
Comparative Proteomic Analysis of Cotton Fiber Development and Protein Extraction Method Comparison in Late Stage Fibers

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Experimental Section
2.1. Plant Materials
2.2. Growth Conditions
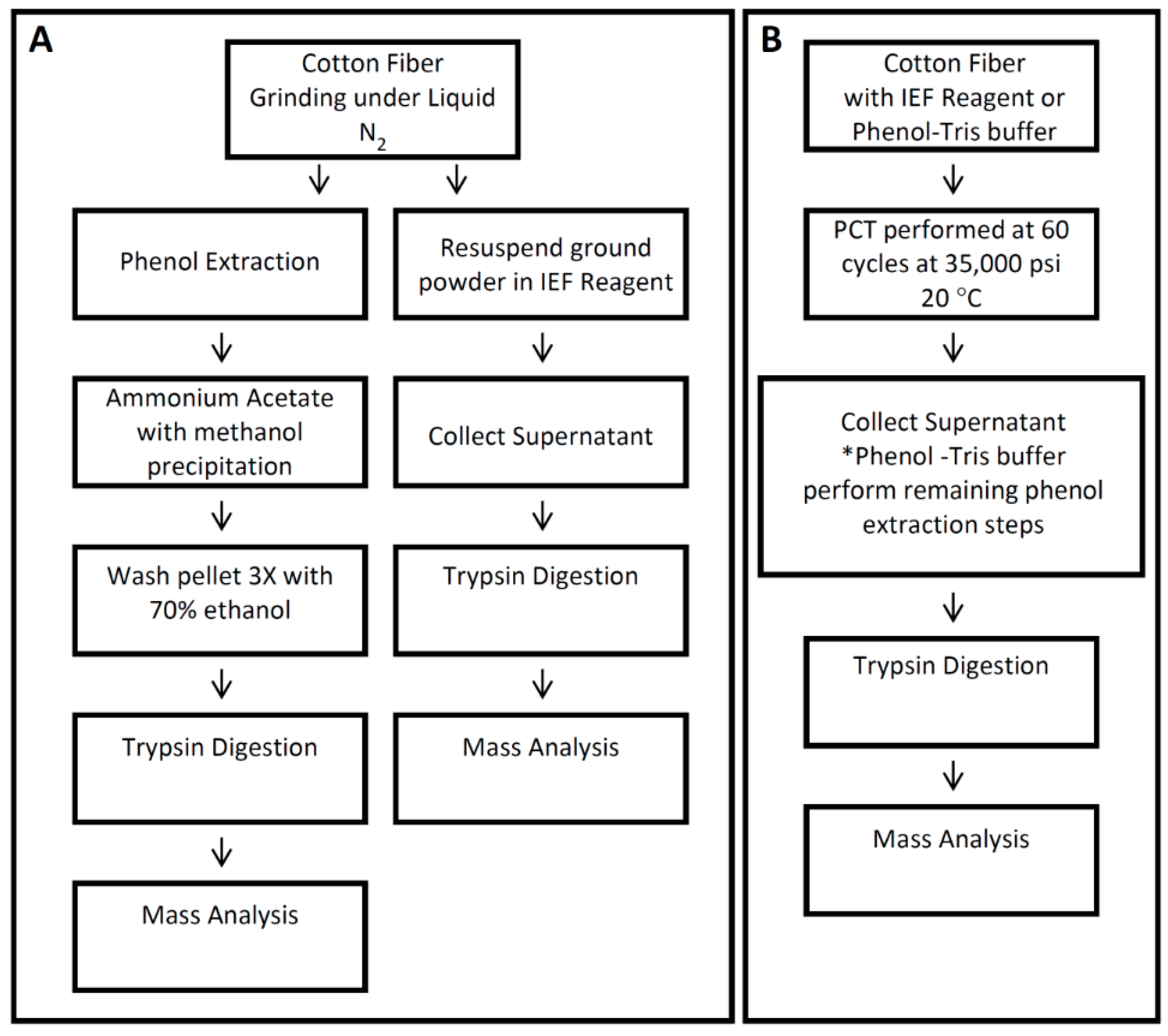
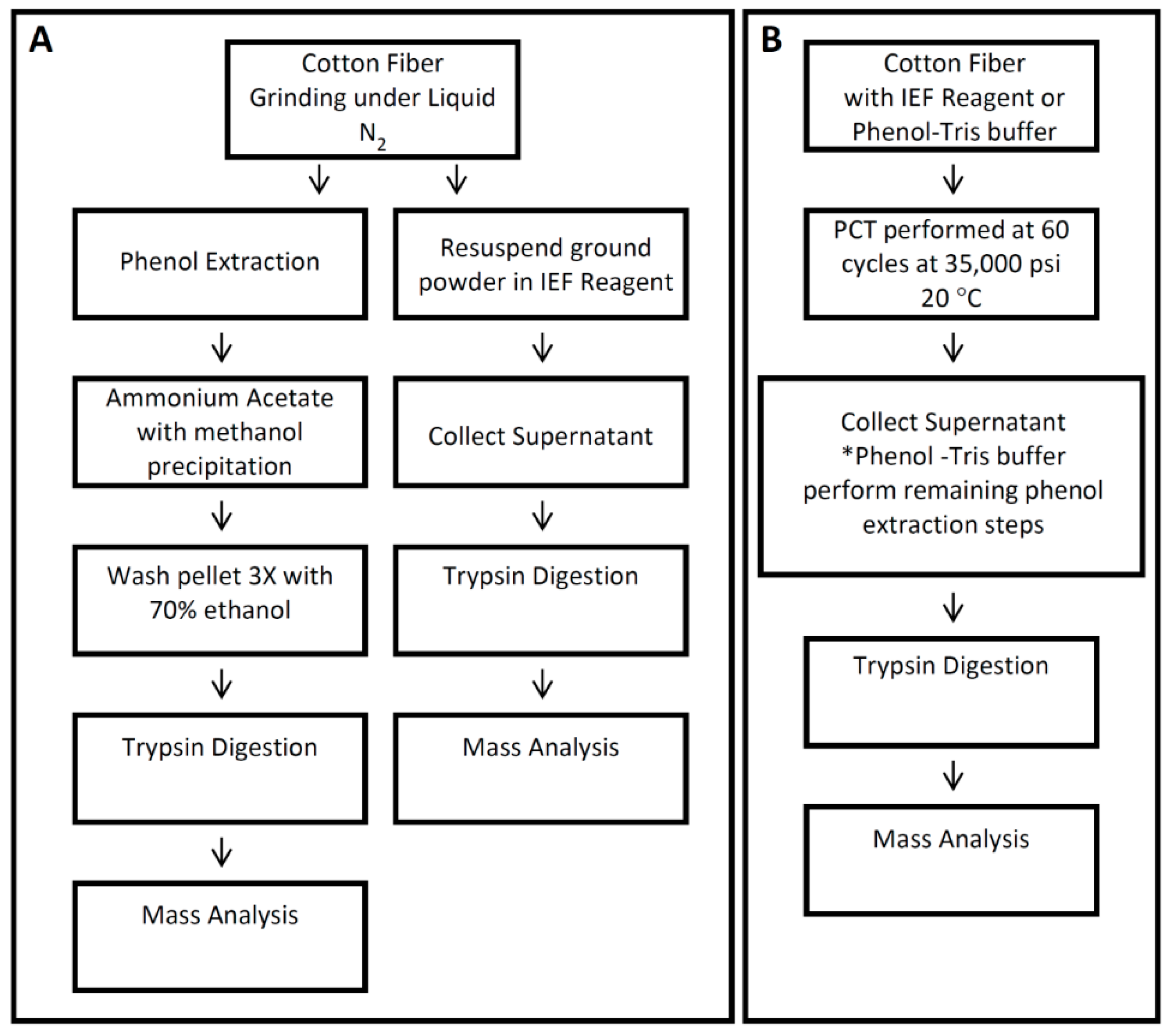
2.3. Protein Extraction with Phenol
2.4. Protein Extraction with Phenol and PCT for 25 dpa Fiber
2.5. Protein Extraction with IEF Reagent for 25 dpa Fiber
2.6. Protein Extraction with IEF Reagent and PCT for 25 dpa and 35 dpa Fiber
2.7. Protein Digestion
2.8. Shotgun Proteomic Analysis
2.9. Protein Identification and Statistical Analysis
2.10. Protein Grouping
2.11. Protein Quantification and Statistical Analysis
3. Results
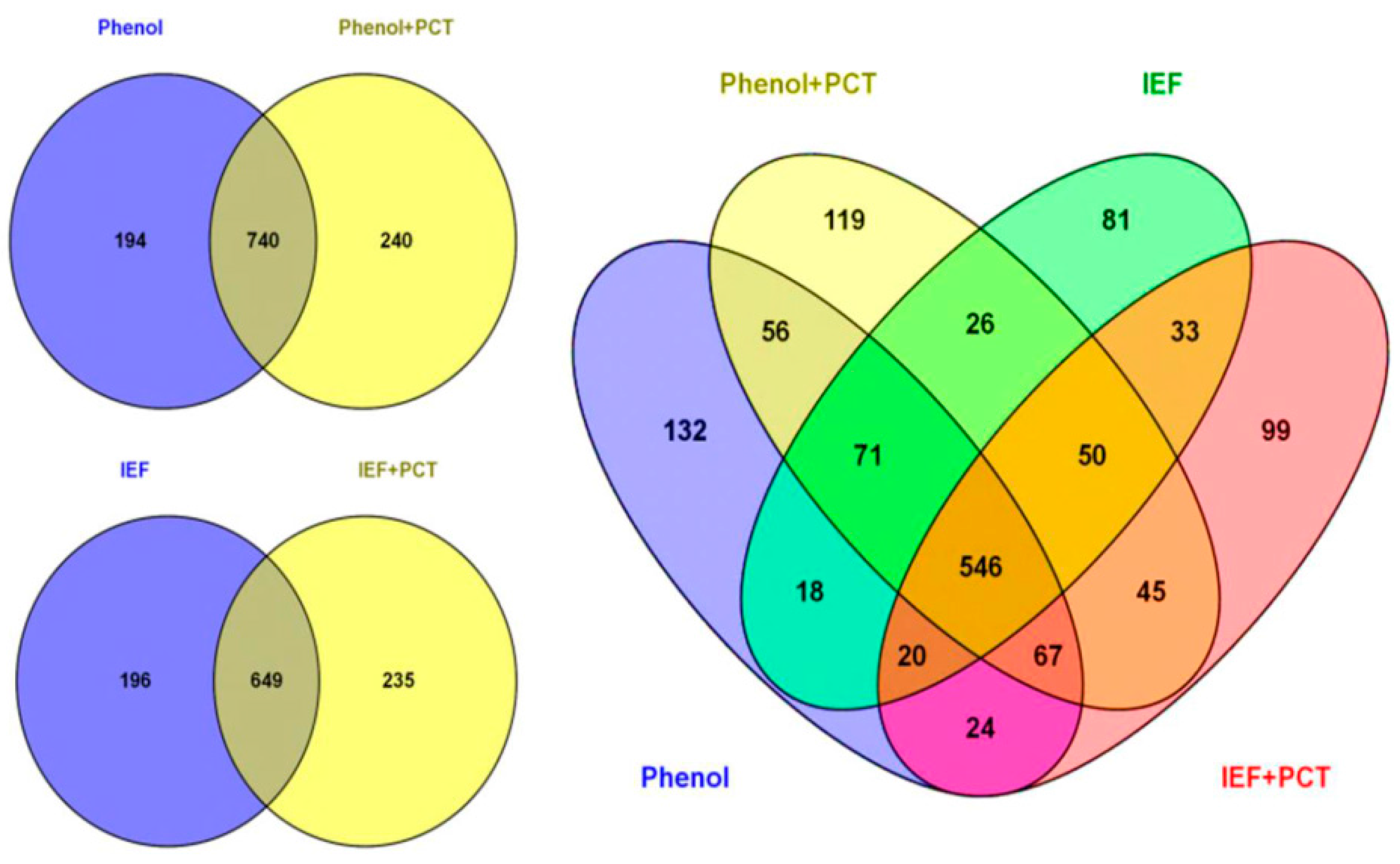
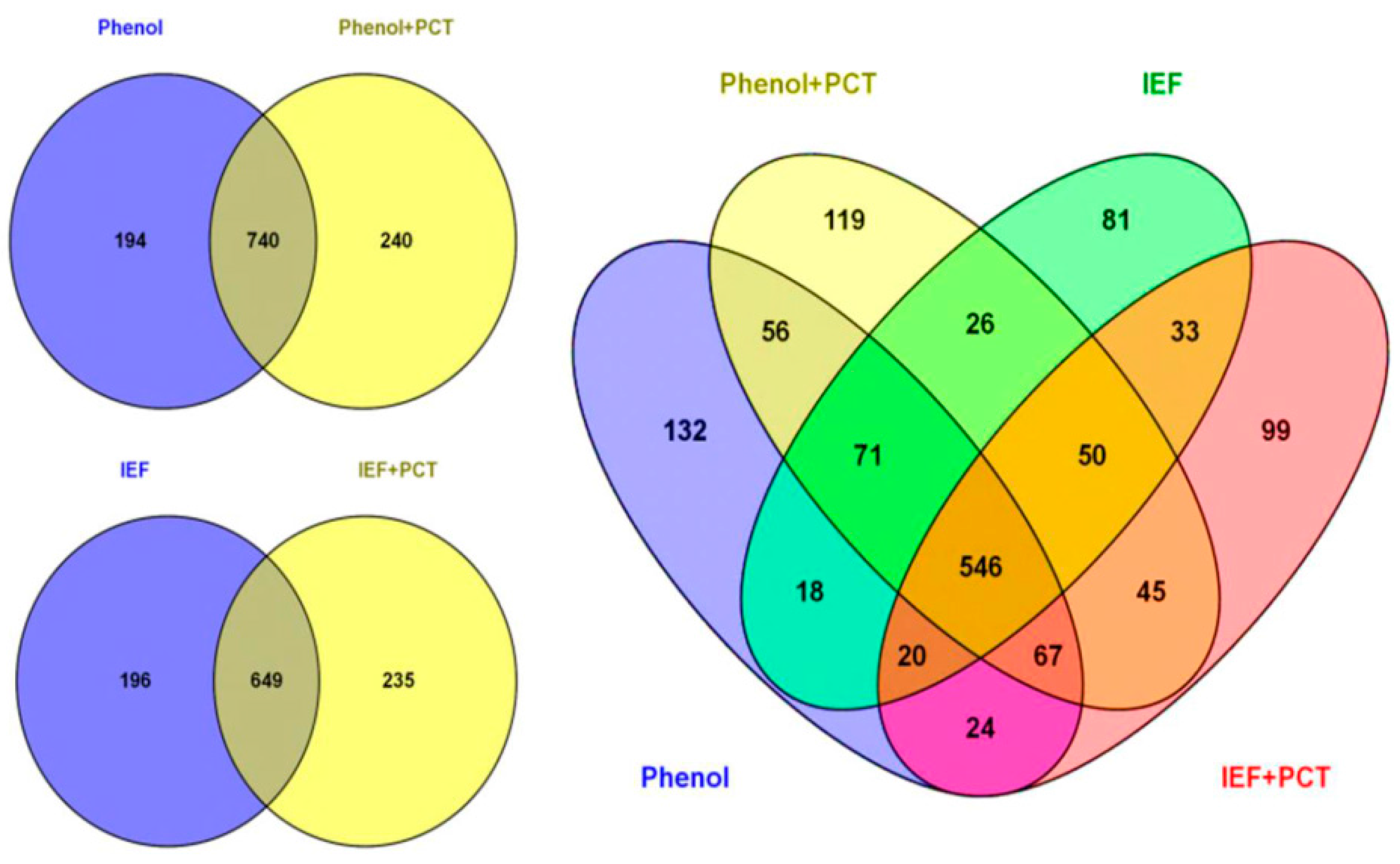
3.1. Comparison of Different Protein Extraction Buffers and Tissue Grinding Methods
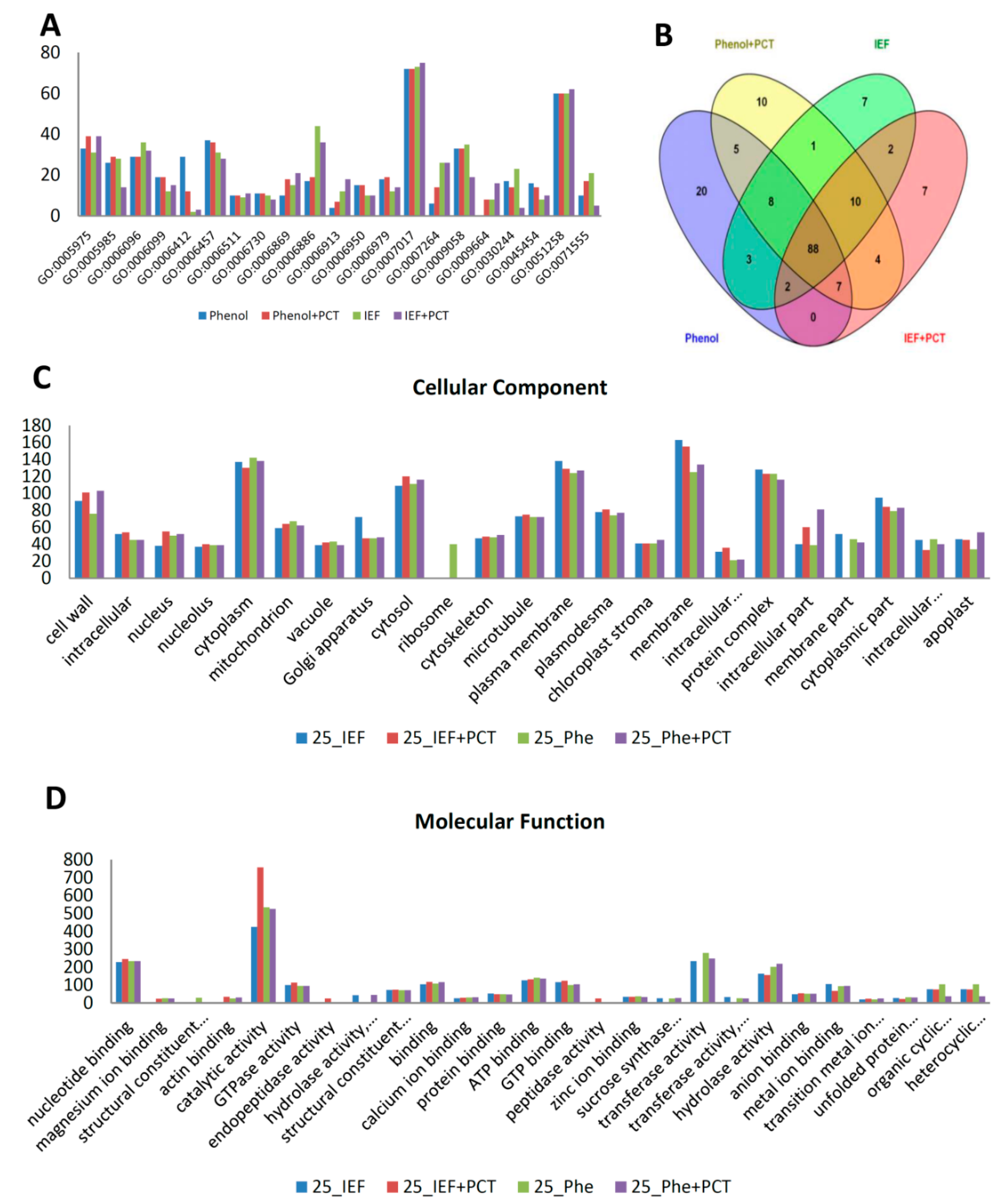
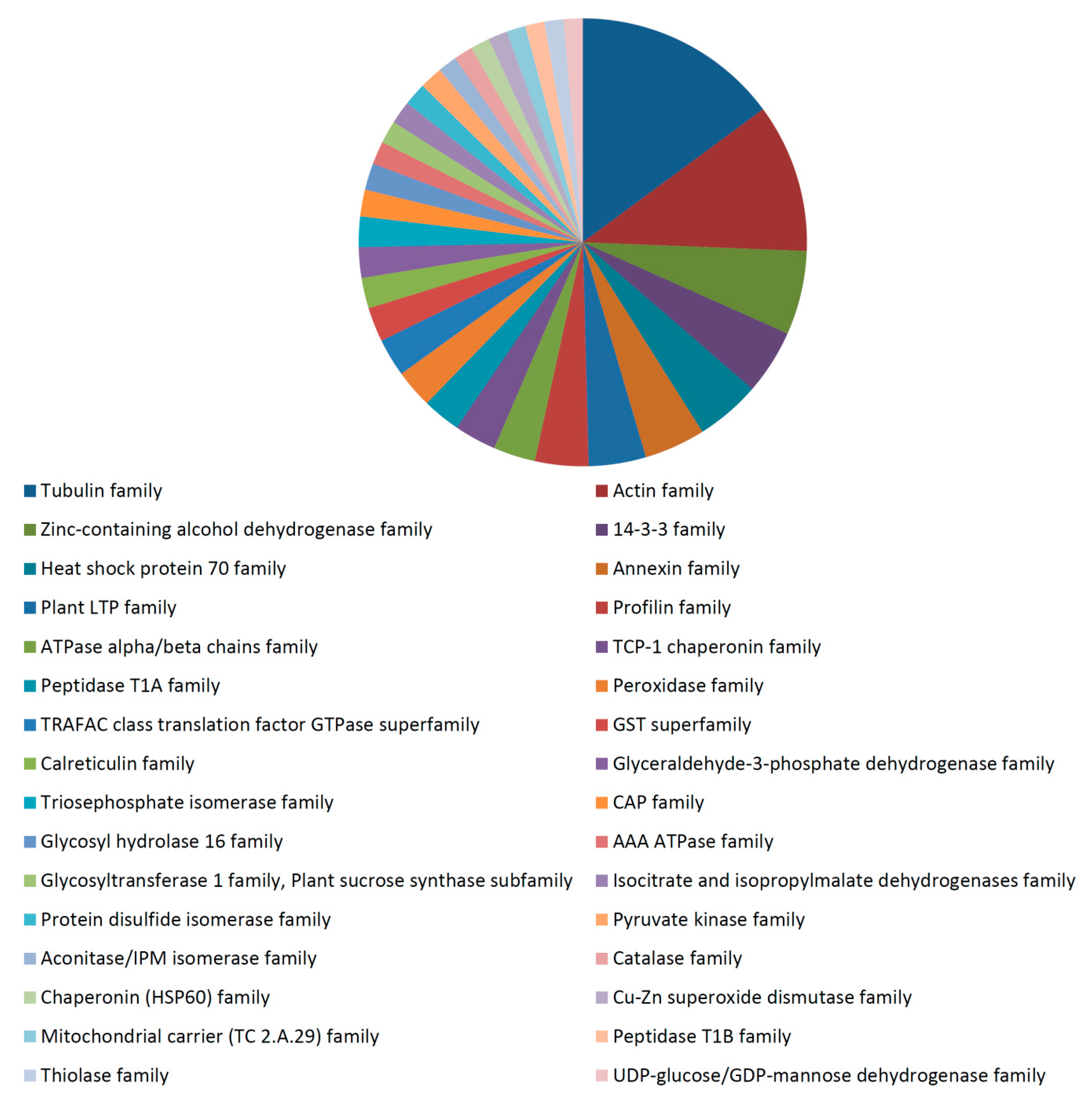
3.2. Fiber Proteome
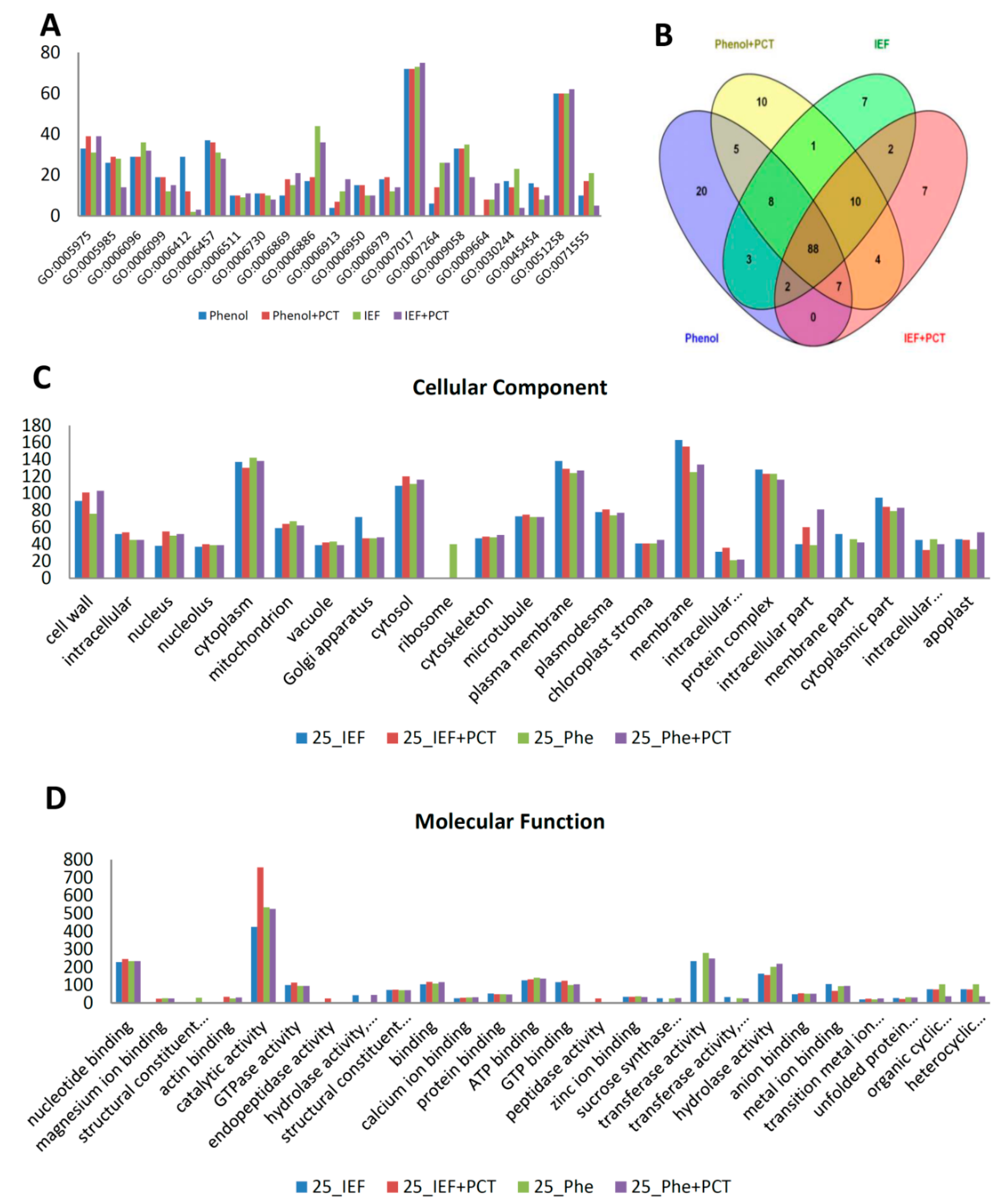
3.3. Functional Analysis of the 35 dpa Proteome
3.4. Differentially-Expressed Proteins during Fiber Development
3.5. Differential Expression of Cytoskeletal Related Proteins
3.6. Differential Expression of Cell Wall-Related Proteins
3.7. Differential Expression of Energy/Carbohydrate Metabolism Proteins
4. Discussion
4.1. Comparison of Protein Extraction Methods for Recalcitrant Cotton Fiber
4.2. Proteome Studies on 35 dpa Fiber
4.3. Proteome Differential Regulation during Cotton Fiber Development
4.4. Cytoskeletal Dynamics
4.5. Cell Wall Dynamics
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, Z.J.; Scheffler, B.E.; Dennis, E.; Triplett, B.A.; Zhang, T.; Guo, W.; Chen, X.; Stelly, D.M.; Rabinowicz, P.D.; Town, C.D.; et al. Toward sequencing cotton (Gossypium) genomes. Plant Physiol. 2007, 145, 1303–1310. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Yang, Y.W.; Zhang, Y.; Liu, J.Y. A high-confidence reference dataset of differentially expressed proteins in elongating cotton fiber cells. Proteomics 2013, 13, 1159–1163. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Triplett, B.A. Cotton fiber growth in planta and in vitro. Models for plant cell elongation and cell wall biogenesis. Plant Physiol. 2001, 127, 1361–1366. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Avci, U.; Eichler Inwood, S.E.; Grimson, M.J.; Landgraf, J.; Mohnen, D.; Sorensen, I.; Wilkerson, C.G.; Willats, W.G.; Haigler, C.H. A specialized outer layer of the primary cell wall joins elongating cotton fibers into tissue-like bundles. Plant Physiol. 2009, 150, 684–699. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.; Liu, J.Y. Mass spectrometric identification of in vivo phosphorylation sites of differentially expressed proteins in elongating cotton fiber cells. PLoS ONE 2013, 8, e58758. [Google Scholar] [CrossRef] [PubMed]
- Pang, C.Y.; Wang, H.; Pang, Y.; Xu, C.; Jiao, Y.; Qin, Y.M.; Western, T.L.; Yu, S.X.; Zhu, Y.X. Comparative proteomics indicates that biosynthesis of pectic precursors is important for cotton fiber and arabidopsis root hair elongation. Mol. Cell Proteomics 2010, 9, 2019–2033. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Yang, Y.W.; Liu, J.Y. An efficient protein preparation for proteomic analysis of developing cotton fibers by 2-de. Electrophoresis 2006, 27, 4559–4569. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.W.; Bian, S.M.; Yao, Y.; Liu, J.Y. Comparative proteomic analysis provides new insights into the fiber elongating process in cotton. J. Proteome Res. 2008, 7, 4623–4637. [Google Scholar] [CrossRef] [PubMed]
- Haigler, C.H.; Betancur, L.; Stiff, M.R.; Tuttle, J.R. Cotton fiber: A powerful single-cell model for cell wall and cellulose research. Front. Plant Sci. 2012, 3, 104. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Wang, F.X.; Wang, Y.Q.; Liu, Y.C.; Zhang, X.Y.; Sun, Y.Q.; Sun, J. Identification of the proteins in green cotton fiber using a proteomics-based approach. Biotechnol. Lett. 2013, 35, 1519–1523. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.J.; Zhang, X.Y.; Wang, F.X.; Yang, C.L.; Liu, F.; Xia, G.X.; Sun, J. A comparative proteomic analysis provides insights into pigment biosynthesis in brown color fiber. J. Proteomics 2013, 78, 374–388. [Google Scholar] [CrossRef] [PubMed]
- Arpat, A.B.; Waugh, M.; Sullivan, J.P.; Gonzales, M.; Frisch, D.; Main, D.; Wood, T.; Leslie, A.; Wing, R.A.; Wilkins, T.A. Functional genomics of cell elongation in developing cotton fibers. Plant Mol. Biol. 2004, 54, 911–929. [Google Scholar] [CrossRef] [PubMed]
- Zhao, P.M.; Wang, L.L.; Han, L.B.; Wang, J.; Yao, Y.; Wang, H.Y.; Du, X.M.; Luo, Y.M.; Xia, G.X. Proteomic identification of differentially expressed proteins in the ligon lintless mutant of upland cotton (Gossypium hirsutum L.). J. Proteome Res. 2010, 9, 1076–1087. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.J.; Hassan, O.S.; Gao, W.; Wei, N.E.; Kohel, R.J.; Chen, X.Y.; Payton, P.; Sze, S.H.; Stelly, D.M.; Chen, Z.J. Developmental and gene expression analyses of a cotton naked seed mutant. Planta 2006, 223, 418–432. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Machado, A.C.; White, R.G.; Llewellyn, D.J.; Dennis, E.S. Expression profiling identifies genes expressed early during lint fibre initiation in cotton. Plant Cell Physiol. 2006, 47, 107–127. [Google Scholar] [CrossRef] [PubMed]
- Liang, W.; Fang, L.; Xiang, D.; Hu, Y.; Feng, H.; Chang, L.; Zhang, T. Transcriptome analysis of short fiber mutant ligon lintless-1 (li1) reveals critical genes and key pathways in cotton fiber elongation and leaf development. PLoS ONE 2015, 10, e0143503. [Google Scholar] [CrossRef] [PubMed]
- Yao, Y.; Zhang, B.; Dong, C.J.; Du, Y.; Jiang, L.; Liu, J.Y. Comparative proteomic and biochemical analyses reveal different molecular events occurring in the process of fiber initiation between wild-type allotetraploid cotton and its fuzzless-lintless mutant. PLoS ONE 2015, 10, e0117049. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.Q.; Liu, F.; Chen, X.S.; Ma, X.J.; Zeng, H.Q.; Yang, Z.M. Transcriptome profiling of early developing cotton fiber by deep-sequencing reveals significantly differential expression of genes in a fuzzless/lintless mutant. Genomics 2010, 96, 369–376. [Google Scholar] [CrossRef] [PubMed]
- Padmalatha, K.V.; Patil, D.P.; Kumar, K.; Dhandapani, G.; Kanakachari, M.; Phanindra, M.L.; Kumar, S.; Mohan, T.C.; Jain, N.; Prakash, A.H.; et al. Functional genomics of fuzzless-lintless mutant of Gossypium hirsutum L. Cv. Mcu5 reveal key genes and pathways involved in cotton fibre initiation and elongation. BMC Genomics 2012, 13, 624. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Han, M.; Zhang, C.; Yao, L.; Sun, J.; Zhang, T. Comparative proteomic analysis reveals the mechanisms governing cotton fiber differentiation and initiation. J. Proteomics 2012, 75, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Liu, K.; Sun, J.; Yao, L.; Yuan, Y. Transcriptome analysis reveals critical genes and key pathways for early cotton fiber elongation in ligon lintless-1 mutant. Genomics 2012, 100, 42–50. [Google Scholar] [CrossRef] [PubMed]
- Du, S.J.; Dong, C.J.; Zhang, B.; Lai, T.F.; Du, X.M.; Liu, J.Y. Comparative proteomic analysis reveals differentially expressed proteins correlated with fuzz fiber initiation in diploid cotton (Gossypium arboreum L.). J. Proteomics 2013, 82, 113–129. [Google Scholar] [CrossRef] [PubMed]
- Graves, D.A.; Stewart, J.M. Analysis of the protein constituency of developing cotton fibres. J. Exp. Bot. 1988, 39, 59–69. [Google Scholar] [CrossRef]
- Hurkman, W.J.; Tanaka, C.K. Solubilization of plant membrane proteins for analysis by two-dimensional gel electrophoresis. Plant Physiol. 1986, 81, 802–806. [Google Scholar] [CrossRef] [PubMed]
- Turley, R.B.; Ferguson, D.L. Changes of ovule proteins during early fiber development in a normal and a fiberless line of cotton (Gossypium hirsutum L.). J. Plant. Physiol. 1996, 149, 695–702. [Google Scholar] [CrossRef]
- Tao, F.; Lawrence, N.P.; Miller, W.W.; Li, C.; Tuzmen, P.; Behnke, J.; Nakhai, B.; Kakita, A.; Christian, T.; Reed, D.; et al. Biological sample preparation system using Pressure Cycling Technology (PCT). In Advances in High Pressure Bioscience and Biotechnology II; Winter, R., Ed.; Springer: Dortmund, Germany, 2003; pp. 413–417. [Google Scholar]
- Tao, F.; Schumacher, R.T.; Lawrence, N.P.; Gross, V.; Smejkal, G.; Lazarev, A.; Han, R. Extraction of Biomolecular Complexes Assisted by Alternating Hydrostatic Pressure. United States Patent US 2009/0286301, 9 November 2009. Available online: http://www.freepatentsonline.com/y2009/0286301.html (accessed on 6 July 2015). [Google Scholar]
- Freeman, E.; Ivanov, A.R. Proteomics under pressure: Development of essential sample preparation techniques in proteomics using ultrahigh hydrostatic pressure. J. Proteome Res. 2011, 10, 5536–5546. [Google Scholar] [CrossRef] [PubMed]
- Smejkal, G.B.; Robinson, M.H.; Lawrence, N.P.; Tao, F.; Saravis, C.A.; Schumacher, R.T. Increased protein yields from Escherichia coli using pressure-cycling technology. J. Biomol. Tech. 2006, 17, 173–175. [Google Scholar] [PubMed]
- Ringham, H.; Bell, R.L.; Smejkal, G.B.; Behnke, J.; Witzmann, F.A. Application of pressure cycling technology to tissue sample preparation for 2-DE. Electrophoresis 2007, 28, 1022–1024. [Google Scholar] [CrossRef] [PubMed]
- Smejkal, G.; Witzmann, F.; Ringham, H.; Small, D.; Chase, S.; Behnke, J.; Ting, E. Sample preparation for two-dimensional gel electrophoresis using pressure cycling technology. Anal Biochem. 2007, 363, 309–311. [Google Scholar] [CrossRef] [PubMed]
- Szabo, Z.; Guttman, A.; Karger, B.L. Rapid release of n-linked glycans from glycoproteins by pressure-cycling technology. Anal. Chem. 2010, 82, 2588–2593. [Google Scholar] [CrossRef] [PubMed]
- Meersman, F.; Dobson, C.M.; Heremans, K. Protein unfolding, amyloid fibril formation and configurational energy landscapes under high pressure conditions. Chem. Soc. Rev. 2006, 35, 908–917. [Google Scholar] [CrossRef] [PubMed]
- Guttman, A.; Chen, F.T.; Evangelista, R.A.; Cooke, N. High-resolution capillary gel electrophoresis of reducing oligosaccharides labeled with 1-aminopyrene-3,6,8-trisulfonate. Anal. Biochem. 1996, 233, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Reddy, K.R.; Hodges, H.; Read, J.; McKinion, J.; Baker, J.; Tarpley, L.; Reddy, V. Soil-plant-atmosphere-research (spar) facility: A tool for plant research and modeling. Biotronics 2001, 30, 27–50. [Google Scholar]
- Hewitt, E.J. Sand and Water Culture Methods used in the Study of Plant Nutrition. Technical Communication 22, 1952; Commonwealth Agriculture Bureaux, Farham Royal: Bucks, UK.
- Chitteti, B.R.; Peng, Z. Proteome and phosphoproteome differential expression under salinity stress in rice (Oryza sativa) roots. J. Proteome Res. 2007, 6, 1718–1727. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Zhang, K.; Mujahid, H.; Verma, D.P.; Peng, Z. Differential histone modification and protein expression associated with cell wall removal and regeneration in rice (Oryza sativa). J. Proteome Res. 2011, 10, 551–563. [Google Scholar] [CrossRef] [PubMed]
- Mujahid, H.; Tan, F.; Zhang, J.; Nallamilli, B.R.; Pendarvis, K.; Peng, Z. Nuclear proteome response to cell wall removal in rice (Oryza sativa). Proteome Sci. 2013, 11, 26. [Google Scholar] [CrossRef] [PubMed]
- Shah, P.; Nanduri, B.; Swiatlo, E.; Ma, Y.; Pendarvis, K. Polyamine biosynthesis and transport mechanisms are crucial for fitness and pathogenesis of streptococcus pneumoniae. Microbiology 2011, 157, 504–515. [Google Scholar] [CrossRef] [PubMed]
- Tan, F.; Li, G.; Chitteti, B.R.; Peng, Z. Proteome and phosphoproteome analysis of chromatin associated proteins in rice (Oryza sativa). Proteomics 2007, 7, 4511–4527. [Google Scholar] [CrossRef] [PubMed]
- Chitteti, B.R.; Tan, F.; Mujahid, H.; Magee, B.G.; Bridges, S.M.; Peng, Z. Comparative analysis of proteome differential regulation during cell dedifferentiation in arabidopsis. Proteomics 2008, 8, 4303–4316. [Google Scholar] [CrossRef] [PubMed]
- Kessner, D.; Chambers, M.; Burke, R.; Agus, D.; Mallick, P. Proteowizard: Open source software for rapid proteomics tools development. Bioinformatics 2008, 24, 2534–2536. [Google Scholar] [CrossRef] [PubMed]
- Craig, R.; Beavis, R.C. Tandem: Matching proteins with tandem mass spectra. Bioinformatics 2004, 20, 1466–1467. [Google Scholar] [CrossRef] [PubMed]
- Protein Grouping. Available online: https://github.com/jsreddy82/ProteinGrouping.git (accessed on 3 May 2015).
- Wan, C.Y.; Wilkins, T.A. A modified hot borate method significantly enhances the yield of high-quality RNA from cotton (Gossypium hirsutum L.). Anal. Biochem. 1994, 223, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Oliveros, J.C. (2007–2015) Venny. An interactive tool for comparing lists with Venn’s diagrams. Available online: http://bioinfogp.cnb.csic.es/tools/venny/index.html (accessed on 20 May 2015).
- Davis, M.J.; Sehgal, M.S.; Ragan, M.A. Automatic, context-specific generation of gene ontology slims. BMC Bioinform. 2010, 11, 498. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghazi, Y.; Bourot, S.; Arioli, T.; Dennis, E.S.; Llewellyn, D.J. Transcript profiling during fiber development identifies pathways in secondary metabolism and cell wall structure that may contribute to cotton fiber quality. Plant Cell Physiol. 2009, 50, 1364–1381. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.; Schmidt, T.B.; Nanduri, B.; Pendarvis, K.; Pittman, J.R.; Thornton, J.A.; Grissett, J.; Donaldson, J.R. Proteomic analysis of the response of listeria monocytogenes to bile salts under anaerobic conditions. J. Med. Microbiol. 2013, 62, 25–35. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, F.M.; Wang, N.; Magee, G.B.; Nanduri, B.; Lawrence, M.L.; Camon, E.B.; Barrell, D.G.; Hill, D.P.; Dolan, M.E.; Williams, W.P.; et al. Agbase: A functional genomics resource for agriculture. BMC Genomics 2006, 7, 229. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, F.M.; Bridges, S.M.; Wang, N.; Magee, G.B.; Williams, W.P.; Luthe, D.S.; Burgess, S.C. Agbase: A unified resource for functional analysis in agriculture. Nucleic Acids Res. 2007, 35, D599–D603. [Google Scholar] [CrossRef] [PubMed]
- Ji, S.J.; Lu, Y.C.; Feng, J.X.; Wei, G.; Li, J.; Shi, Y.H.; Fu, Q.; Liu, D.; Luo, J.C.; Zhu, Y.X. Isolation and analyses of genes preferentially expressed during early cotton fiber development by subtractive PCR and CDNA array. Nucleic Acids Res. 2003, 31, 2534–2543. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Yu, Y.; Chen, Z.L.; Xia, G.X. Functional characterization of Gossypium hirsutum profilin 1 gene (ghpfn1) in tobacco suspension cells. Characterization of in vivo functions of a cotton profilin gene. Planta 2005, 222, 594–603. [Google Scholar] [CrossRef] [PubMed]
- Bao, Y.; Hu, G.; Flagel, L.E.; Salmon, A.; Bezanilla, M.; Paterson, A.H.; Wang, Z.; Wendel, J.F. Parallel up-regulation of the profilin gene family following independent domestication of diploid and allopolyploid cotton (Gossypium). Proc. Natl. Acad. Sci. USA 2011, 108, 21152–21157. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Munoz, M.; Hamilton, S.L.; Kaetzel, M.A.; Hazarika, P.; Dedman, J.R. Modulation of Ca2+ release channel activity from sarcoplasmic reticulum by annexin vi (67-kda calcimedin). J. Biol. Chem. 1990, 265, 15894–15899. [Google Scholar] [PubMed]
- Alvarez-Martinez, M.T.; Porte, F.; Liautard, J.P.; Sri Widada, J. Effects of profilin-annexin I association on some properties of both profilin and annexin I: Modification of the inhibitory activity of profilin on actin polymerization and inhibition of the self-association of annexin I and its interactions with liposomes. Biochim. Biophys. Acta 1997, 1339, 331–340. [Google Scholar] [CrossRef]
- Carroll, A.D.; Moyen, C.; van Kesteren, P.; Tooke, F.; Battey, N.H.; Brownlee, C. Ca2+, annexins, and GTP modulate exocytosis from maize root cap protoplasts. Plant Cell 1998, 10, 1267–1276. [Google Scholar] [CrossRef] [PubMed]
- Lafont, F.; Lecat, S.; Verkade, P.; Simons, K. Annexin xiiib associates with lipid microdomains to function in apical delivery. J. Cell Biol. 1998, 142, 1413–1427. [Google Scholar] [CrossRef] [PubMed]
- Reddy, V.S.; Reddy, A.S. Proteomics of calcium-signaling components in plants. Phytochemistry 2004, 65, 1745–1776. [Google Scholar] [CrossRef] [PubMed]
- Konopka-Postupolska, D. Annexins: Putative linkers in dynamic membrane-cytoskeleton interactions in plant cells. Protoplasma 2007, 230, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Morel, E.; Gruenberg, J. Annexin a2 binding to endosomes and functions in endosomal transport are regulated by tyrosine 23 phosphorylation. J. Biol. Chem. 2009, 284, 1604–1611. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Wang, J.; Zhang, L.; Zuo, K. A cotton annexin protein anxgb6 regulates fiber elongation through its interaction with actin 1. PLoS ONE 2013, 8, e66160. [Google Scholar] [CrossRef] [PubMed]
- Brill, E.; van Thournout, M.; White, R.G.; Llewellyn, D.; Campbell, P.M.; Engelen, S.; Ruan, Y.L.; Arioli, T.; Furbank, R.T. A novel isoform of sucrose synthase is targeted to the cell wall during secondary cell wall synthesis in cotton fiber. Plant Physiol. 2011, 157, 40–54. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, M.J. Physiological roles for secondary metabolites in plants: Some progress, many outstanding problems. Plant Mol. Biol. 1994, 24, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Sebastiana, M.; Figueiredo, A.; Monteiro, F.; Martins, J.; Franco, C.; Coelho, A.V.; Vaz, F.; Simoes, T.; Penque, D.; Pais, M.S.; et al. A possible approach for gel-based proteomic studies in recalcitrant woody plants. Springerplus 2013, 2, 210. [Google Scholar] [CrossRef] [PubMed]
- Granier, F. Extraction of plant proteins for two-dimensional electrophoresis. Electrophoresis 1988, 9, 712–718. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.; Wang, J.; Yu, W.; He, Z. Protein inference: A review. Brief Bioinform. 2012, 13, 586–614. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wang, H.Y.; Zhao, P.M.; Han, L.B.; Jiao, G.L.; Zheng, Y.Y.; Huang, S.J.; Xia, G.X. Overexpression of a profilin (ghpfn2) promotes the progression of developmental phases in cotton fibers. Plant Cell Physiol. 2010, 51, 1276–1290. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.Y.; Wang, J.; Gao, P.; Jiao, G.L.; Zhao, P.M.; Li, Y.; Wang, G.L.; Xia, G.X. Down-regulation of ghadf1 gene expression affects cotton fibre properties. Plant Biotechnol. J. 2009, 7, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.; Burns, T.H.; Light, G.; Sun, Y.; Fokar, M.; Kasukabe, Y.; Fujisawa, K.; Maekawa, Y.; Allen, R.D. Xyloglucan endotransglycosylase/hydrolase genes in cotton and their role in fiber elongation. Planta 2010, 232, 1191–1205. [Google Scholar] [CrossRef] [PubMed]
- Stiff, M.R.; Haigler, C.H. Recent advances in cotton fiber development. In Flowering and Fruiting in Cotton; Oosterhuis, D.M., Cothren, J.T., Eds.; The Cotton Foundation: Cordova, TN, USA, 2012; Volume 8, pp. 163–192. [Google Scholar]
- Pu, L.; Li, Q.; Fan, X.; Yang, W.; Xue, Y. The r2r3 MYB transcription factor ghmyb109 is required for cotton fiber development. Genetics 2008, 180, 811–820. [Google Scholar] [CrossRef] [PubMed]
- Sampedro, J.; Cosgrove, D.J. The expansin superfamily. Genome Biol. 2005, 6, 242. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cosgrove, D.J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6, 850–861. [Google Scholar] [CrossRef] [PubMed]
- Harmer, S.E.; Orford, S.J.; Timmis, J.N. Characterisation of six alpha-expansin genes in Gossypium hirsutum (upland cotton). Mol. Genet. Genomics 2002, 268, 1–9. [Google Scholar] [PubMed]
- An, C.; Saha, S.; Jenkins, J.N.; Scheffler, B.E.; Wilkins, T.A.; Stelly, D.M. Transcriptome profiling, sequence characterization, and snp-based chromosomal assignment of the expansin genes in cotton. Mol. Genet. Genomics 2007, 278, 539–553. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, D.J. Relaxation in a high-stress environment: The molecular bases of extensible cell walls and cell enlargement. Plant Cell 1997, 9, 1031–1041. [Google Scholar] [CrossRef] [PubMed]
- Ruan, Y.L.; Llewellyn, D.J.; Furbank, R.T. The control of single-celled cotton fiber elongation by developmentally reversible gating of plasmodesmata and coordinated expression of sucrose and K+ transporters and expansin. Plant Cell 2001, 13, 47–60. [Google Scholar] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mujahid, H.; Pendarvis, K.; Reddy, J.S.; Nallamilli, B.R.R.; Reddy, K.R.; Nanduri, B.; Peng, Z. Comparative Proteomic Analysis of Cotton Fiber Development and Protein Extraction Method Comparison in Late Stage Fibers. Proteomes 2016, 4, 7. https://doi.org/10.3390/proteomes4010007
Mujahid H, Pendarvis K, Reddy JS, Nallamilli BRR, Reddy KR, Nanduri B, Peng Z. Comparative Proteomic Analysis of Cotton Fiber Development and Protein Extraction Method Comparison in Late Stage Fibers. Proteomes. 2016; 4(1):7. https://doi.org/10.3390/proteomes4010007
Chicago/Turabian StyleMujahid, Hana, Ken Pendarvis, Joseph S. Reddy, Babi Ramesh Reddy Nallamilli, K. R. Reddy, Bindu Nanduri, and Zhaohua Peng. 2016. "Comparative Proteomic Analysis of Cotton Fiber Development and Protein Extraction Method Comparison in Late Stage Fibers" Proteomes 4, no. 1: 7. https://doi.org/10.3390/proteomes4010007
APA StyleMujahid, H., Pendarvis, K., Reddy, J. S., Nallamilli, B. R. R., Reddy, K. R., Nanduri, B., & Peng, Z. (2016). Comparative Proteomic Analysis of Cotton Fiber Development and Protein Extraction Method Comparison in Late Stage Fibers. Proteomes, 4(1), 7. https://doi.org/10.3390/proteomes4010007