
Comparing Simplification Strategies for the Skeletal Muscle Proteome

Abstract
:1. Introduction
2. Experimental Section
2.1. Experimental Animals
2.2. Sample Preparation
2.3. 1-D SDS-PAGE
2.4. ProteoMiner Protein Equalization
2.5. OFFGEL Isoelectric Focusing
2.6. Filter Aided Sample Preparation (FASP)
2.7. Analysis of Peptides by LC-MS/MS
2.8. Protein Identifications
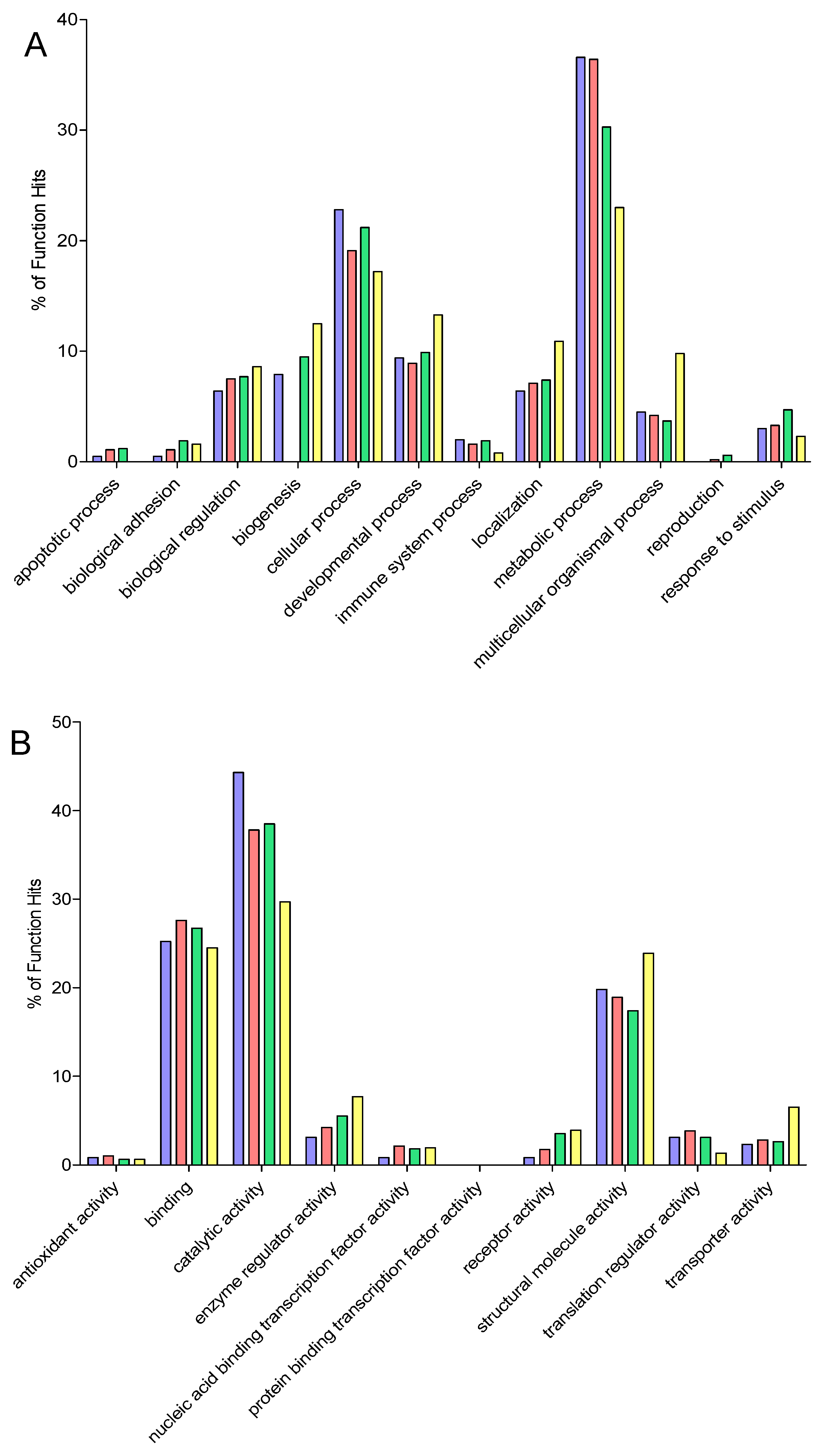
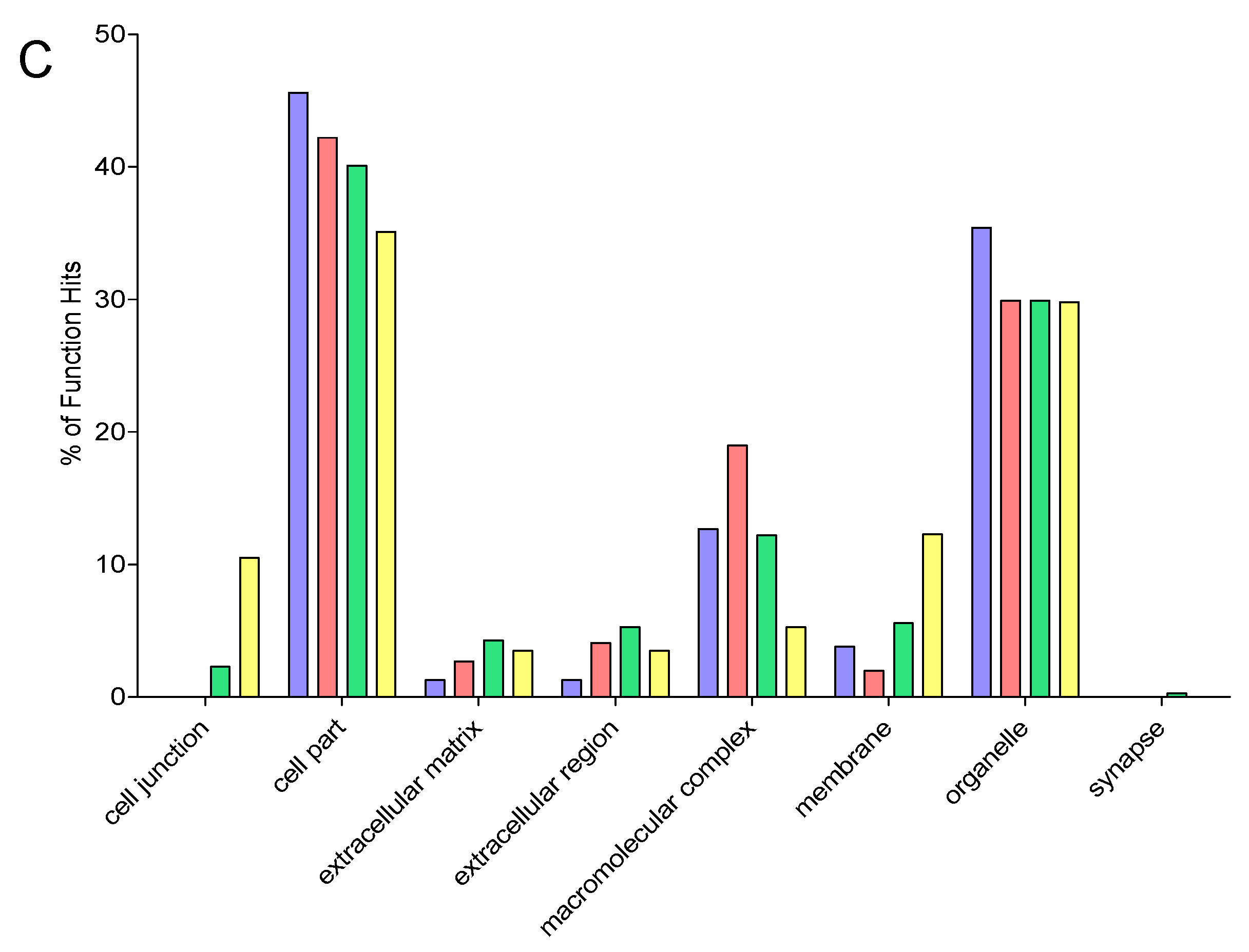
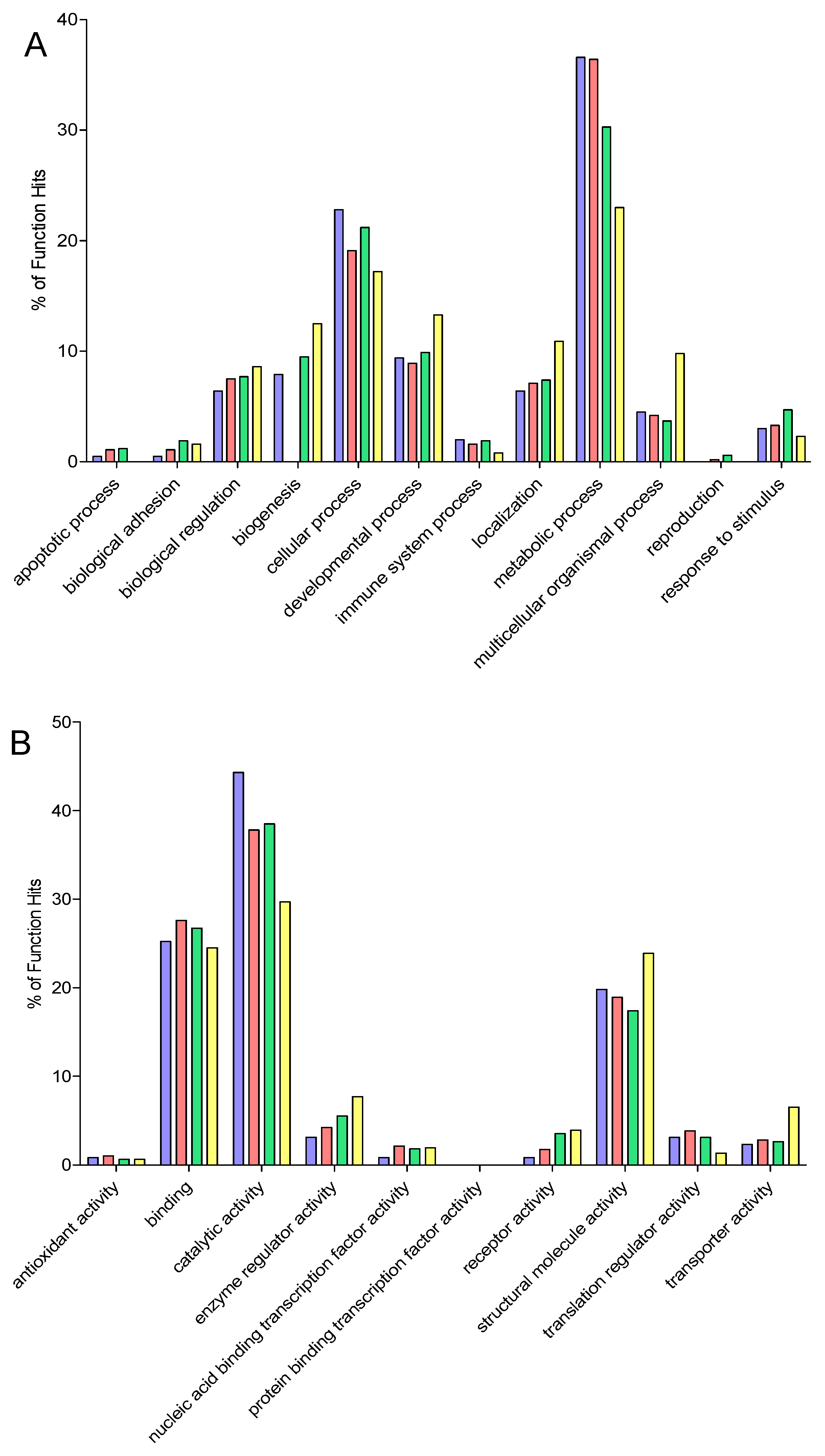
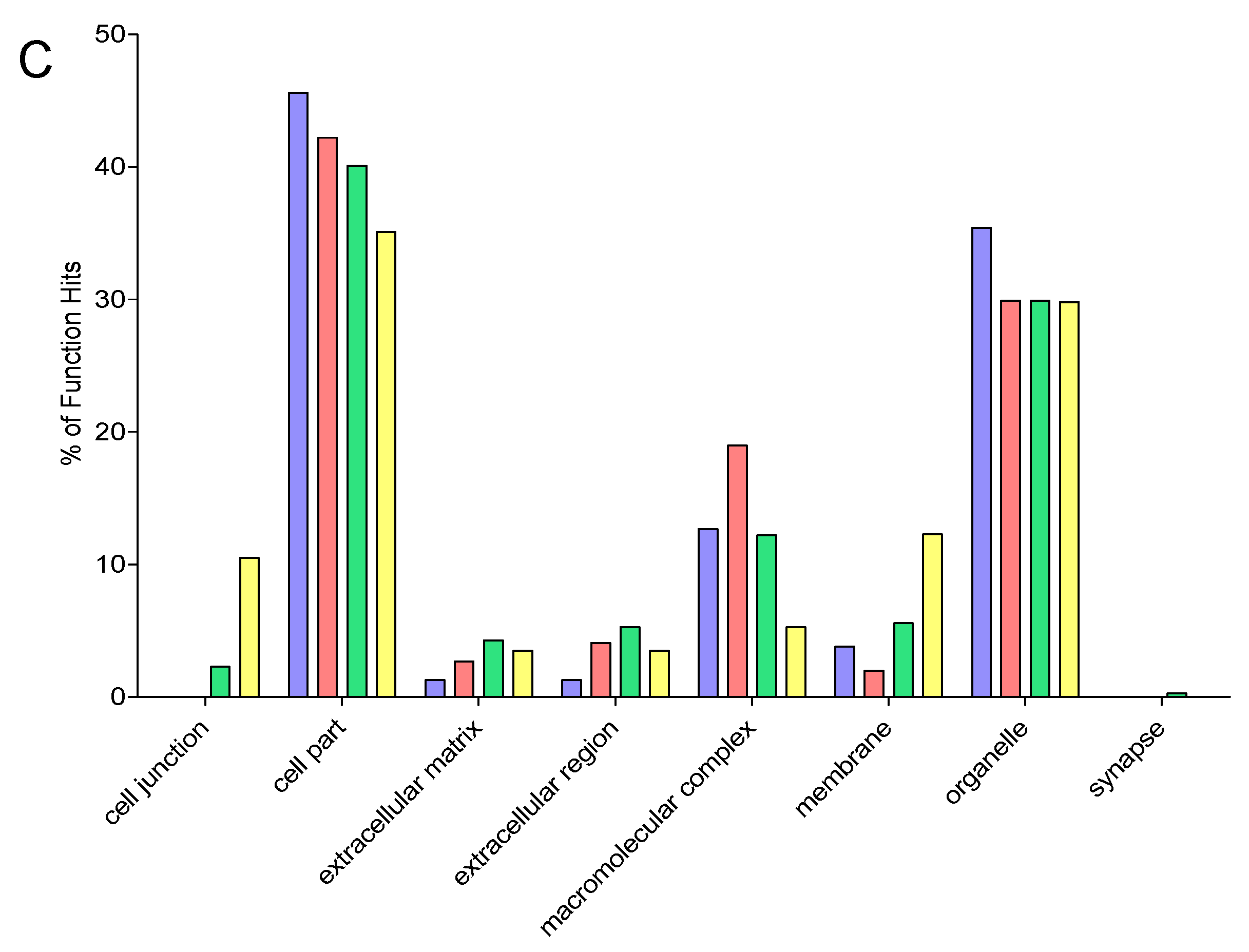
2.9. Gene Ontology
3. Results and Discussion
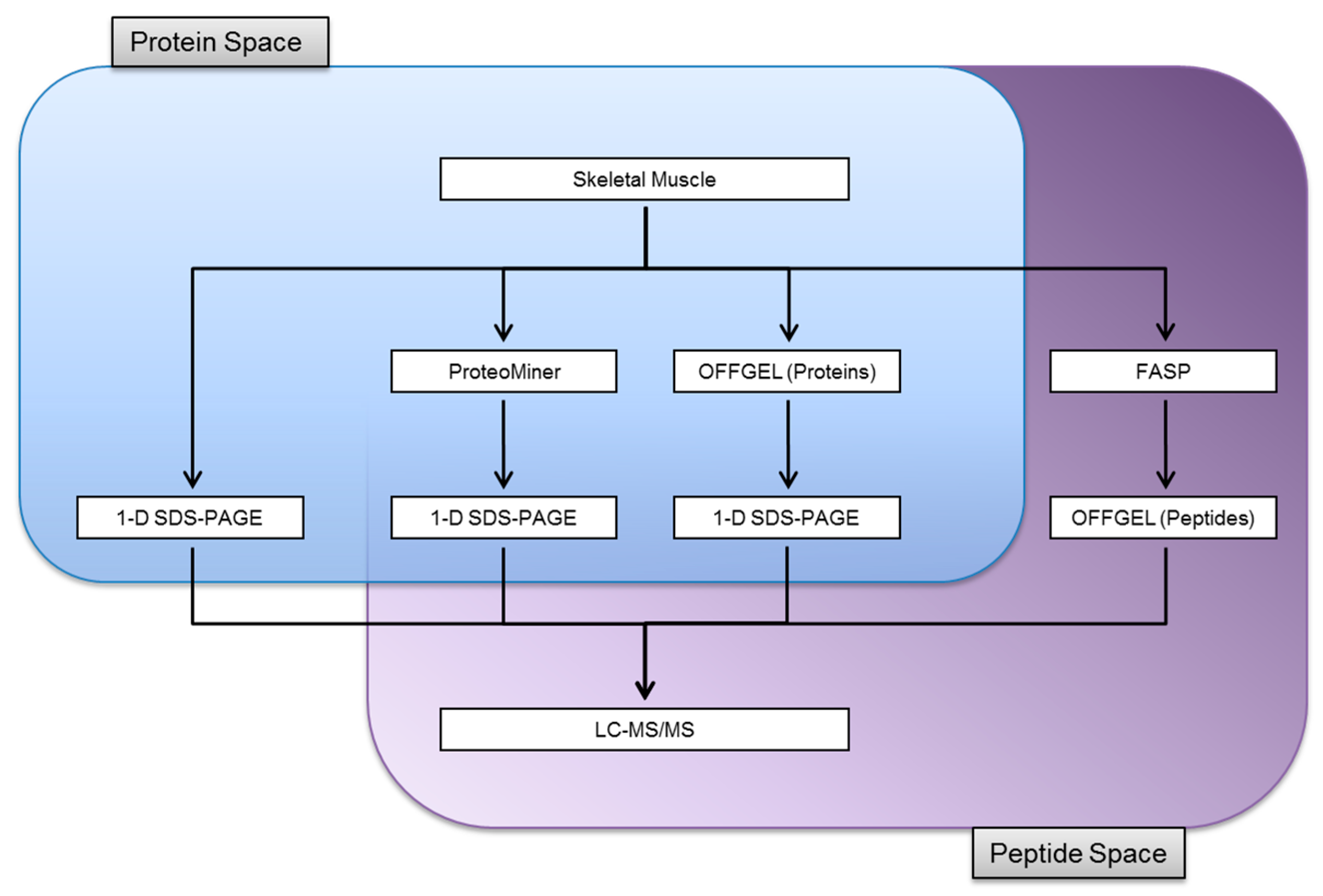
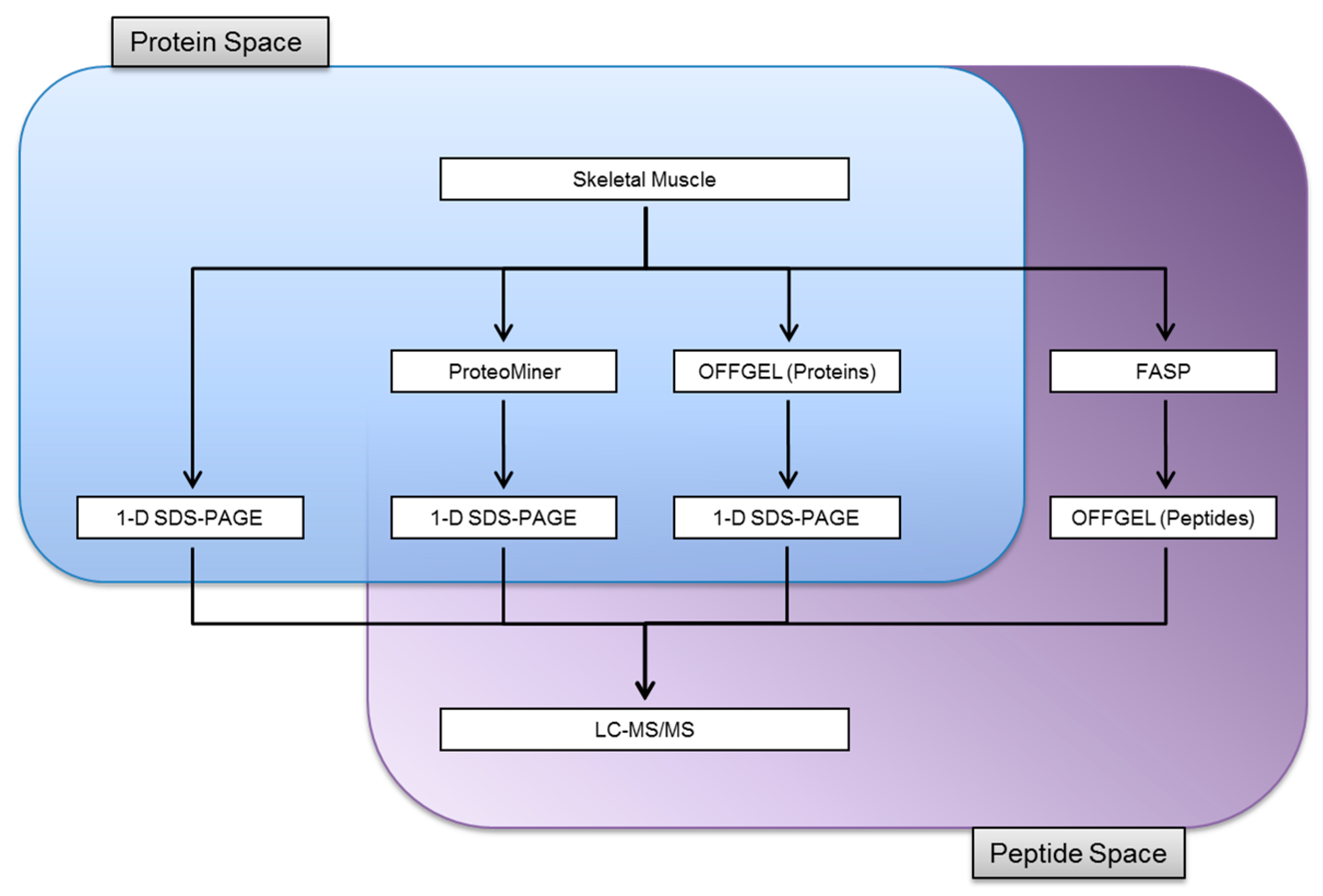
3.1. Experimental Design
3.2. Protein Identifications in Skeletal Muscle by 1-D SDS-PAGE
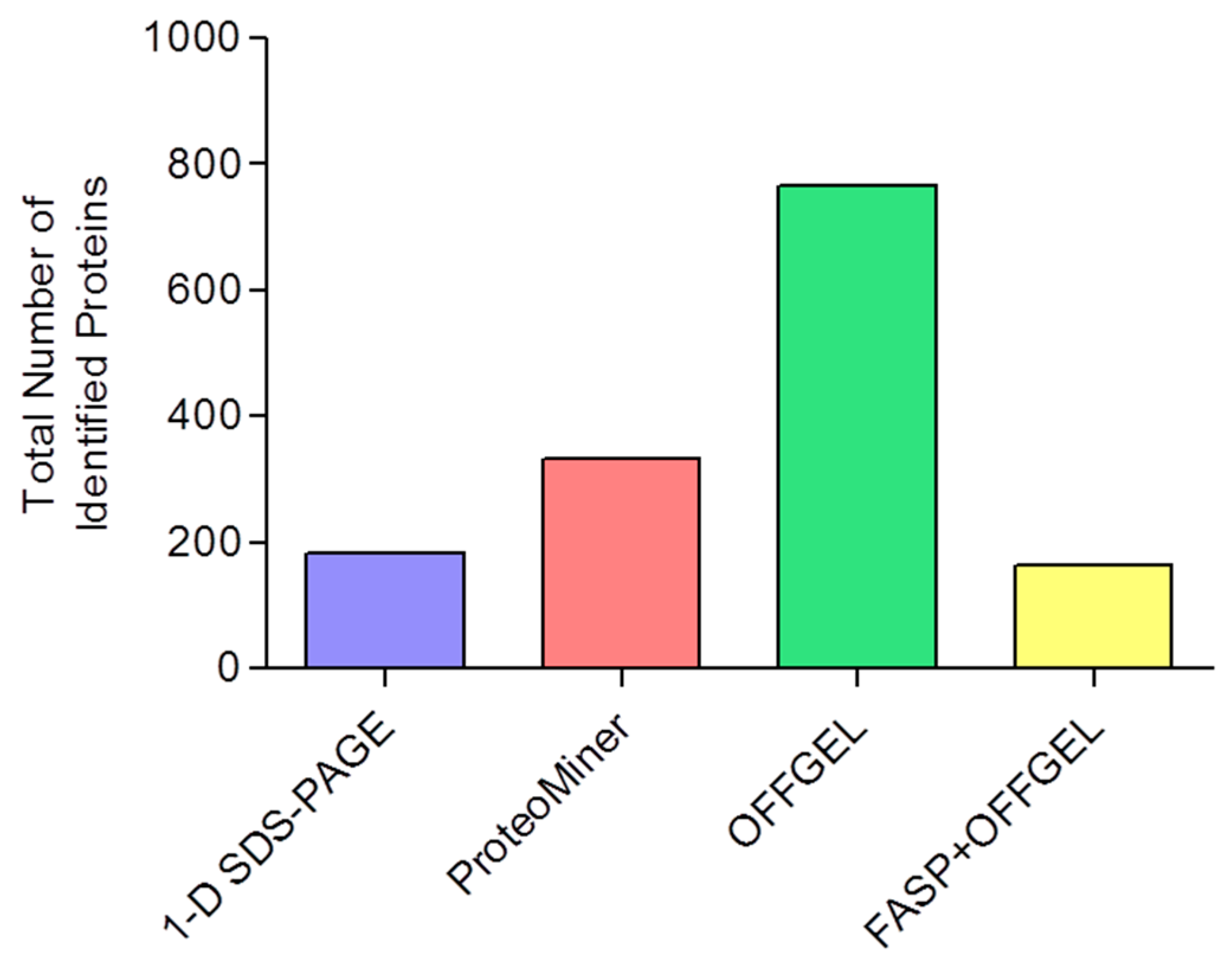
3.3. Proteome Equalization Increases the Number of Protein Identifications
3.4. Extending Proteome Coverage Through In-Solution Isoelectric Focusing
3.5. Improving the Confidence of Protein Identifications by FASP
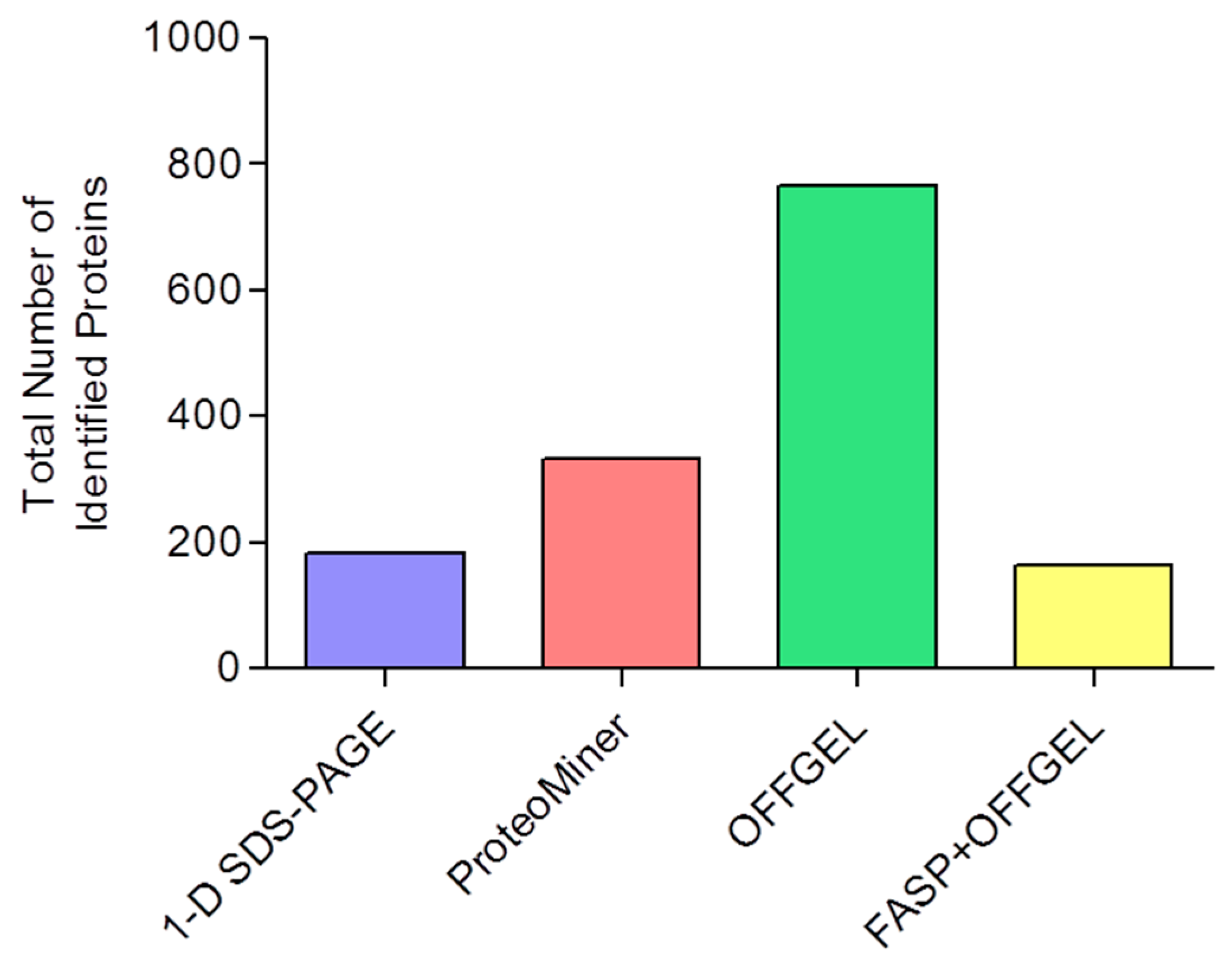
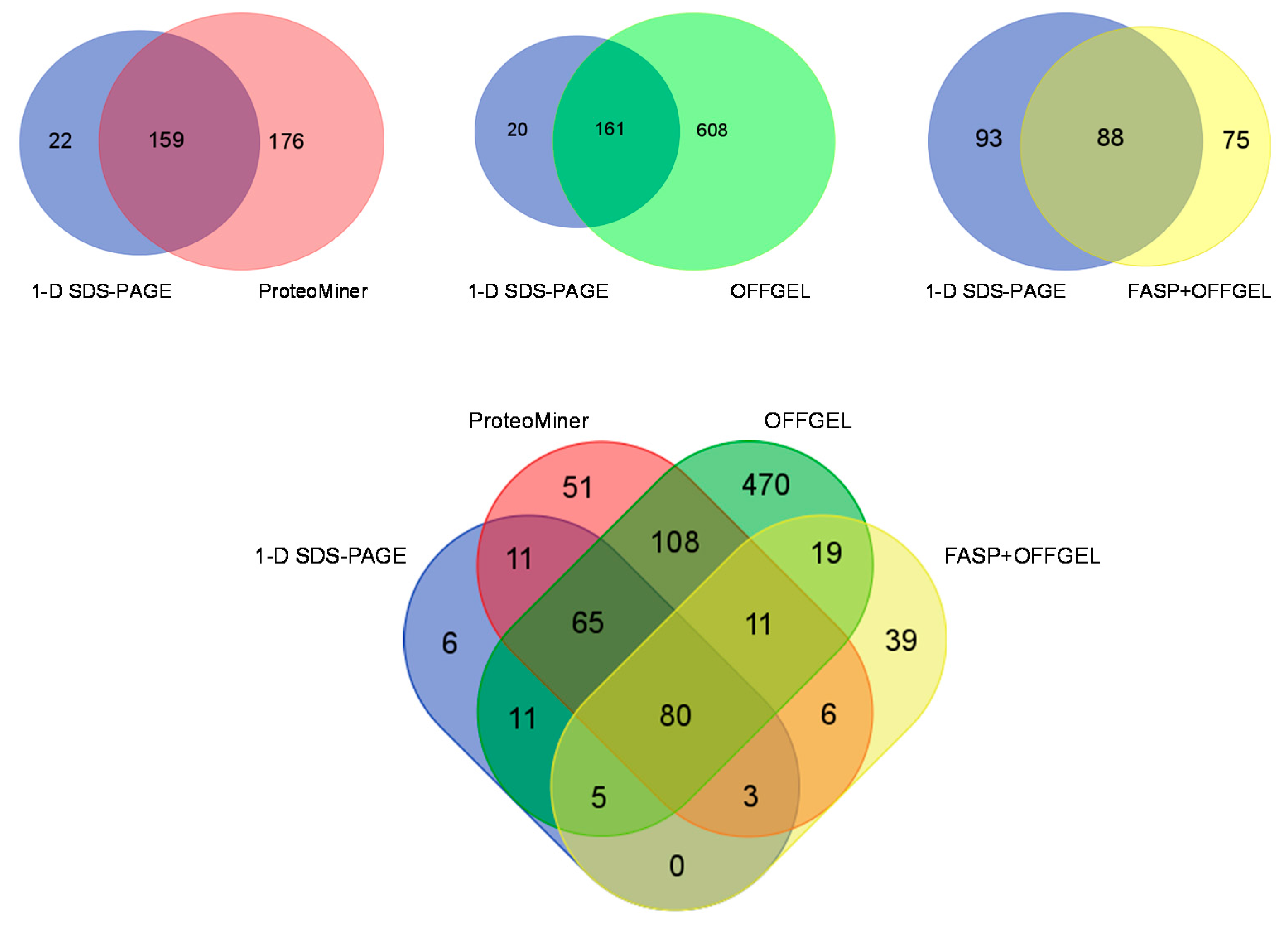
3.6. Comparison of Identification and Overlaps
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gelfi, C.; Vasso, M.; Cerretelli, P. Diversity of human skeletal muscle in health and disease: Contribution of proteomics. J. Proteomics 2011, 74, 774–795. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.K.; McLean, L.; Hayter, J.R.; Pratt, J.M.; Robertson, D.H.; El-Shafei, A.; Gaskell, S.J.; Beynon, R.J. The proteome of chicken skeletal muscle: Changes in soluble protein expression during growth in a layer strain. Proteomics 2004, 4, 2082–2093. [Google Scholar] [CrossRef] [PubMed]
- Parker, K.C.; Walsh, R.J.; Salajegheh, M.; Amato, A.A.; Krastins, B.; Sarracino, D.A.; Greenberg, S.A. Characterization of human skeletal muscle biopsy samples using shotgun proteomics. J. Proteome Res. 2009, 8, 3265–3277. [Google Scholar] [CrossRef] [PubMed]
- Burniston, J.G.; Connolly, J.; Kainulainen, H.; Britton, S.L.; Koch, L.G. Label-free profiling of skeletal muscle using high-definition mass spectrometry. Proteomics 2014, 14, 2339–2344. [Google Scholar] [CrossRef] [PubMed]
- Zubarev, R.A. The challenge of the proteome dynamic range and its implications for in-depth proteomics. Proteomics 2013, 13, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Ohlendieck, K. Skeletal muscle proteomics: Current approaches, technical challenges and emerging techniques. Skelet. Muscle 2011, 1, 6. [Google Scholar] [CrossRef] [PubMed]
- Dunkley, T.P.; Watson, R.; Griffin, J.L.; Dupree, P.; Lilley, K.S. Localization of organelle proteins by isotope tagging (LOPIT). Mol. Cell. Proteomics 2004, 3, 1128–1134. [Google Scholar] [CrossRef] [PubMed]
- Ly, L.; Wasinger, V.C. Protein and peptide fractionation, enrichment and depletion: Tools for the complex proteome. Proteomics 2011, 11, 513–534. [Google Scholar] [CrossRef] [PubMed]
- Chromy, B.A.; Gonzales, A.D.; Perkins, J.; Choi, M.W.; Corzett, M.H.; Chang, B.C.; Corzett, C.H.; McCutchen-Maloney, S.L. Proteomic analysis of human serum by two-dimensional differential gel electrophoresis after depletion of high-abundant proteins. J. Proteome Res. 2004, 3, 1120–1127. [Google Scholar] [CrossRef] [PubMed]
- Granger, J.; Siddiqui, J.; Copeland, S.; Remick, D. Albumin depletion of human plasma also removes low abundance proteins including the cytokines. Proteomics 2005, 5, 4713–4718. [Google Scholar] [CrossRef] [PubMed]
- Tu, C.; Rudnick, P.A.; Martinez, M.Y.; Cheek, K.L.; Stein, S.E.; Slebos, R.J.; Liebler, D.C. Depletion of abundant plasma proteins and limitations of plasma proteomics. J. Proteome Res. 2010, 9, 4982–4991. [Google Scholar] [CrossRef] [PubMed]
- Thulasiraman, V.; Lin, S.; Gheorghiu, L.; Lathrop, J.; Lomas, L.; Hammond, D.; Boschetti, E. Reduction of the concentration difference of proteins in biological liquids using a library of combinatorial ligands. Electrophoresis 2005, 26, 3561–3571. [Google Scholar] [CrossRef] [PubMed]
- Guerrier, L.; Thulasiraman, V.; Castagna, A.; Fortis, F.; Lin, S.; Lomas, L.; Righetti, P.G.; Boschetti, E. Reducing protein concentration range of biological samples using solid-phase ligand libraries. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2006, 833, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Shores, K.S.; Udugamasooriya, D.G.; Kodadek, T.; Knapp, D.R. Use of peptide analogue diversity library beads for increased depth of proteomic analysis: Application to cerebrospinal fluid. J. Proteome Res. 2008, 7, 1922–1931. [Google Scholar] [CrossRef] [PubMed]
- Rivers, J.; Hughes, C.; McKenna, T.; Woolerton, Y.; Vissers, J.P.C.; Langridge, J.I.; Beynon, R.J. Asymmetric proteome equalization of the skeletal muscle proteome using a combinatorial hexapeptide library. PLoS ONE 2011, 6, e28902. [Google Scholar] [CrossRef] [PubMed]
- Candiano, G.; Santucci, L.; Bruschi, M.; Petretto, A.; D’Ambrosio, C.; Scaloni, A.; Righetti, P.G.; Ghiggeri, G.M. “Cheek-to-cheek” urinary proteome profiling via combinatorial peptide ligand libraries: A novel, unexpected elution system. J. Proteomics 2012, 75, 796–805. [Google Scholar] [CrossRef] [PubMed]
- Johnston, I.A.; Fleming, J.D.; Crockford, T. Thermal acclimation and muscle contractile properties in cyprinid fish. Am. J. Physiol. 1990, 259, R231–R236. [Google Scholar] [PubMed]
- Gracey, A.Y.; Fraser, E.J.; Li, W.; Fang, Y.; Taylor, R.R.; Rogers, J.; Brass, A.; Cossins, A.R. Coping with cold: An integrative, multitissue analysis of the transcriptome of a poikilothermic vertebrate. Proc. Natl. Acad. Sci. USA 2004, 101, 16970–16975. [Google Scholar] [CrossRef] [PubMed]
- Fuentes, E.N.; Zuloaga, R.; Nardocci, G.; Fernandez de la Reguera, C.; Simonet, N.; Fumeron, R. Skeletal muscle plasticity induced by seasonal acclimatization in carp involves differential expression of rRNA and molecules that epigenetically regulate its synthesis. Comp. Biochem. Physiol. Part B Biochem. Mol. Biol. 2014, 172–173, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.K.; Brownridge, P.; Owen, M.A.; Davies, S.J.; Young, I.S.; Whitfield, P.D. A proteomics strategy for determining the synthesis and degradation rates of individual proteins in fish. J. Proteomics 2012, 75, 4471–4477. [Google Scholar] [CrossRef] [PubMed]
- Doherty, M.K.; Owen, M.A.; Davies, S.J.; Young, I.S.; Whitfield, P.D. Assessment of global proteome dynamics in carp: A model for investigating environmental stress. J. Proteome Res. 2013, 12, 5246–5252. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Mann, M. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 2008, 26, 1367–1372. [Google Scholar] [CrossRef] [PubMed]
- Cox, J.; Neuhauser, N.; Michalski, A.; Scheltema, R.A.; Olsen, J.V.; Mann, M. Andromeda: A peptide search engine integrated into the MaxQuant environment. J. Proteome Res. 2011, 10, 1794–1805. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Thomas, P.D. PANTHER in 2013: Modeling the evolution of gene function, and other gene attributes, in the context of phylogenetic trees. Nucleic Acids Res. 2013, 41, D377–D386. [Google Scholar] [CrossRef] [PubMed]
- Mi, H.; Muruganujan, A.; Casagrande, J.T.; Thomas, P.D. Large-scale gene function analysis with the PANTHER classification system. Nat. Protoc. 2013, 8, 1551–1566. [Google Scholar] [CrossRef] [PubMed]
- McLean, L.; Young, I.S.; Doherty, M.K.; Robertson, D.H.; Cossins, A.R.; Gracey, A.Y.; Beynon, R.J.; Whitfield, P.D. Global cooling: Cold acclimation and the expression of soluble proteins in carp skeletal muscle. Proteomics 2007, 7, 2667–2681. [Google Scholar] [CrossRef] [PubMed]
- Deshmukh, A.S.; Murgia, M.; Nagaraj, N.; Treebak, J.T.; Cox, J.; Mann, M. Deep proteomics of mouse skeletal muscle enables quantitation of protein isoforms, metabolic pathways, and transcription factors. Mol. Cell. Proteomics 2015, 14, 841–853. [Google Scholar] [CrossRef] [PubMed]
- Murgia, M.; Nagaraj, N.; Deshmukh, A.S.; Zeiler, M.; Cancellara, P.; Moretti, I.; Reggiani, C.; Schiaffino, S.; Mann, M. Single muscle fiber proteomics reveals unexpected mitochondrial specialization. EMBO Rep. 2015, 16, 387–395. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Technique a | Total Number of Identified Proteins | Mean Number of Identified Proteins | Mean Number of Peptides per Protein | Mean Sequence Coverage (%) |
|---|---|---|---|---|
| 1-D SDS-PAGE | 182 | 120 ± 1 | 7.5 ± 0.2 | 20 ± 2 |
| ProteoMiner | 334 | 209 ± 15 | 7.3 ± 0.4 | 18 ± 4 |
| OFFGEL | 766 | 425 ± 62 | 6.4 ± 0.1 | 17 ± 0.2 |
| FASP+OFFGEL | 163 | 118 ± 9 | 20.5 ± 0.3 | 29 ± 1 |
| Method | 1-D SDS-PAGE | ProteoMiner | OFFGEL | FASP |
|---|---|---|---|---|
| Time from tissue homogenisation to protein digest | 2 days | 3 days | 4 days+ | 3 days |
| Number of protein digests per tissue sample | 12 | 12 | 144 | 12 |
| Overlap between replicate identifications | 66 | 57 | 46 | 66 |
| Total experimental time | 4 days | 5 days | 25 days | 4 days |
| File size demands | Low | Low | Very high | Low |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Geary, B.; Young, I.S.; Cash, P.; Whitfield, P.D.; Doherty, M.K. Comparing Simplification Strategies for the Skeletal Muscle Proteome. Proteomes 2016, 4, 10. https://doi.org/10.3390/proteomes4010010
Geary B, Young IS, Cash P, Whitfield PD, Doherty MK. Comparing Simplification Strategies for the Skeletal Muscle Proteome. Proteomes. 2016; 4(1):10. https://doi.org/10.3390/proteomes4010010
Chicago/Turabian StyleGeary, Bethany, Iain S. Young, Phillip Cash, Phillip D. Whitfield, and Mary K. Doherty. 2016. "Comparing Simplification Strategies for the Skeletal Muscle Proteome" Proteomes 4, no. 1: 10. https://doi.org/10.3390/proteomes4010010
APA StyleGeary, B., Young, I. S., Cash, P., Whitfield, P. D., & Doherty, M. K. (2016). Comparing Simplification Strategies for the Skeletal Muscle Proteome. Proteomes, 4(1), 10. https://doi.org/10.3390/proteomes4010010