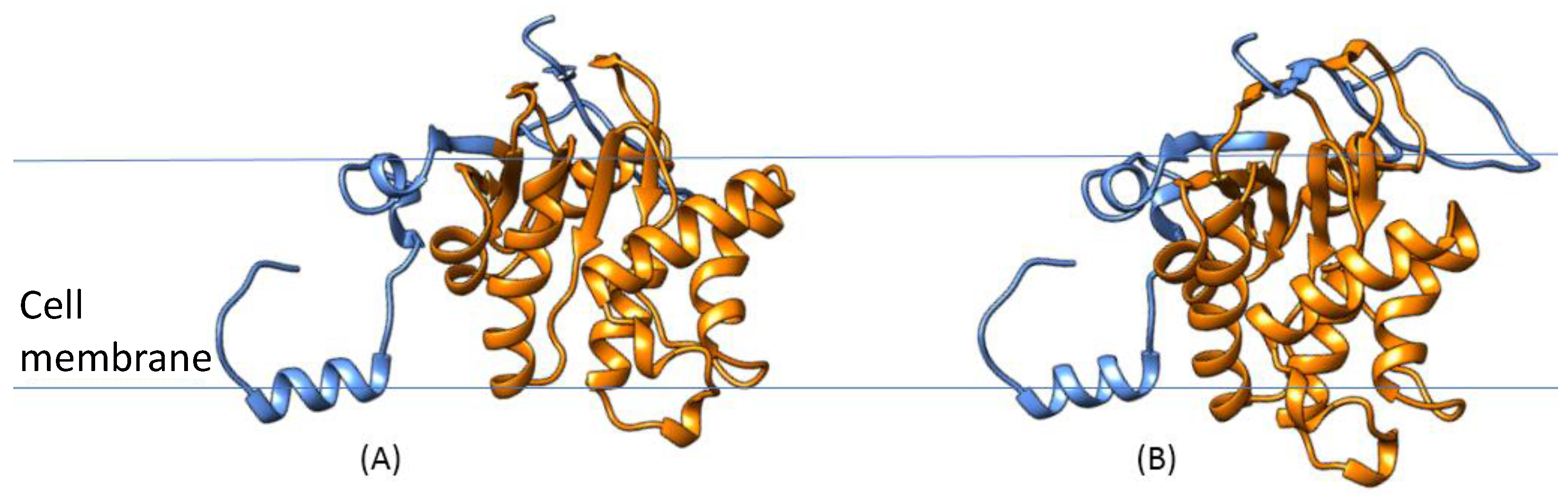
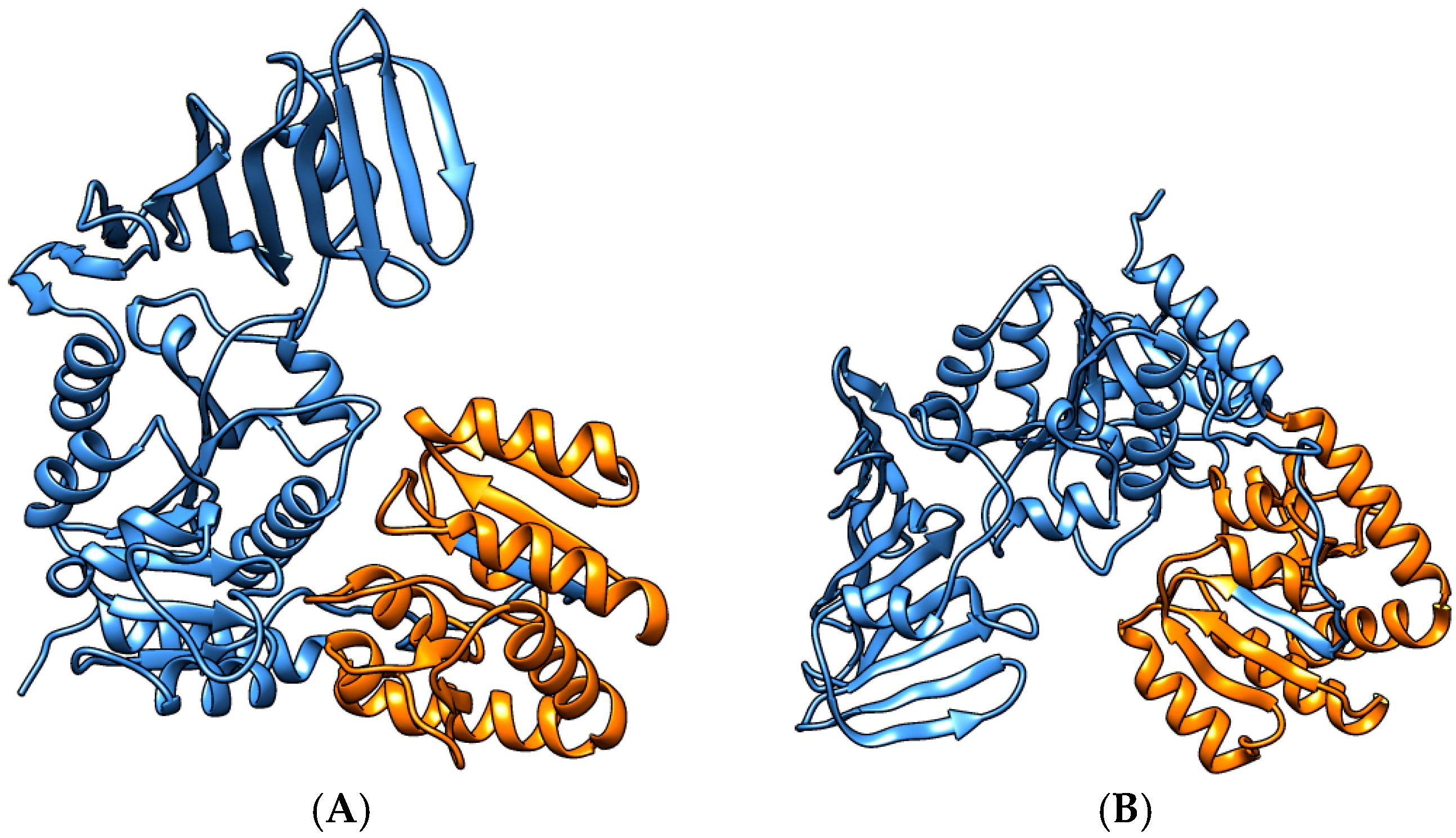
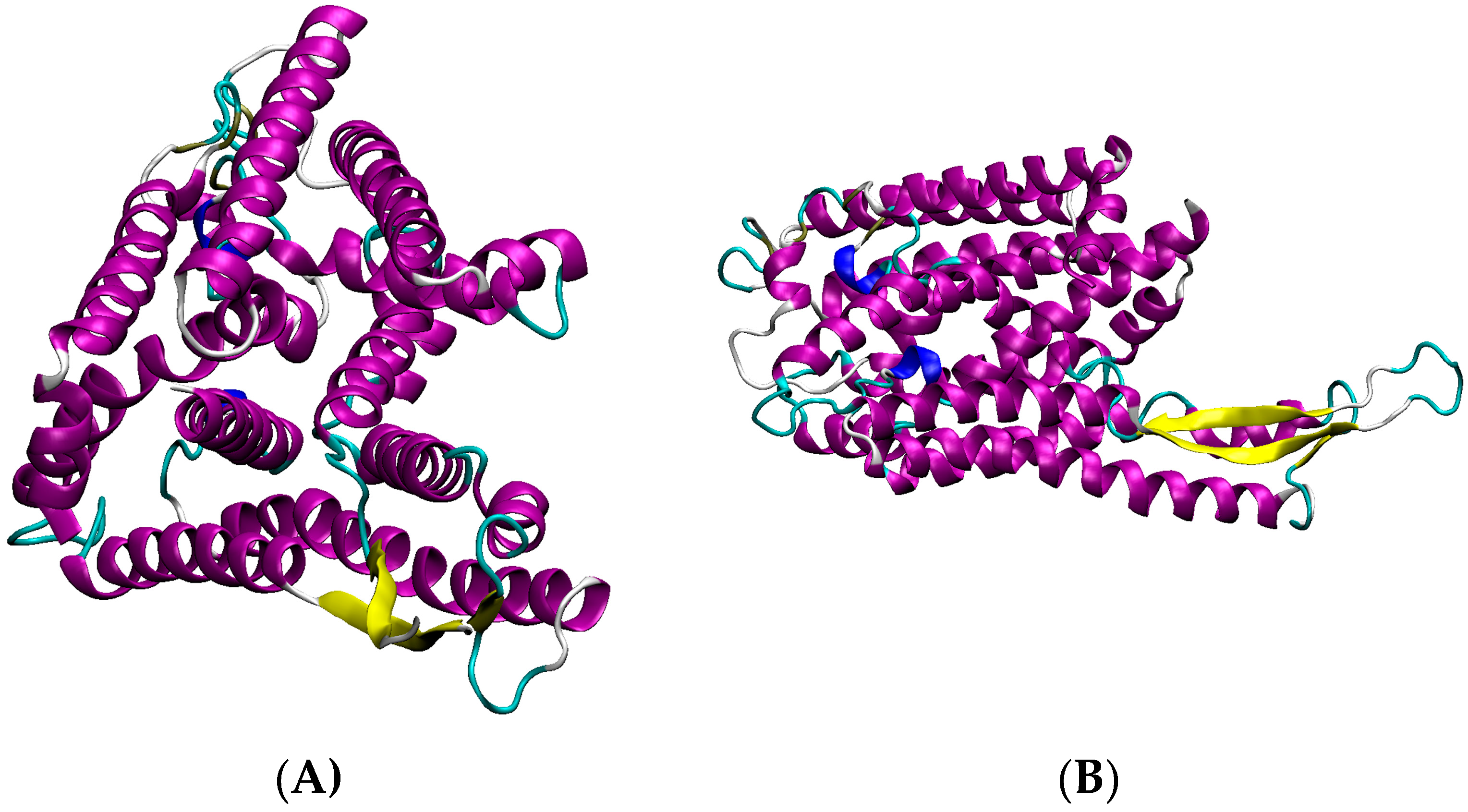
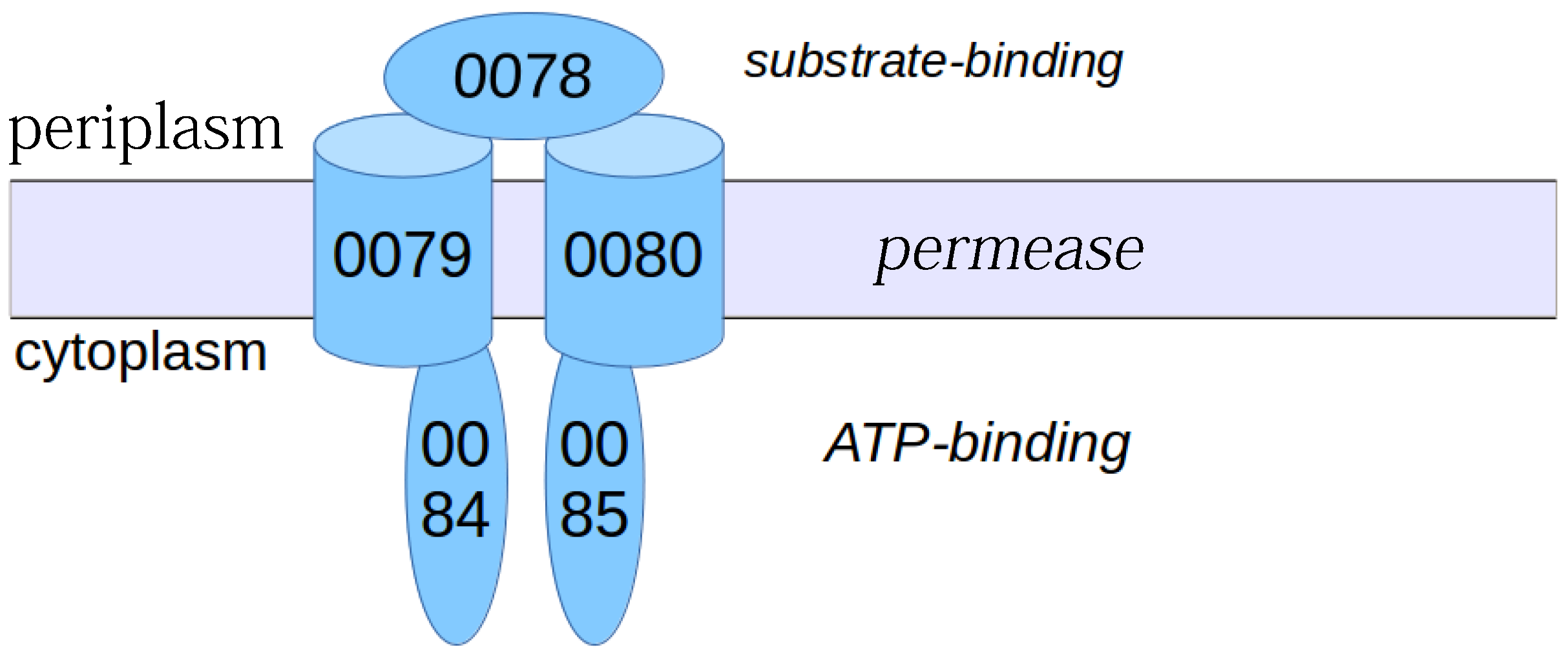
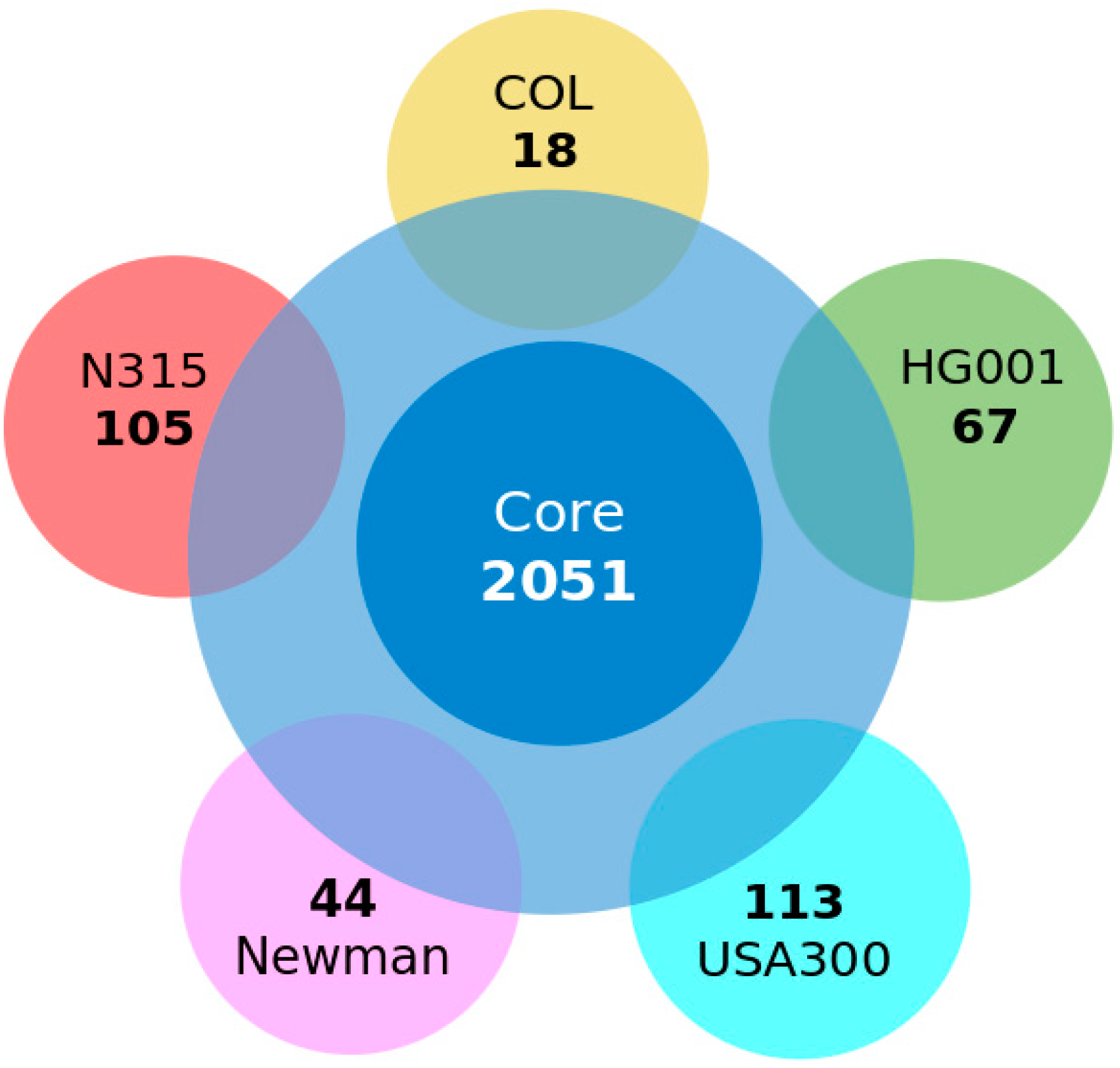
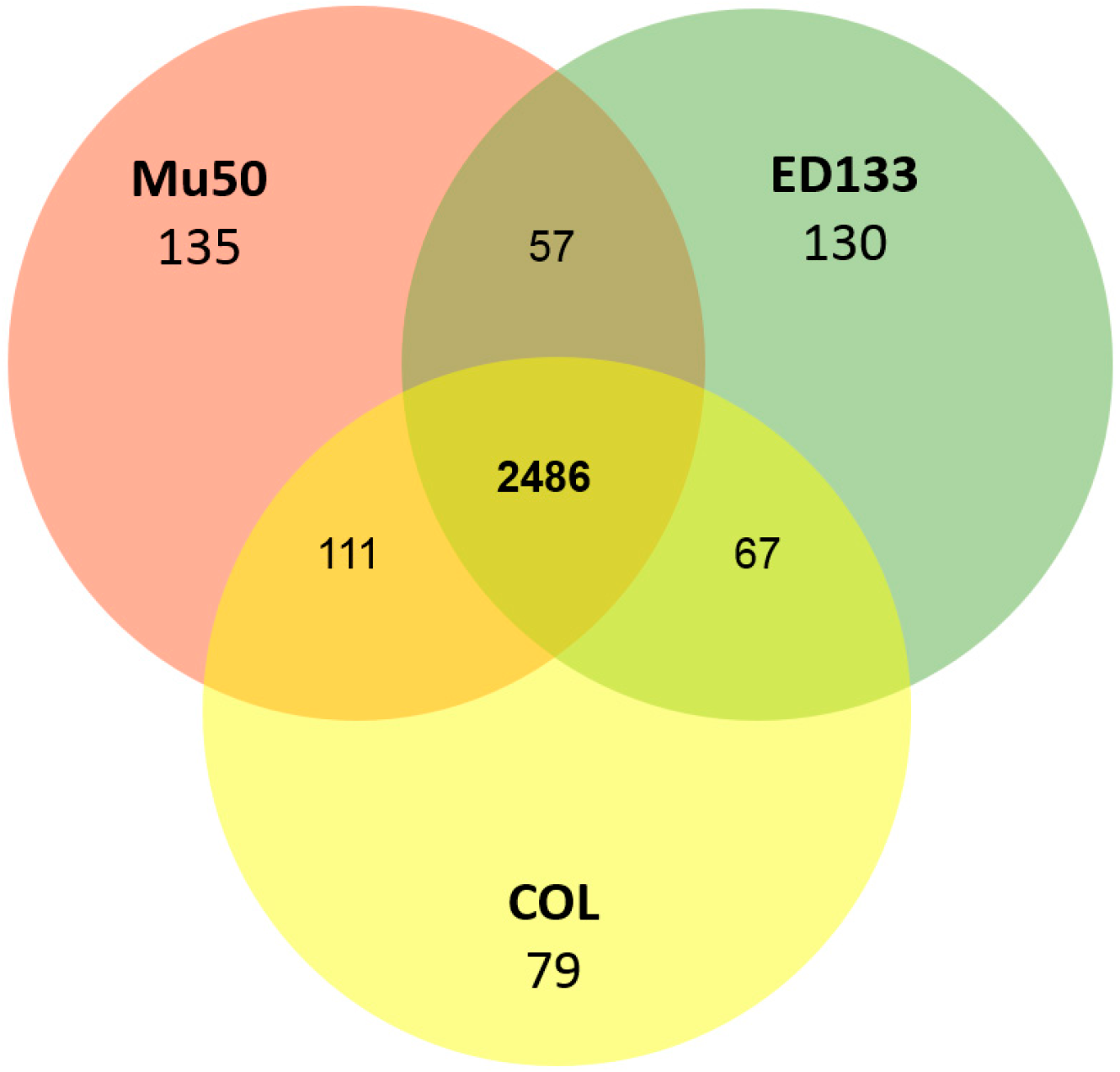
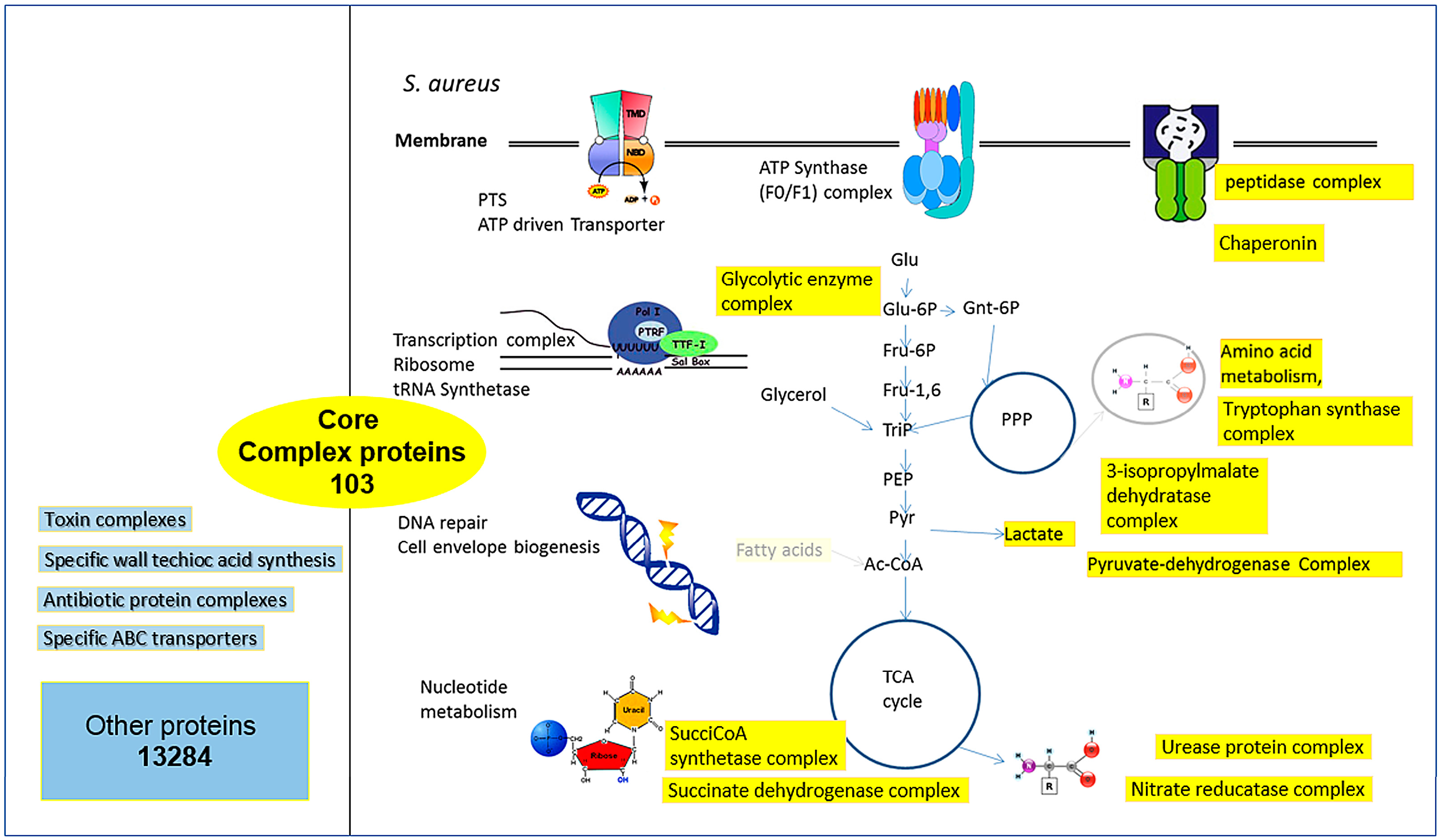
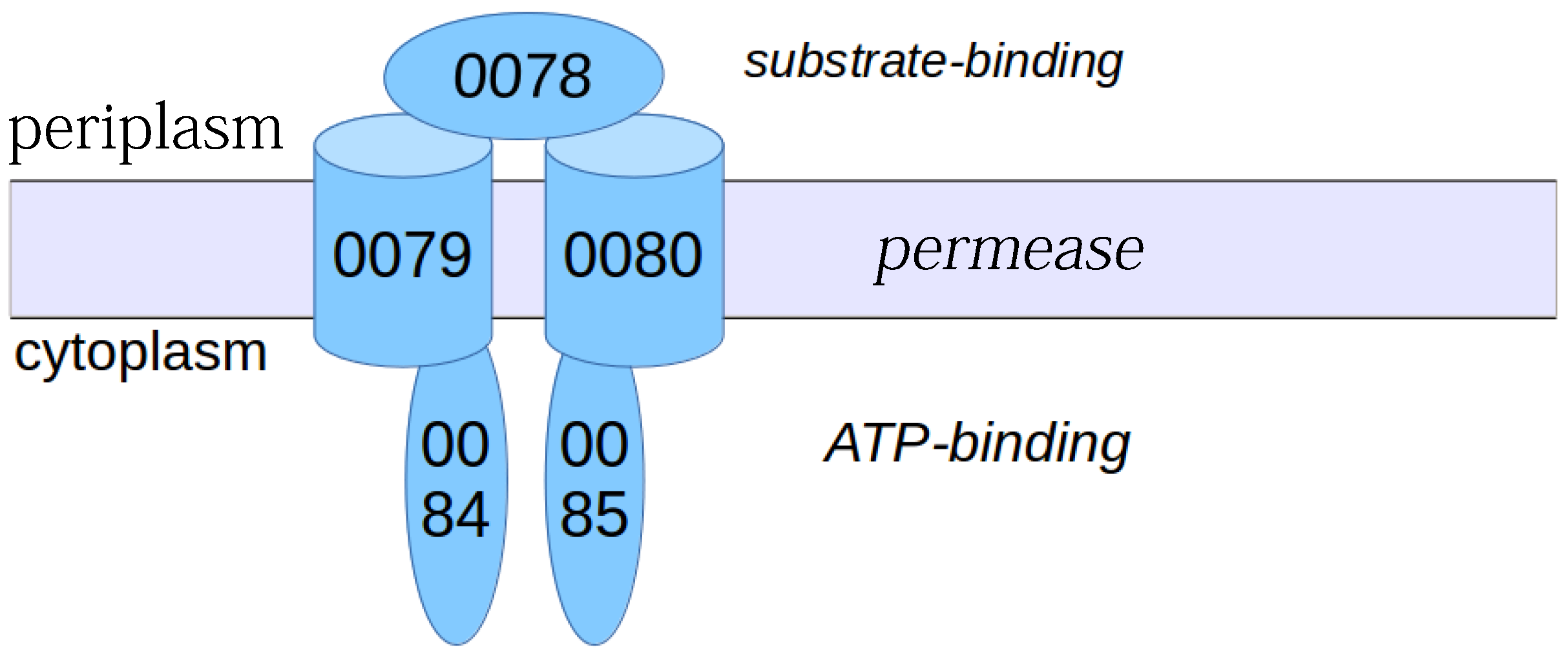
3.2. Conserved Protein Complexes and Strain-Specific Proteins
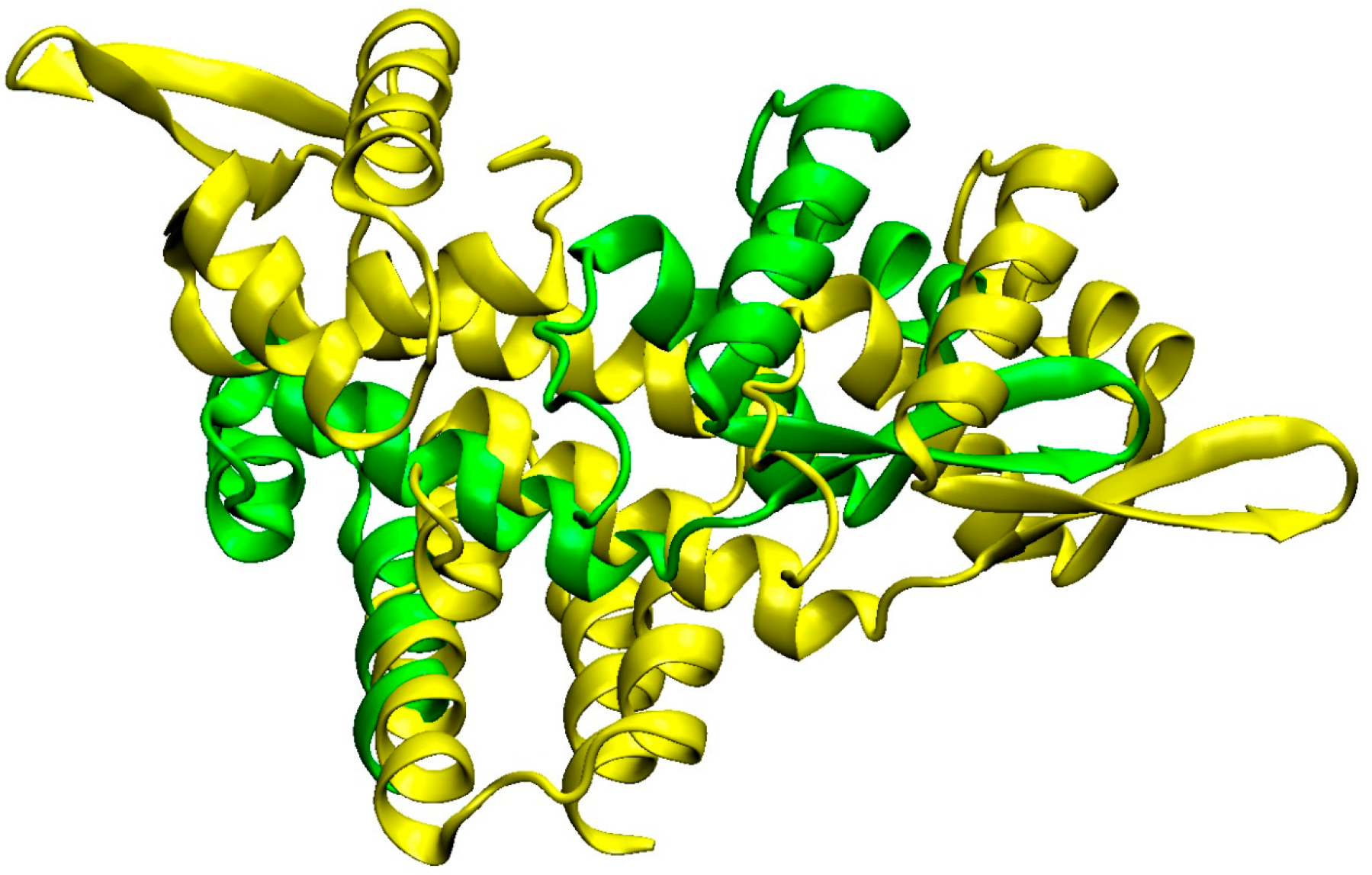
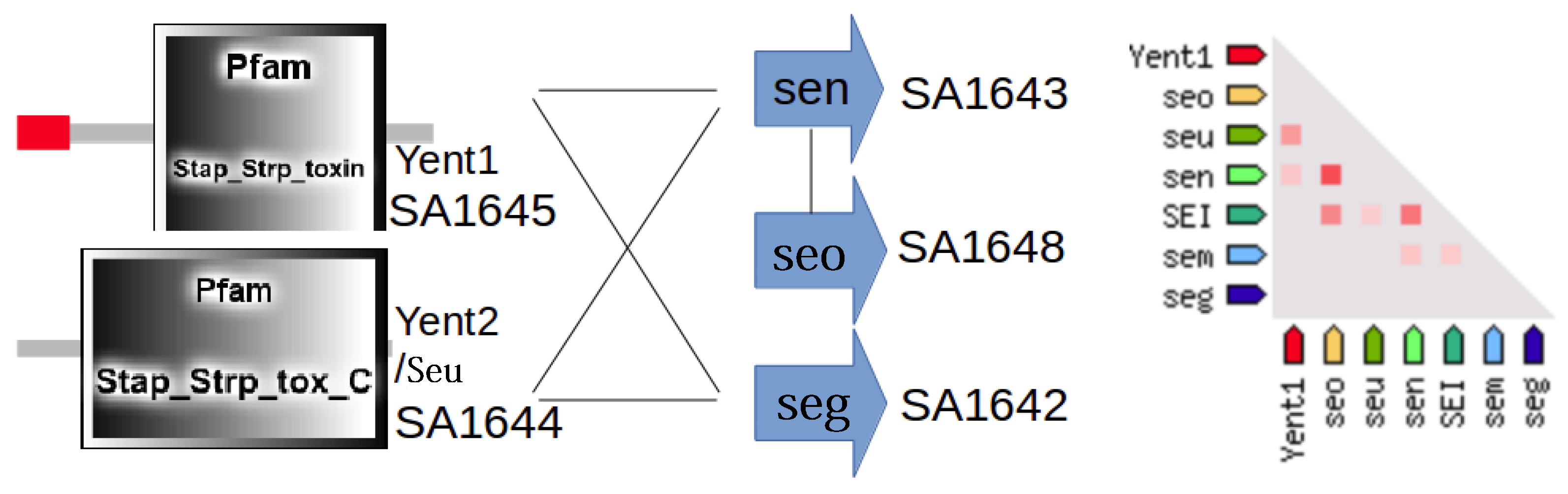
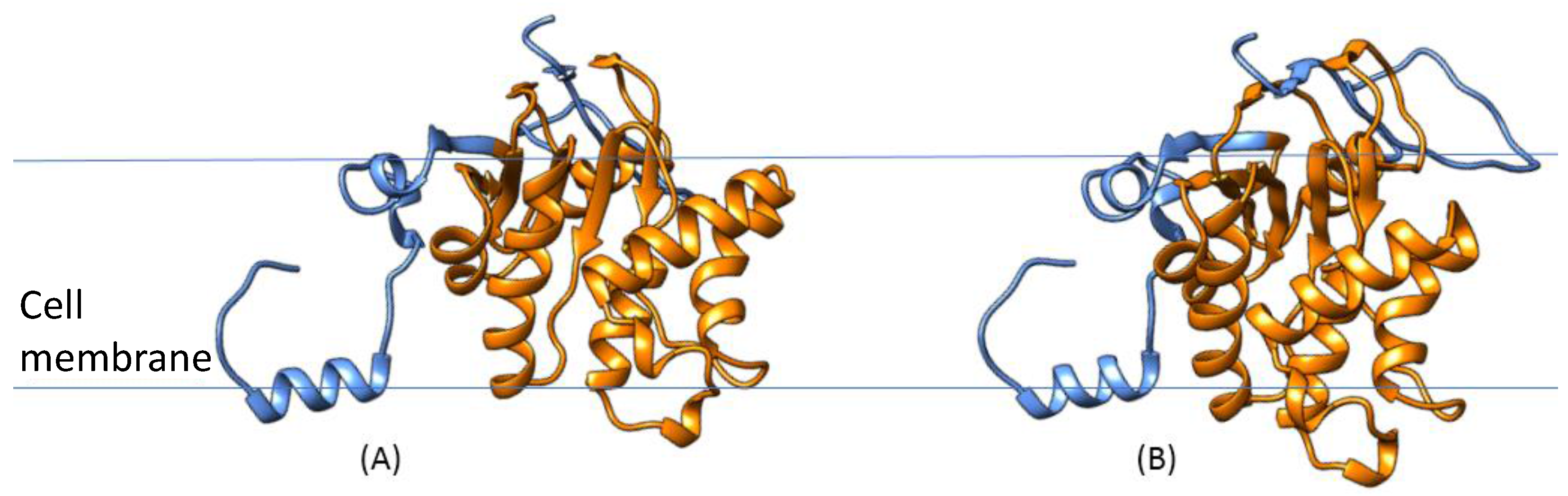
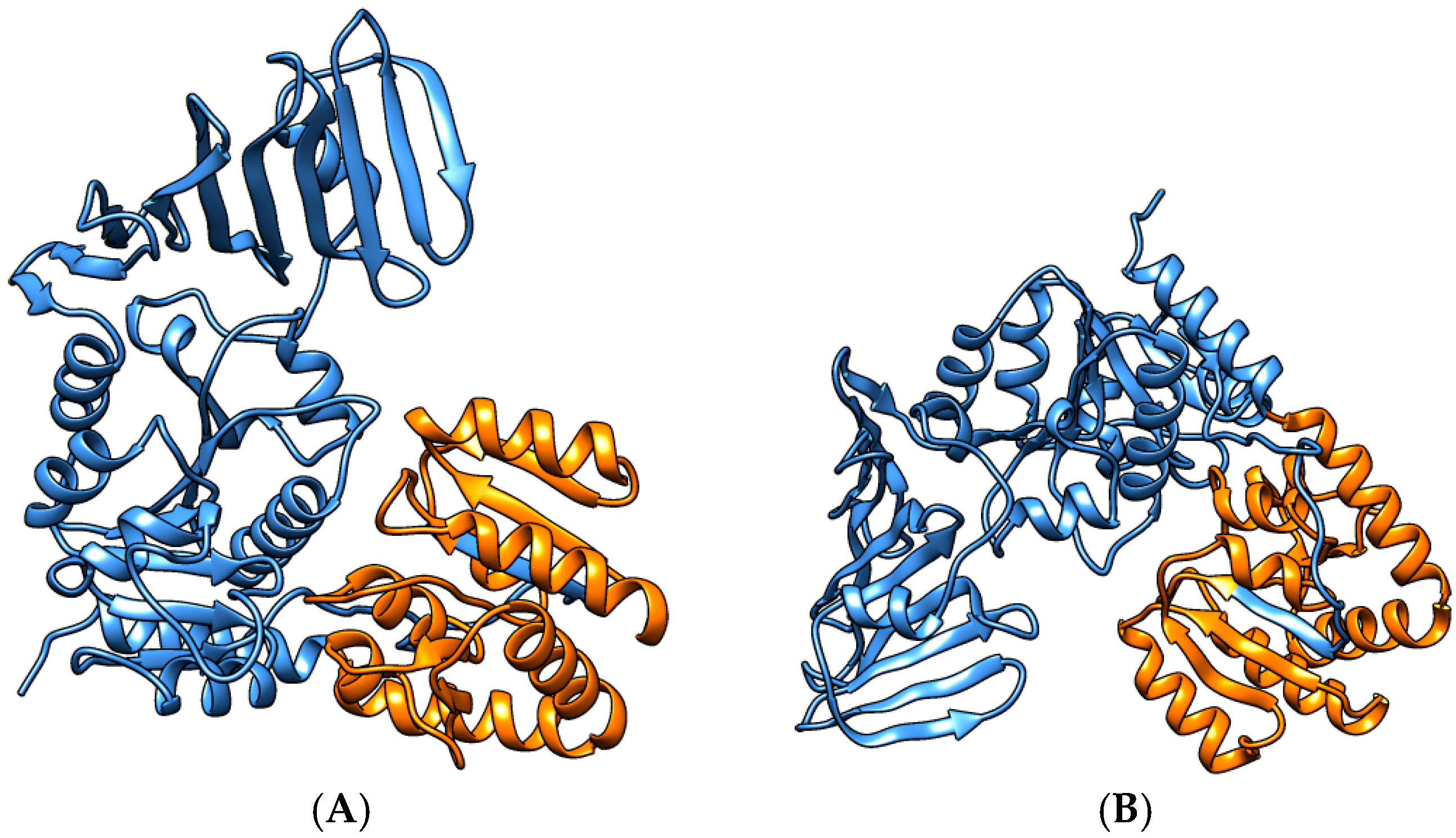
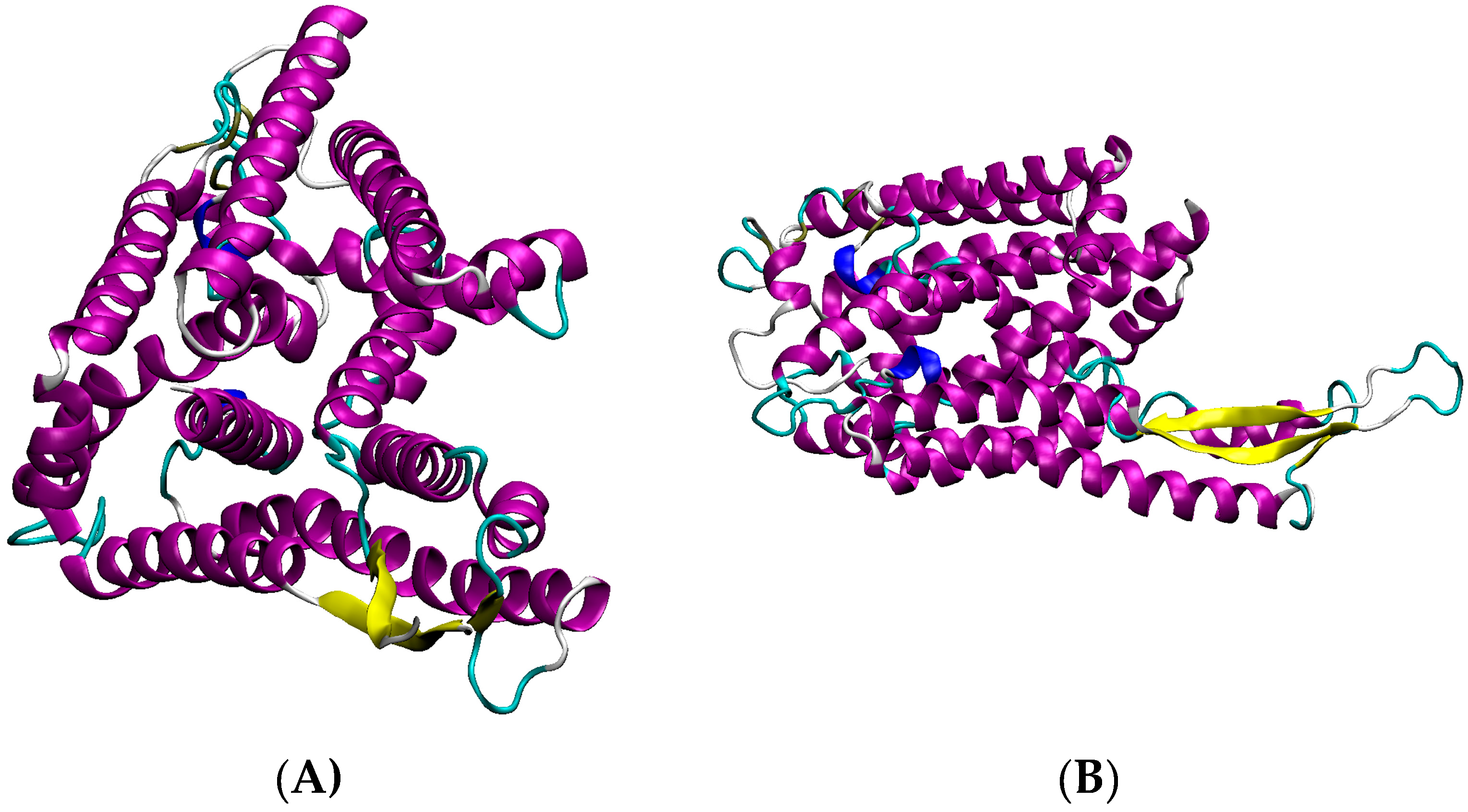
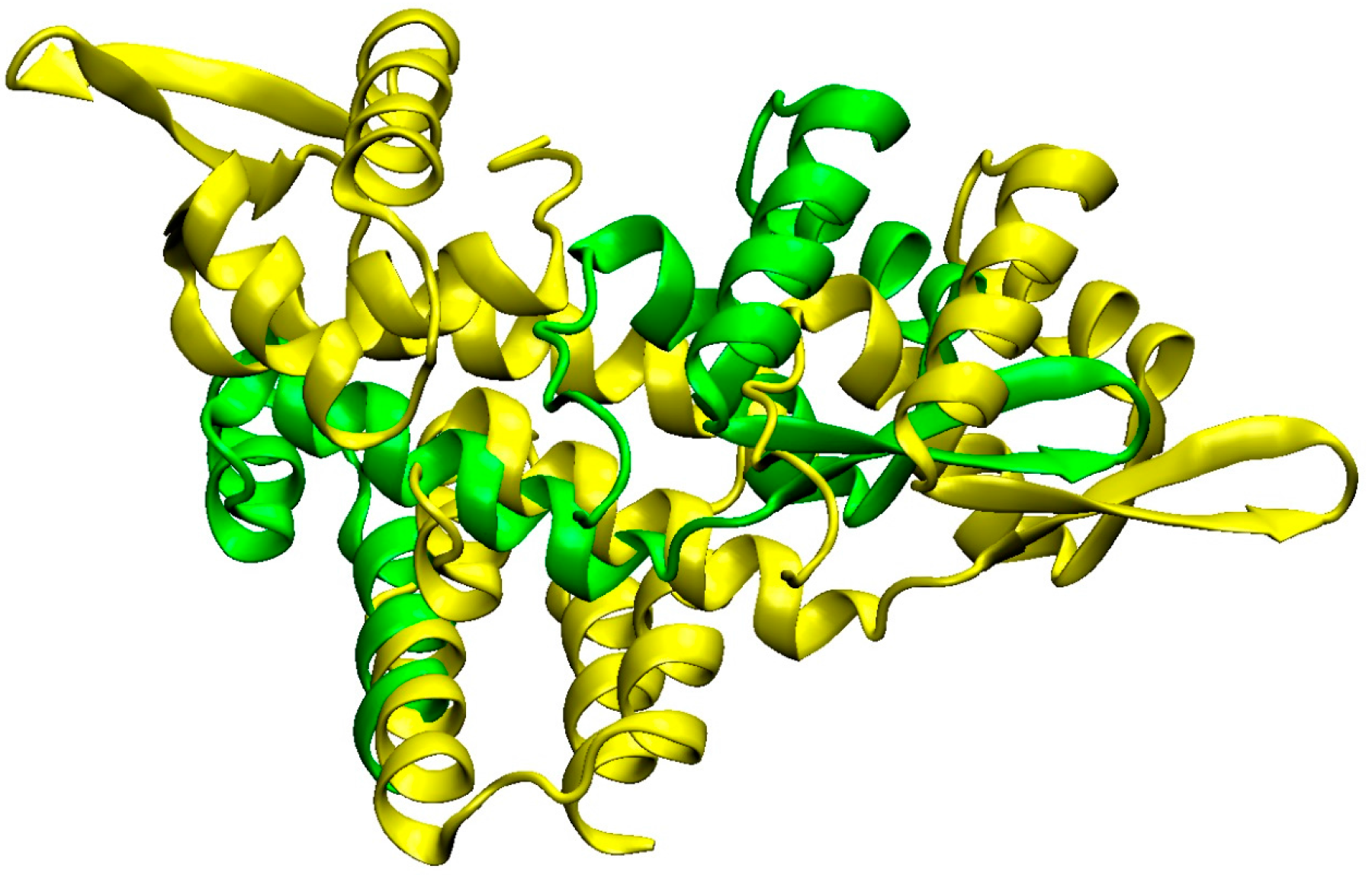
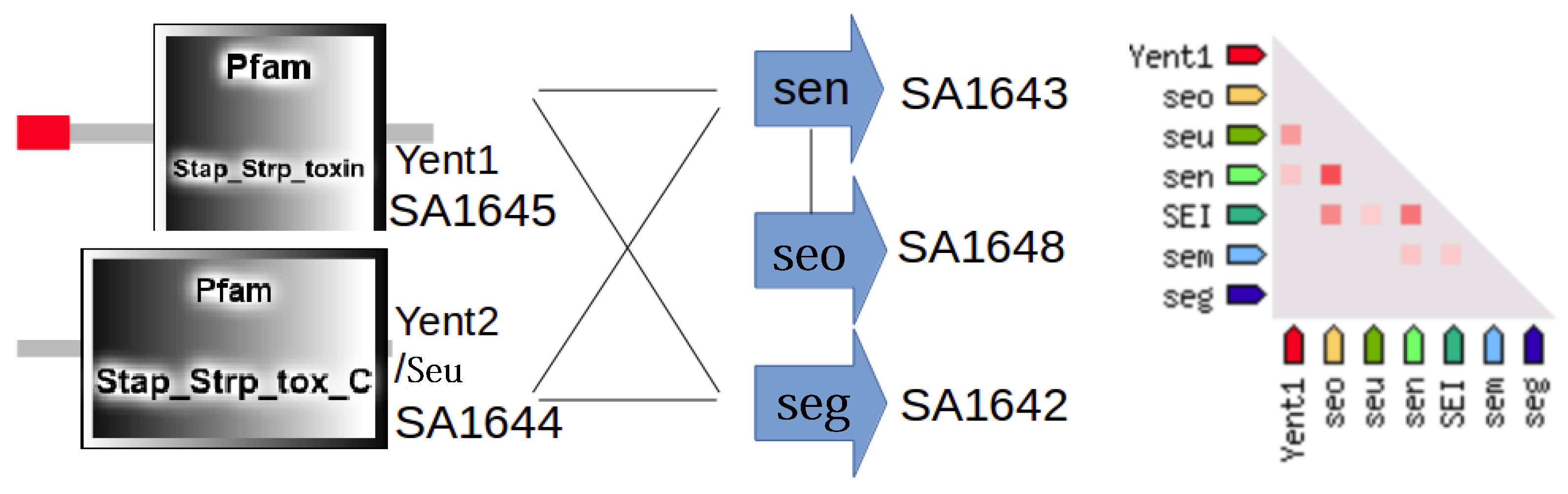
3.3. Detailed Analysis of S. aureus Strain-Specific Proteins
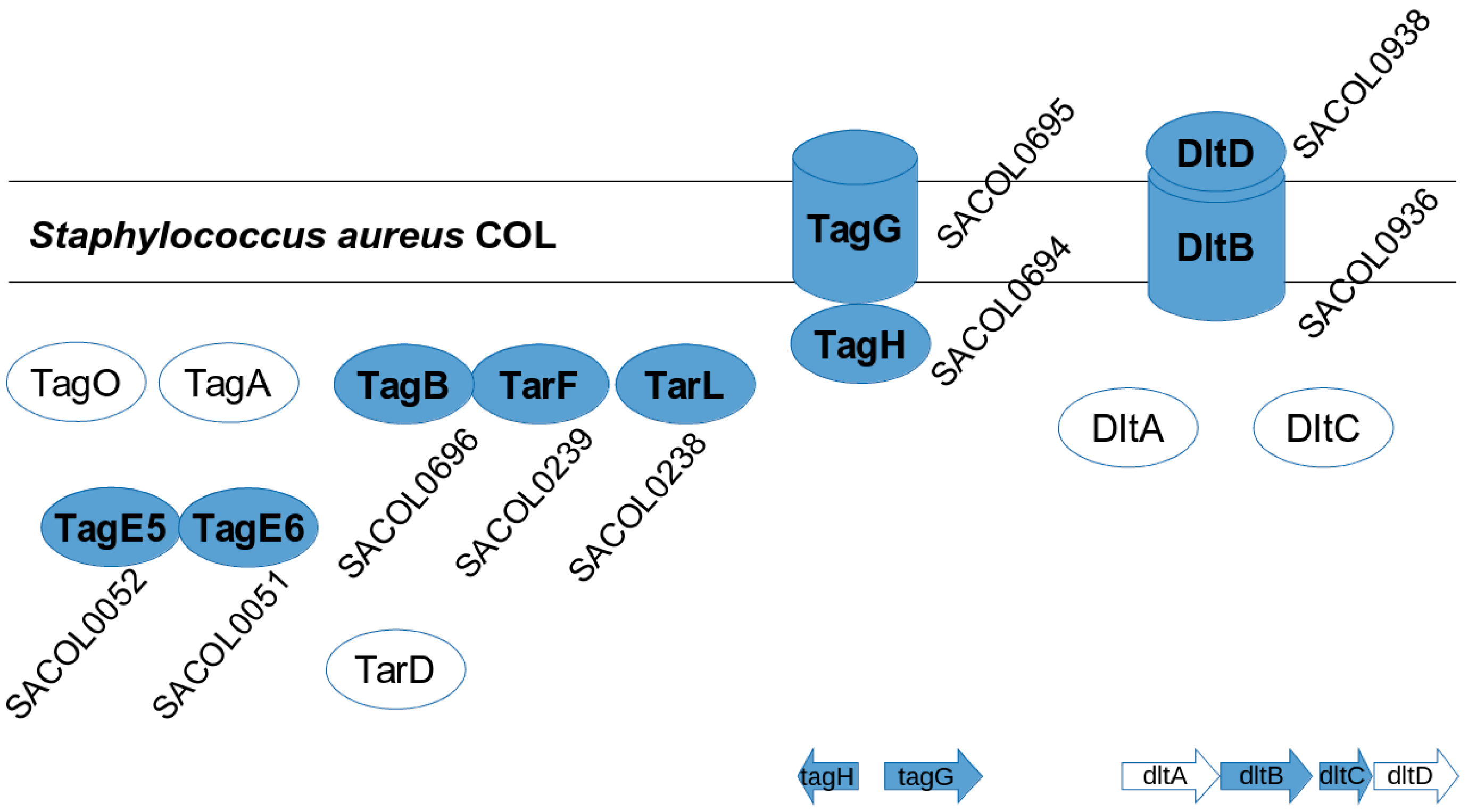
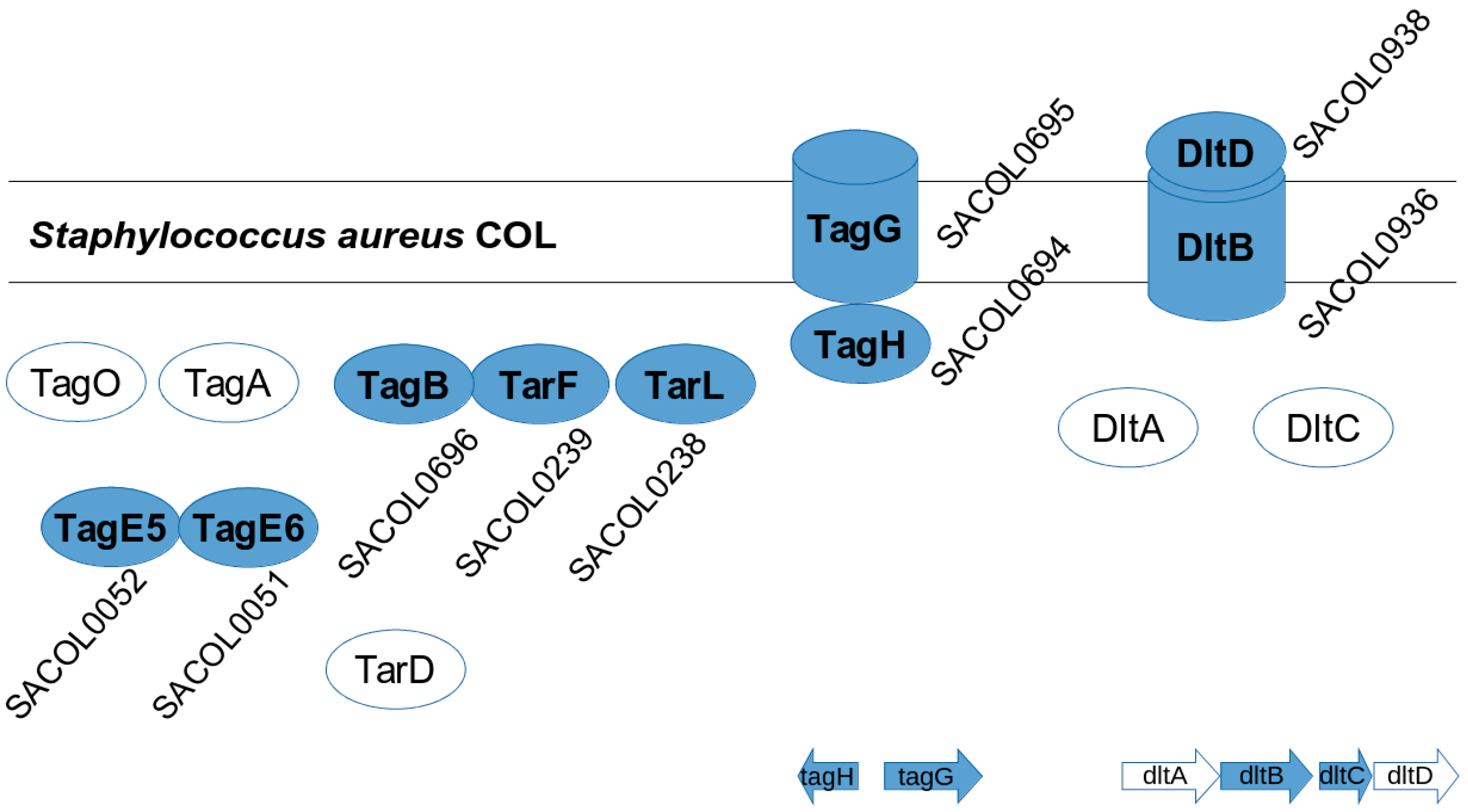
3.4. S. aureus COL Proteins
3.5. S. aureus N315 Proteins
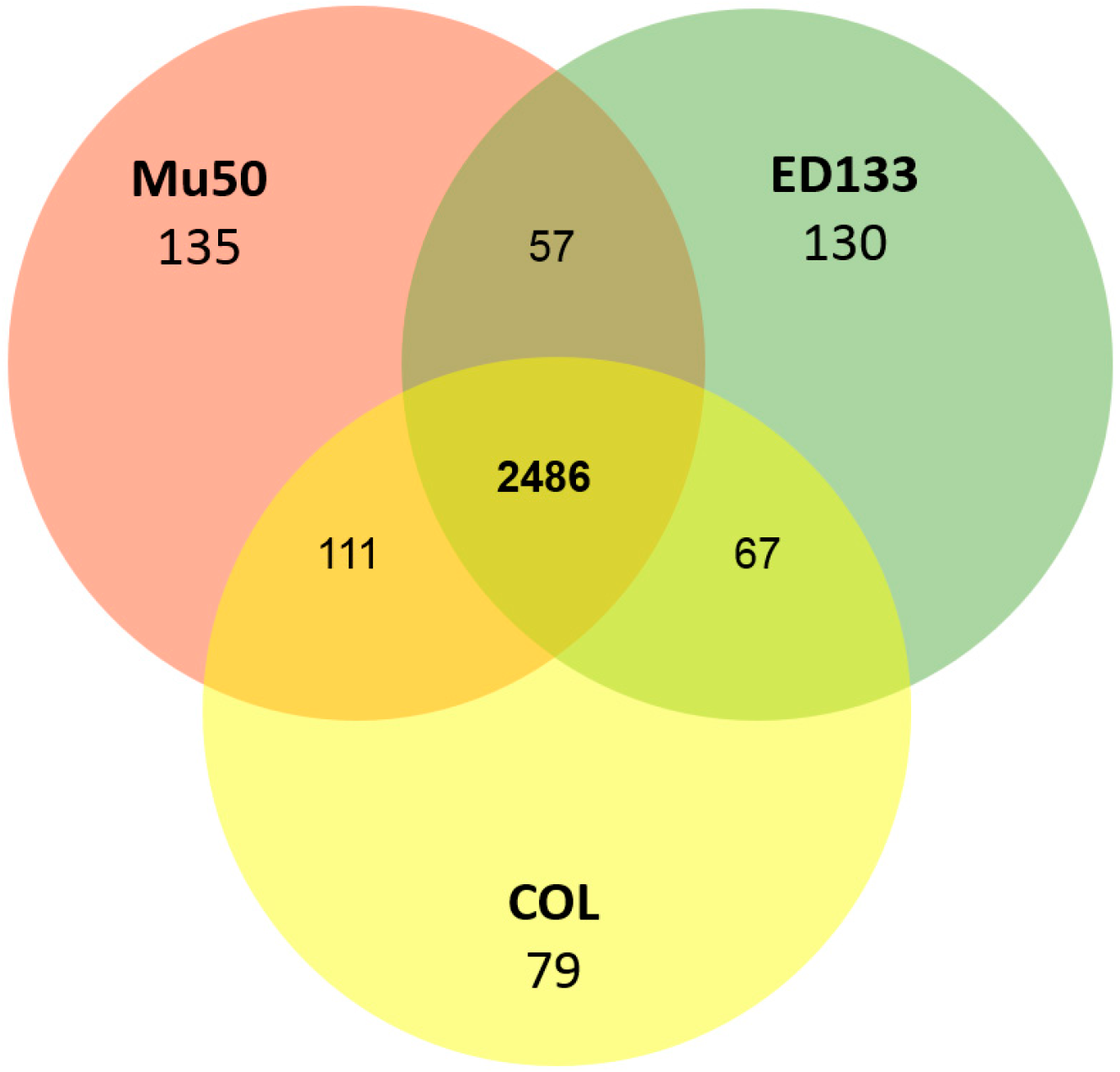
3.6. Other S. aureus Strains (Clade B, Clade C)
4. Discussion
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Liang, C.; Liebeke, M.; Schwarz, R.; Zühlke, D.; Fuchs, S.; Menschner, L.; Engelmann, S.; Wolz, C.; Jaglitz, S.; Bernhardt, J.; et al. Staphylococcus aureus physiological growth limitations: Insights from flux calculations built on proteomics and external metabolite data. Proteomics 2011, 11, 1915–1935. [Google Scholar] [CrossRef] [PubMed]
- Keseler, I.M.; Mackie, A.; Peralta-Gil, M.; Santos-Zavaleta, A.; Gama-Castro, S.; Bonavides-Martínez, C.; Fulcher, C.; Huerta, A.M.; Kothari, A.; Krummenacker, M.; et al. EcoCyc: Fusing model organism databases with systems biology. Nucleic Acids Res. 2013, 41, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Cafarelli, T.M.; Rands, T.J.; Godoy, V.G. The DinB•RecA complex of Escherichia coli mediates an efficient and high-fidelity response to ubiquitous alkylation lesions. Environ. Mol. Mutagen. 2014, 55, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Krüger, B.; Liang, C.; Prell, F.; Fieselmann, A.; Moya, A.; Schuster, S.; Völker, U.; Dandekar, T. Metabolic adaptation and protein complexes in prokaryotes. Metabolites 2012, 2, 940–958. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Becher, D.; Fuchs, S.; Engelmann, S. A proteomic view of cell physiology and virulence of Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Reder, A.; Fuchs, S.; Pagels, M.; Engelmann, S. Physiological proteomics and stress/starvation responses in Bacillus subtilis and Staphylococcus aureus. Res. Microbiol. 2009, 160, 245–258. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Kühner, S.; van Noort, V.; Betts, M.J.; Leo-Macias, A.; Batisse, C.; Rode, M.; Yamada, T.; Maier, T.; Bader, S.; Beltran-Alvarez, P.; et al. Proteome organization in a genome-reduced bacterium. Science 2009, 326, 1235–1240. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Bork, P.; Dandekar, T. A versatile structural domain analysis server using profile weight matrices. J. Chem. Inf. Comput. Sci. 2002, 42, 405–407. [Google Scholar] [CrossRef] [PubMed]
- Liang, C.; Krüger, B.; Dandekar, T. GoSynthetic database tool to analyse natural and engineered molecular processes. Database (Oxford) 2013. [Google Scholar] [CrossRef] [PubMed]
- Cecil, A.; Ohlsen, K.; Menzel, T.; François, P.; Schrenzel, J.; Fischer, A.; Dörries, K.; Selle, M.; Lalk, M.; Hantzschmann, J.; et al. Modelling antibiotic and cytotoxic isoquinoline effects in Staphylococcus aureus, Staphylococcus epidermidis and mammalian cells. Int. J. Med. Microbiol. 2015, 305, 96–109. [Google Scholar] [CrossRef] [PubMed]
- Cecil, A.; Rikanović, C.; Ohlsen, K.; Liang, C.; Bernhardt, J.; Oelschlaeger, T.A.; Gulder, T.; Bringmann, G.; Holzgrabe, U.; Unger, M.; et al. Modeling antibiotic and cytotoxic effects of the dimeric isoquinoline IQ-143 on metabolism and its regulation in Staphylococcus aureus, Staphylococcus epidermidis and human cells. Genome Biol. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, A.; Howorth, D.; Brenner, S.E.; Hubbard, T.J.P.; Chothia, C.; Murzin, A.G. SCOP database in 2004: Refinements integrate structure and sequence family data. Nucleic Acids Res. 2004, 32, D226–D229. [Google Scholar] [CrossRef] [PubMed]
- Schäffer, A.A.; Wolf, Y.I.; Ponting, C.P.; Koonin, E.V.; Aravind, L.; Altschul, S.F. IMPALA: Matching a protein sequence against a collection of PSI-BLAST-constructed position-specific score matrices. Bioinformatics 1999, 15, 1000–1011. [Google Scholar] [CrossRef] [PubMed]
- FTP site of National Center for Biotechnology Information. Available online: ftp://ftp.ncbi.nih.gov (accessed on 1 February 2016).
- Smith, T.F.; Waterman, M.S. Identification of Common Molecular Subsequences. J. Mol. Biol. 1981, 147, 195–197. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Tong, S.Y.; Schaumburg, F.; Ellington, M.J.; Corander, J.; Pichon, B.; Leendertz, F.; Bentley, S.D.; Parkhill, J.; Holt, D.C.; Peters, G.; et al. Novel staphylococcal species that form part of a Staphylococcus aureus-related complex: The non-pigmented Staphylococcus argenteus sp. nov. and the non-human primate-associated Staphylococcus schweitzeri sp. nov. Int. J. Syst. Evol. Microbiol. 2015, 65 (Pt 1), 15–22. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Bae, T.; Schneewind, O.; Takeuchi, F.; Hiramatsu, K. Genome sequence of Staphylococcus aureus strain Newman and comparative analysis of staphylococcal genomes: Polymorphism and evolution of two major pathogenicity islands. J. Bacteriol. 2008, 190, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Bäsell, K.; Otto, A.; Junker, S.; Zühlke, D.; Rappen, G.M.; Schmidt, S.; Hentschker, C.; Macek, B.; Ohlsen, K.; Hecker, M.; et al. The phosphoproteome and its physiological dynamics in Staphylococcus aureus. Int. J. Med. Microbiol. 2014, 304, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Koprivnjak, T.; Mlakar, V.; Swanson, L.; Fournier, B.; Peschel, A.; Weiss, J.P. Cation-Induced Transcriptional Regulation of the dlt Operon of Staphylococcus aureus. J. Bacteriol. 2006, 188, 3622–3630. [Google Scholar] [CrossRef] [PubMed]
- Heymans, F.; Fischer, A.; Stow, N.W.; Girard, M.; Vourexakis, Z.; Des Courtis, A.; Renzi, G.; Huggler, E.; Vlaminck, S.; Bonfils, P.; et al. Screening for staphylococcal superantigen genes shows no correlation with the presence or the severity of chronic rhinosinusitis and nasal polyposis. PLoS ONE 2010, 5, e9525. [Google Scholar] [CrossRef] [PubMed]
- Paterson, G.K.; Harrison, E.M.; Holmes, M.A. The emergence of mecC methicillin-resistant Staphylococcus aureus. Trends Microbiol. 2014, 22, 42–47. [Google Scholar] [CrossRef] [PubMed]
- Harrison, E.M.; Paterson, G.K.; Holden, M.T.; Morgan, F.J.; Larsen, A.R.; Petersen, A.; Leroy, S.; de Vliegher, S.; Perreten, V.; Fox, L.K.; et al. A Staphylococcus xylosus isolate with a new mecC allotype. Antimicrob. Agents Chemother. 2013, 57, 1524–1528. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Beswick, E.J.; Reyes, V.E. Staphylococcal enterotoxins. Toxins (Basel) 2010, 2, 2177–2197. [Google Scholar] [CrossRef] [PubMed]
- Kohler, C.; Wolff, S.; Albrecht, D.; Fuchs, S.; Becher, D.; Büttner, K.; Engelmann, S.; Hecker, M. Proteome analyses of Staphylococcus aureus in growing and non-growing cells: A physiological approach. Int. J. Med. Microbiol. 2005, 295, 547–565. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Zühlke, D.; Pané-Farré, J.; Kusch, H.; Wolf, C.; Reiß, S.; le Binh, T.N.; Albrecht, D.; Riedel, K.; Hecker, M.; et al. Aureolib—A proteome signature library: Towards an understanding of staphylococcus aureus pathophysiology. PLoS ONE 2013, 8, e70669. [Google Scholar] [CrossRef] [PubMed]
- Ohlsen, K.; Donat, S. The impact of serine/threonine phosphorylation in Staphylococcus aureus. Int. J. Med. Microbiol. 2010, 300, 137–141. [Google Scholar] [CrossRef] [PubMed]
- Diep, B.A.; Gill, S.R.; Chang, R.F.; Phan, T.H.; Chen, J.H.; Davidson, M.G.; Lin, F.; Lin, J.; Carleton, H.A.; Mongodin, E.F.; et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet 2006, 367, 731–739. [Google Scholar] [CrossRef]
- Highlander, S.K.; Hultén, K.G.; Qin, X.; Jiang, H.; Yerrapragada, S.; Mason, E.O., Jr.; Shang, Y.; Williams, T.M.; Fortunov, R.M.; Liu, Y.; et al. Subtle genetic changes enhance virulence of methicillin resistant and sensitive Staphylococcus aureus. BMC Microbiol. 2007, 7. [Google Scholar] [CrossRef] [PubMed]
- Altman, D.R.; Sebra, R.; Hand, J.; Attie, O.; Deikus, G.; Carpini, K.W.; Patel, G.; Rana, M.; Arvelakis, A.; Grewal, P.; et al. Transmission of methicillin-resistant Staphylococcus aureus via deceased donor liver transplantation confirmed by whole genome sequencing. Am. J. Transplant. 2014, 14, 2640–2644. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain Name | Accession | Genome Size | Proteins 1 | Protein Complexes 2 |
|---|---|---|---|---|
| N315 | NC_002745 | 2814816 | 2714 | 37 |
| COL | NC_002951 | 2809422 | 2764 | 36 |
| HG001 (NCTC 8325) | NC_007795 | 2821361 | 2767 | 34 |
| USA300_TCH1516 | NC_010079 | 2872915 | 2660 | 36 |
| Newman | NC_009641 | 2878897 | 2894 | 35 |
| ED133 | NC_017337 | 2832479 | 2740 | (See results) |
| Mu50 | NC_02758 | 2878530 | 2812 | |
| MRSA252 | NC_002952 | 2902620 | 2781 |
| Annotation | COL (A) | N315(B) | HG001(A) | Newman(A) | USA300_TCH1516(A) |
|---|---|---|---|---|---|
| glycosyltransferase tuaA | SACOL0114 | SA0124 | SAOUHSC_00089 | NWMN_0073 | SAUSA300_0131 |
| glycosyltransferase epsF | SACOL0115 | SA0125 | SAOUHSC_00090 | NWMN_0074 | SAUSA300_0132 |
| glycosyltransferase fam.1 | SACOL0147 | SA0155 | SAOUHSC_00125 | NWMN_0106 | SAUSA300_0163 |
| glycosyltransferase tarS | SACOL0243 | SA0248 | SAOUHSC_00228 | NWMN_0192 | SAUSA300_0252 |
| glycosyltransferase tagE1 | SACOL0611 | SA0522 | SAOUHSC_00547 | NWMN_0526 | SAUSA300_0549 |
| glycosyltransferase tagE2 | SACOL0612 | SA0523 | SAOUHSC_00548 | NWMN_0527 | SAUSA300_0550 |
| Glycosyltransferase tagX | SACOL0697 | SA0596 | SAOUHSC_00644 | NWMN_0610 | SAUSA300_0627 |
| glycosyltransferase | SACOL0764 | SA0659 | SAOUHSC_00713 | NWMN_0673 | SAUSA300_0689 |
| glycosyltransferase tarM | SACOL1043 | - | - | NWMN_0906 | SAUSA300_0939 |
| glycosyltransferase | SACOL1498 | SA1291 | SAOUHSC_01475 | NWMN_1369 | SAUSA300_1349 |
| glycosyltransferase | SACOL1932 | SA1691 | SAOUHSC_02012 | NWMN_1766 | SAUSA300_1855 |
| 4,4-diaponeurosporenoate glycosyltransferase | SACOL2578 | SA2350 | SAOUHSC_02880 | NWMN_2463 | SAUSA300_2500 |
| accessory Sec system glycosyltransferase GtfB | SACOL2669 | SA2440 | SAOUHSC_02983 | NWMN_2546 | SAUSA300_2582 |
| accessory Sec system glycosyltransferase GtfA | SACOL2670 | SA2441 | SAOUHSC_02984 | NWMN_2547 | SAUSA300_2583 |
| N-glycosyltransferase icaA | SACOL2689 | SA2459 | SAOUHSC_03002 | NWMN_2565 | SAUSA300_2600 |
| Annotation | COL (A) | Mu50 (B) | ED133 (C) |
|---|---|---|---|
| virulence factor (esxA) | SACOL_RS01375 | SAV_RS01590 | SAOV_RS01175 |
| virulence factor (esxB) | SACOL_RS01410 | SAV_RS01625 | SAOV_RS01210 |
| virulence factor B (cvfB) | SACOL_RS07265 | SAV_RS07495 | SAOV_RS07490 |
| virulence factor C | SACOL_RS07475 | SAV_RS07700 | |
| putative enterotoxin | SACOL_RS02230 | SAV_RS02045 | SAOV_RS02015 |
| leucotoxin LukDv (lukD) | SACOL_RS09650 | SAV_RS09765 | SAOV_RS09495 |
| Enterotoxin1 (sek) | SACOL_RS04550 | SAV_RS09795 | SAOV_RS02205 |
| enterotoxin (sei) | SACOL_RS04555 | SAV_RS09800 | |
| enterotoxin (seb) | SACOL_RS04655 | SAV_RS09805 | SAOV_RS02280 |
| enterotoxin | SAOV_RS05800 | ||
| enterotoxin type A | SACOL_RS08455 | SAV_RS09810 | SAOV_RS08355 |
| enterotoxin (epiD) | SACOL_RS09620 | SAV_RS09815 | SAOV_RS09460 |
| enterotoxin (sem) | SAV_RS09820 | ||
| enterotoxin (seo) | SAV_RS09825 | ||
| enterotoxin (sep) | SAV_RS10675 | ||
| enterotoxin (sel) | SAV_RS10975 | SAOV_RS02210 | |
| enterotoxin (sec3) | SAV_RS10980 | ||
| antitoxin MazE (mazE) | SACOL_RS10780 | SAV_RS11325 | SAOV_RS11100 |
| toxin | SACOL_RS11560 | SAV_RS12060 | SAOV_RS11860 |
| Antitoxin RelB (relB) | SACOL_RS12615 | SAV_RS13100 | SAOV_RS12895 |
| MarF (marF) | SACOL_RS01610 |
| Gene | Annotation | N315 (B) | COL (A) | HG001 (A) | Newman (A) | USA300TCH1516 (B) |
|---|---|---|---|---|---|---|
| stk | serine/threonine kinase | SA1063 | Partial (388/664) | SAOUHSC_01187 | NWMN_1130 | SAUSA300_1113 |
| stp | serine/threonine phosphatase | SA1062 | SACOL1231 | SAOUHSC_01186 | NWMN_1129 | SAUSA300_1112 |
| 2, 3-cyclic nucleotide 2-phosphodiesterase | SA0140 | SACOL0130 | SAOUHSC_00107 | NWMN_0088 | SAUSA300_0147 | |
| DNA repair exonuclease | SA1662 | SACOL1900 | SAOUHSC_01975 | NWMN_1736 | SAUSA300_1793 | |
| phosphohydrolase | SA2225 | SACOL2440 | SAOUHSC_02728 | NWMN_2336 | SAUSA300_2382 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liang, C.; Schaack, D.; Srivastava, M.; Gupta, S.K.; Sarukhanyan, E.; Giese, A.; Pagels, M.; Romanov, N.; Pané-Farré, J.; Fuchs, S.; et al. A Staphylococcus aureus Proteome Overview: Shared and Specific Proteins and Protein Complexes from Representative Strains of All Three Clades. Proteomes 2016, 4, 8. https://doi.org/10.3390/proteomes4010008
Liang C, Schaack D, Srivastava M, Gupta SK, Sarukhanyan E, Giese A, Pagels M, Romanov N, Pané-Farré J, Fuchs S, et al. A Staphylococcus aureus Proteome Overview: Shared and Specific Proteins and Protein Complexes from Representative Strains of All Three Clades. Proteomes. 2016; 4(1):8. https://doi.org/10.3390/proteomes4010008
Chicago/Turabian StyleLiang, Chunguang, Dominik Schaack, Mugdha Srivastava, Shishir K. Gupta, Edita Sarukhanyan, Anne Giese, Martin Pagels, Natalie Romanov, Jan Pané-Farré, Stephan Fuchs, and et al. 2016. "A Staphylococcus aureus Proteome Overview: Shared and Specific Proteins and Protein Complexes from Representative Strains of All Three Clades" Proteomes 4, no. 1: 8. https://doi.org/10.3390/proteomes4010008
APA StyleLiang, C., Schaack, D., Srivastava, M., Gupta, S. K., Sarukhanyan, E., Giese, A., Pagels, M., Romanov, N., Pané-Farré, J., Fuchs, S., & Dandekar, T. (2016). A Staphylococcus aureus Proteome Overview: Shared and Specific Proteins and Protein Complexes from Representative Strains of All Three Clades. Proteomes, 4(1), 8. https://doi.org/10.3390/proteomes4010008