
Urine-HILIC: Automated Sample Preparation for Bottom-Up Urinary Proteome Profiling in Clinical Proteomics


Abstract
1. Introduction
2. Materials and Methods
2.1. Urine Sample Collection Protocol and Pilot Study Cohort
2.2. Sample Preparation
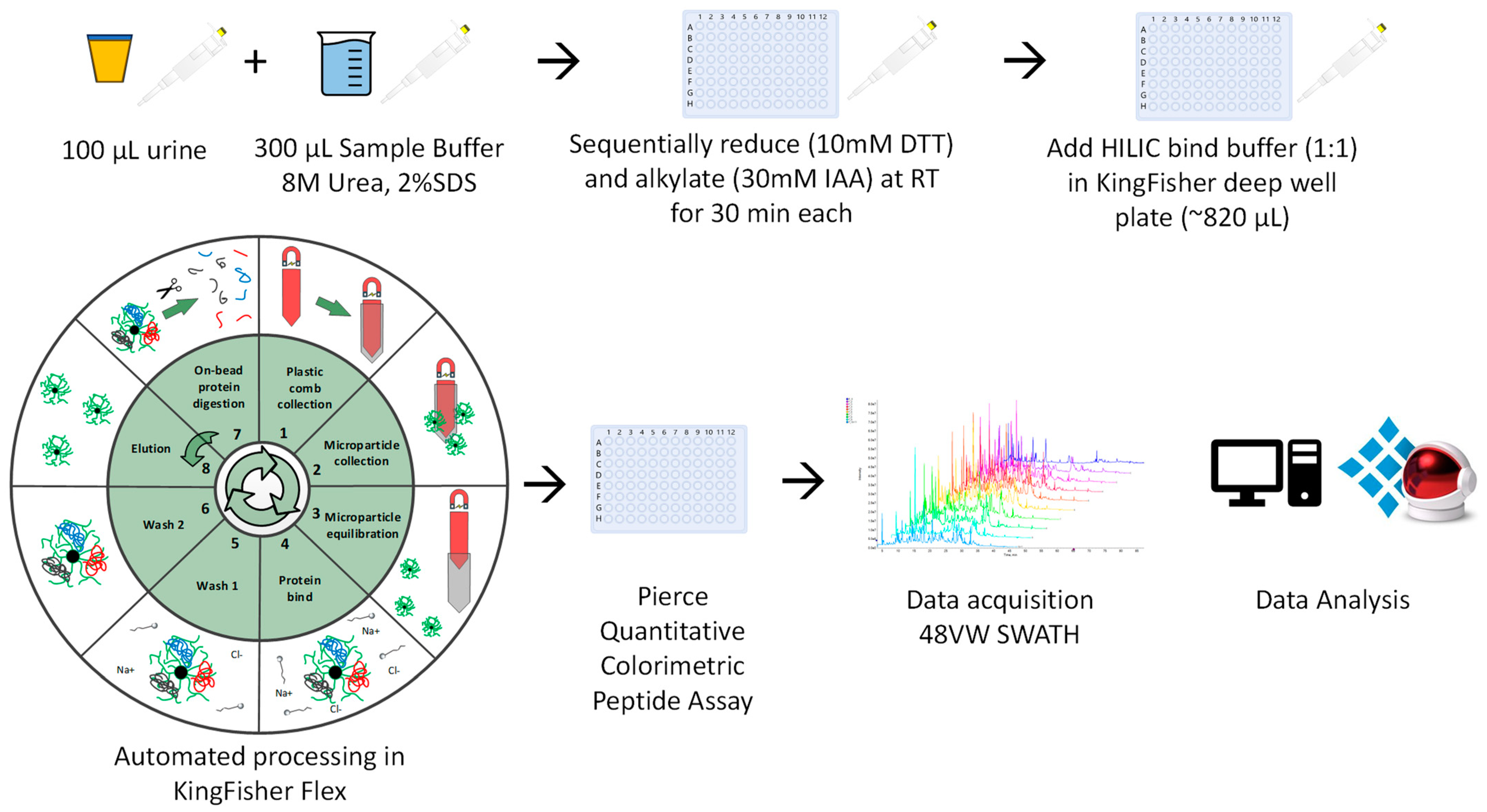
2.2.1. Automated Urine-HILIC Workflow
2.2.2. On-Membrane Workflow Based on MStern Blot
2.3. LC SWATH-MS Data Acquisition
2.4. Data Processing
2.5. Bioinformatic and Clincial Data Analysis
3. Results
3.1. Workflow Time Comparisons
3.2. Peptide Yield
3.3. Peptides and Proteins Identified
3.4. Protein Properties and Dynamic Range Comparison
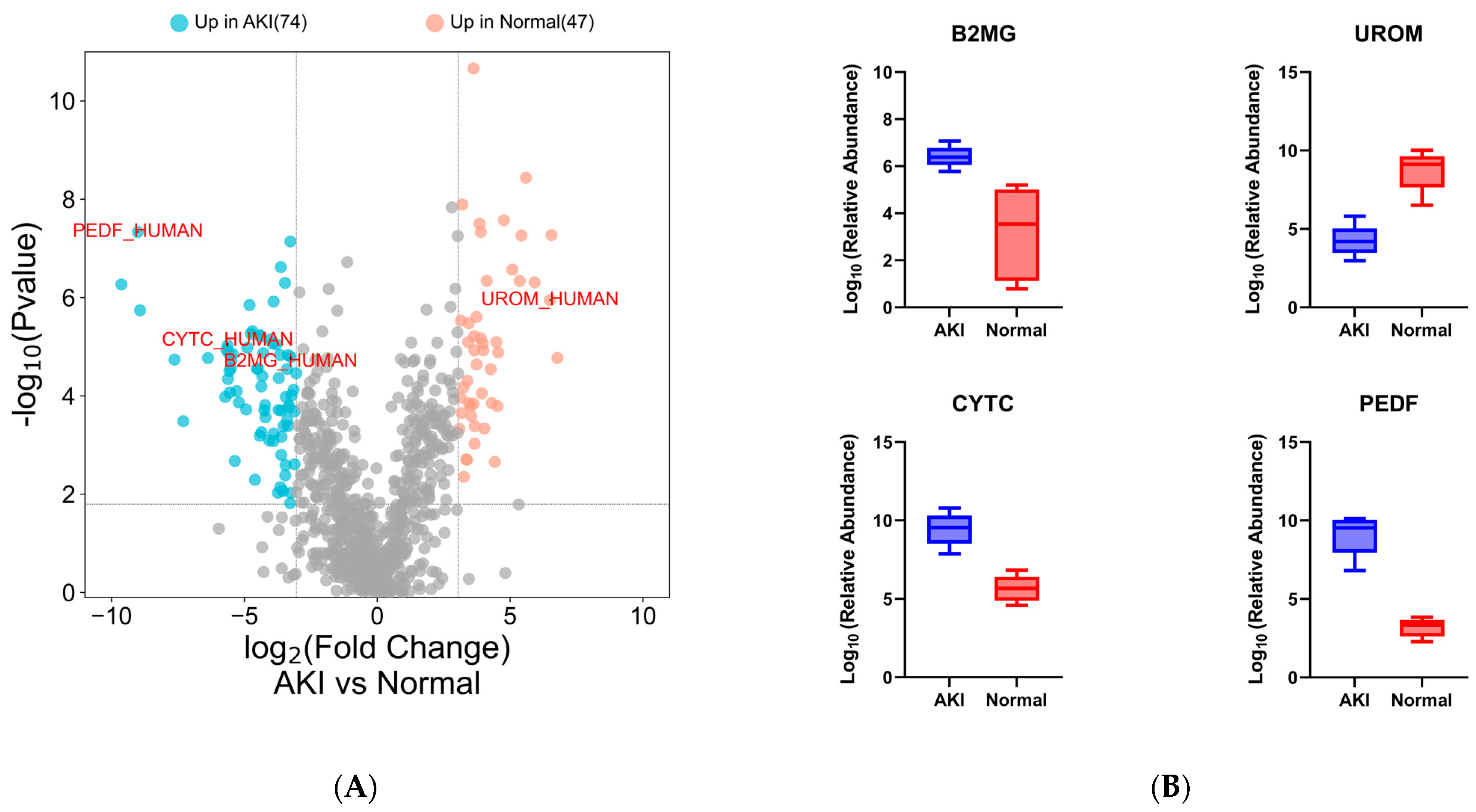
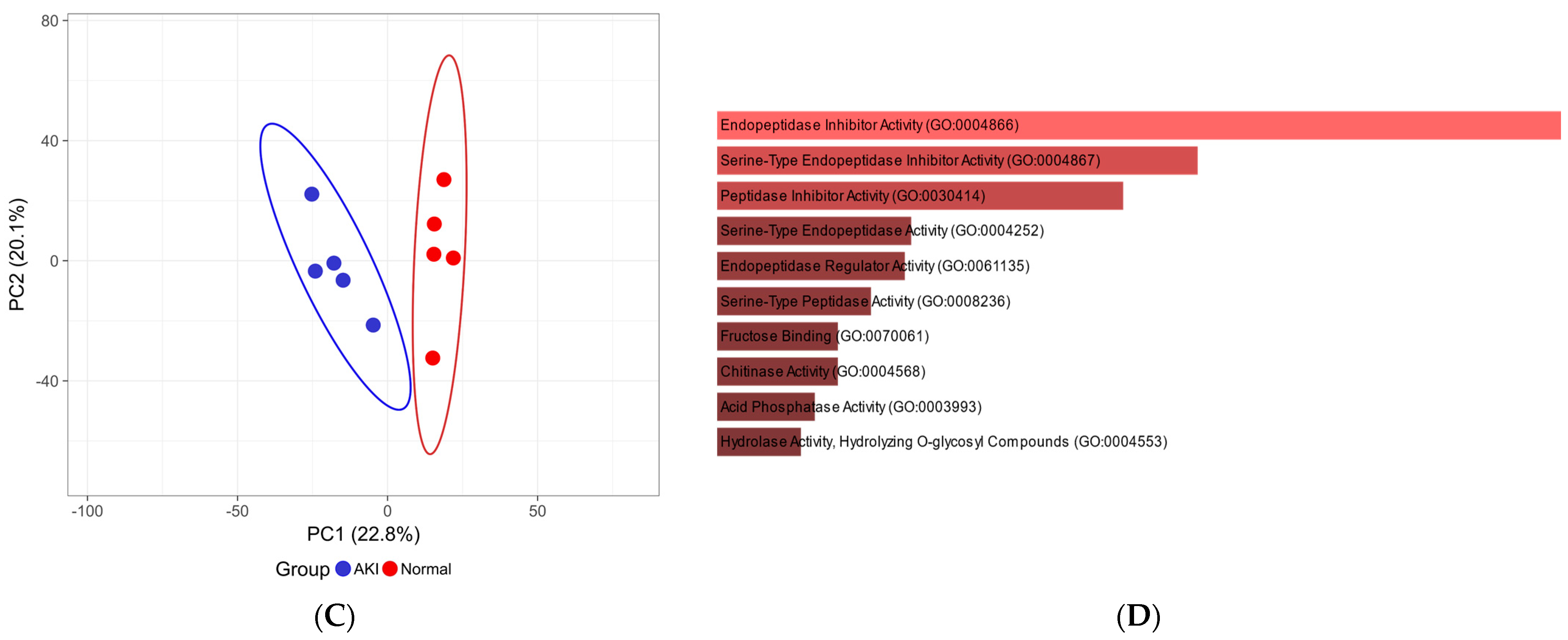
3.5. Pilot Study Clinical Data
3.6. Pilot Study: Data-Independent Analysis for Clinical Samples
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Prunotto, M.; Ghiggeri, G.M.; Candiano, G.; Lescuyer, P.; Hochstrasser, D.; Moll, S. Urinary Proteomics and Drug Discovery in Chronic Kidney Disease: A New Perspective. J. Proteome Res. 2011, 10, 126–132. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Hao, L.; Ricke, W.A.; Li, L. Biomarker Discovery in Mass Spectrometry-Based Urinary Proteomics. Proteom. Clin. Appl. 2016, 10, 358–370. [Google Scholar] [CrossRef] [PubMed]
- Decramer, S.; de Peredo, A.G.; Breuil, B.; Mischak, H.; Monsarrat, B.; Bascands, J.L.; Schanstra, J.P. Urine in Clinical Proteomics. Mol. Cell. Proteom. 2008, 7, 1850–1862. [Google Scholar] [CrossRef] [PubMed]
- Beasley-Green, A. Urine Proteomics in the Era of Mass Spectrometry. Int. Neurourol. J. 2016, 20, S70–S75. [Google Scholar] [CrossRef]
- Ignjatovic, V.; Geyer, P.E.; Palaniappan, K.K.; Chaaban, J.E.; Omenn, G.S.; Baker, M.S.; Deutsch, E.W.; Schwenk, J.M. Mass Spectrometry-Based Plasma Proteomics: Considerations from Sample Collection to Achieving Translational Data. J. Proteome Res. 2019, 18, 4085. [Google Scholar] [CrossRef]
- Hortin, G.L.; Sviridov, D. The Dynamic Range Problem in the Analysis of the Plasma Proteome. J. Proteom. 2010, 73, 629–636. [Google Scholar] [CrossRef]
- Kalantari, S.; Jafari, A.; Moradpoor, R.; Ghasemi, E.; Khalkhal, E. Human Urine Proteomics: Analytical Techniques and Clinical Applications in Renal Diseases. Int. J. Proteom. 2015, 2015, 782798. [Google Scholar] [CrossRef]
- Hu, S.; Loo, J.A.; Wong, D.T. Human Body Fluid Proteome Analysis. Proteomics 2006, 6, 6326–6353. [Google Scholar] [CrossRef]
- Gao, Y. Urine Is a Better Biomarker Source than Blood Especially for Kidney Diseases. Adv. Exp. Med. Biol. 2015, 845, 3–12. [Google Scholar] [CrossRef]
- Zou, L.; Sun, W. Human Urine Proteome: A Powerful Source for Clinical Research. Adv. Exp. Med. Biol. 2015, 845, 31–42. [Google Scholar] [CrossRef]
- Beretov, J.; Wasinger, V.C.; Schwartz, P.; Graham, P.H.; Li, Y. A Standardized and Reproducible Urine Preparation Protocol for Cancer Biomarkers Discovery. Biomark Cancer 2014, 6, BIC.S17991. [Google Scholar] [CrossRef]
- Beretov, J.; Wasinger, V.C.; Millar, E.K.A.; Schwartz, P.; Graham, P.H.; Li, Y. Proteomic Analysis of Urine to Identify Breast Cancer Biomarker Candidates Using a Label-Free LC-MS/MS Approach. PLoS ONE 2015, 10, e0141876. [Google Scholar] [CrossRef] [PubMed]
- Saito, S.; Hirao, Y.; Quadery, A.F.; Xu, B.; Elguoshy, A.; Fujinaka, H.; Koma, S.; Yamamoto, K.; Yamamoto, T. The Optimized Workflow for Sample Preparation in LC-MS/MS-Based Urine Proteomics. Methods Protoc. 2019, 2, 46. [Google Scholar] [CrossRef]
- Tkáčiková, S.; Talian, I.; Sabo, J. Optimisation of Urine Sample Preparation for Shotgun Proteomics. Open Chem. 2020, 18, 850–856. [Google Scholar] [CrossRef]
- Percy, A.J.; Yang, J.; Hardie, D.B.; Chambers, A.G.; Tamura-Wells, J.; Borchers, C.H. Precise Quantitation of 136 Urinary Proteins by LC/MRM-MS Using Stable Isotope Labeled Peptides as Internal Standards for Biomarker Discovery and/or Verification Studies. Methods 2015, 81, 24–33. [Google Scholar] [CrossRef]
- Winter, S.V.; Karayel, O.; Strauss, M.T.; Padmanabhan, S.; Surface, M.; Merchant, K.; Alcalay, R.N.; Mann, M. Urinary Proteome Profiling for Stratifying Patients with Familial Parkinson’s Disease. EMBO Mol. Med. 2021, 13, e13257. [Google Scholar] [CrossRef]
- Berger, S.T.; Ahmed, S.; Muntel, J.; Polo, N.C.; Bachur, R.; Kentsis, A.; Steen, J.; Steen, H. MStern Blotting–High Throughput Polyvinylidene Fluoride (PVDF) Membrane-Based Proteomic Sample Preparation for 96-Well Plates. Mol. Cell Proteom. 2015, 14, 2814. [Google Scholar] [CrossRef]
- Ding, H.; Fazelinia, H.; Spruce, L.A.; Weiss, D.A.; Zderic, S.A.; Seeholzer, S.H. Urine Proteomics: Evaluation of Different Sample Preparation Workflows for Quantitative, Reproducible, and Improved Depth of Analysis. J. Proteome Res. 2020, 19, 1857–1862. [Google Scholar] [CrossRef]
- Jonscher, K.R.; Osypuk, A.A.; van Bokhoven, A.; Lucia, M.S. Evaluation of Urinary Protein Precipitation Protocols for the Multidisciplinary Approach to the Study of Chronic Pelvic Pain Research Network. J. Biomol. Tech. 2014, 25, 118–126. [Google Scholar] [CrossRef][Green Version]
- Thongboonkerd, V.; McLeish, K.R.; Arthur, J.M.; Klein, J.B. Proteomic Analysis of Normal Human Urinary Proteins Isolated by Acetone Precipitation or Ultracentrifugation. Kidney Int. 2002, 62, 1461–1469. [Google Scholar] [CrossRef]
- Wiśniewski, J.R. Quantitative Evaluation of Filter Aided Sample Preparation (FASP) and Multienzyme Digestion FASP Protocols. Anal. Chem. 2016, 88, 5438–5443. [Google Scholar] [CrossRef] [PubMed]
- Macklin, A.; Khan, S.; Kislinger, T. Recent Advances in Mass Spectrometry Based Clinical Proteomics: Applications to Cancer Research. Clin. Proteom. 2020, 17, 1–25. [Google Scholar] [CrossRef]
- Tang, X.; Xiao, X.; Sun, H.; Zheng, S.; Xiao, X.; Guo, Z.; Liu, X.; Sun, W. 96DRA-Urine: A High Throughput Sample Preparation Method for Urinary Proteome Analysis. J. Proteom. 2022, 257, 104529. [Google Scholar] [CrossRef]
- Dupree, E.J.; Jayathirtha, M.; Yorkey, H.; Mihasan, M.; Petre, B.A.; Darie, C.C. A Critical Review of Bottom-Up Proteomics: The Good, the Bad, and the Future of This Field. Proteomes 2020, 8, 14. [Google Scholar] [CrossRef]
- Nweke, E.E.; Naicker, P.; Aron, S.; Stoychev, S.; Devar, J.; Tabb, D.L.; Omoshoro-Jones, J.; Smith, M.; Candy, G. SWATH-MS Based Proteomic Profiling of Pancreatic Ductal Adenocarcinoma Tumours Reveals the Interplay between the Extracellular Matrix and Related Intracellular Pathways. PLoS ONE 2020, 15, e0240453. [Google Scholar] [CrossRef] [PubMed]
- Baichan, P.; Naicker, P.; Augustine, T.N.; Smith, M.; Candy, G.; Devar, J.; Nweke, E.E. Proteomic Analysis Identifies Dysregulated Proteins and Associated Molecular Pathways in a Cohort of Gallbladder Cancer Patients of African Ancestry. Clin. Proteom. 2023, 20, 8. [Google Scholar] [CrossRef] [PubMed]
- Zuma, K.; Simbayi, L.; Zungu, N.; Moyo, S.; Marinda, E.; Jooste, S.; North, A.; Nadol, P.; Aynalem, G.; Igumbor, E.; et al. The HIV Epidemic in South Africa: Key Findings from 2017 National Population-Based Survey. Int. J. Environ. Res. Public Health 2022, 19, 8125. [Google Scholar] [CrossRef]
- Seedat, F.; Martinson, N.; Motlhaoleng, K.; Abraham, P.; Mancama, D.; Naicker, S.; Variava, E. Acute Kidney Injury, Risk Factors, and Prognosis in Hospitalized HIV-Infected Adults in South Africa, Compared by Tenofovir Exposure. AIDS Res. Hum. Retroviruses 2017, 33, 33–40. [Google Scholar] [CrossRef]
- Ostermann, M.; Joannidis, M. Acute Kidney Injury 2016: Diagnosis and Diagnostic Workup. Crit. Care 2016, 20, 299. [Google Scholar] [CrossRef]
- Makris, K.; Spanou, L. Acute Kidney Injury: Diagnostic Approaches and Controversies. Clin. Biochem. Rev. 2016, 37, 153–175. [Google Scholar]
- Waikar, S.S.; Betensky, R.A.; Bonventre, J.V. Creatinine as the Gold Standard for Kidney Injury Biomarker Studies? Nephrol. Dial. Transplant. 2009, 24, 3263–3265. [Google Scholar] [CrossRef] [PubMed]
- Koh, H.M.; Suresh, K. Tenofovir-Induced Nephrotoxicity: A Retrospective Cohort Study. Med. J. Malays. 2016, 71, 308–312. [Google Scholar]
- Perazella, M.A. Tenofovir-Induced Kidney Disease: An Acquired Renal Tubular Mitochondriopathy. Kidney Int. 2010, 78, 1060–1063. [Google Scholar] [CrossRef]
- Mtisi, T.J.; Ndhlovu, C.E.; Maponga, C.C.; Morse, G.D. Tenofovir-Associated Kidney Disease in Africans: A Systematic Review. AIDS Res. Ther. 2019, 16, 12. [Google Scholar] [CrossRef]
- Ezinga, M.; Wetzels, J.F.M.; Bosch, M.E.W.; Van Der Ven, A.J.A.M.; Burger, D.M. Long-Term Treatment with Tenofovir: Prevalence of Kidney Tubular Dysfunction and Its Association with Tenofovir Plasma Concentration. Antivir. Ther. 2014, 19, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Machado, M.N.; Nakazone, M.A.; Maia, L.N. Acute Kidney Injury Based on KDIGO (Kidney Disease Improving Global Outcomes) Criteria in Patients with Elevated Baseline Serum Creatinine Undergoing Cardiac Surgery. Rev. Bras. Cir. Cardiovasc. 2014, 29, 299–307. [Google Scholar] [CrossRef]
- Röst, H.L.; Rosenberger, G.; Navarro, P.; Gillet, L.; Miladinović, S.M.; Schubert, O.T.; Wolski, W.; Collins, B.C.; Malmström, J.; Malmström, L.; et al. OpenSWATH Enables Automated, Targeted Analysis of Data-Independent Acquisition MS Data. Nat. Biotechnol. 2014, 32, 219–223. [Google Scholar] [CrossRef]
- Reiter, L.; Rinner, O.; Picotti, P.; Hüttenhain, R.; Beck, M.; Brusniak, M.-Y.; Hengartner, M.O.; Aebersold, R. MProphet: Automated Data Processing and Statistical Validation for Large-Scale SRM Experiments. Nat. Methods 2011, 8, 430–435. [Google Scholar] [CrossRef]
- Zhang, B.; Chambers, M.C.; Tabb, D.L. Proteomic Parsimony through Bipartite Graph Analysis Improves Accuracy and Transparency. J. Proteome Res. 2007, 6, 3549–3557. [Google Scholar] [CrossRef]
- Storey, J.D. A Direct Approach to False Discovery Rates. J. R. Stat. Soc. Ser. B Stat. Methodol. 2002, 64, 479–498. [Google Scholar] [CrossRef]
- Choi, M.; Chang, C.-Y.; Clough, T.; Broudy, D.; Killeen, T.; MacLean, B.; Vitek, O. MSstats: An R Package for Statistical Analysis of Quantitative Mass Spectrometry-Based Proteomic Experiments. Bioinformatics 2014, 30, 2524–2526. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, E.; Gattiker, A.; Hoogland, C.; Ivanyi, I.; Appel, R.D.; Bairoch, A. ExPASy: The Proteomics Server for in-Depth Protein Knowledge and Analysis. Nucleic Acids Res. 2003, 31, 3784–3788. [Google Scholar] [CrossRef] [PubMed]
- Metsalu, T.; Vilo, J. ClustVis: A Web Tool for Visualizing Clustering of Multivariate Data Using Principal Component Analysis and Heatmap. Nucleic Acids Res. 2015, 43, W566. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’ayan, A. Enrichr: Interactive and Collaborative HTML5 Gene List Enrichment Analysis Tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef]
- Yeung, Y.-G.; Nieves, E.; Angeletti, R.H.; Stanley, E.R. Removal of Detergents from Protein Digests for Mass Spectrometry Analysis. Anal. Biochem. 2008, 382, 135–137. [Google Scholar] [CrossRef]
- Lacroix, C.; Caubet, C.; Gonzalez-de-Peredo, A.; Breuil, B.; Bouyssié, D.; Stella, A.; Garrigues, L.; Le Gall, C.; Raevel, A.; Massoubre, A.; et al. Label-Free Quantitative Urinary Proteomics Identifies the Arginase Pathway as a New Player in Congenital Obstructive Nephropathy. Mol. Cell Proteom. 2014, 13, 3421–3434. [Google Scholar] [CrossRef]
- Potriquet, J.; Laohaviroj, M.; Bethony, J.M.; Mulvenna, J. A Modified FASP Protocol for High-Throughput Preparation of Protein Samples for Mass Spectrometry. PLoS ONE 2017, 12, e0175967. [Google Scholar] [CrossRef]
- Yu, Y.; Pieper, R. Urine Sample Preparation in 96-Well Filter Plates to Characterize Inflammatory and Infectious Diseases of the Urinary Tract; Springer: Dordrecht, The Netherlands, 2015; pp. 77–87. [Google Scholar]
- Schaub, S.; Wilkins, J.A.; Antonovici, M.; Krokhin, O.; Weiler, T.; Rush, D.; Nickerson, P. Proteomic-Based Identification of Cleaved Urinary Beta2-Microglobulin as a Potential Marker for Acute Tubular Injury in Renal Allografts. Am. J. Transplant. 2005, 5, 729–738. [Google Scholar] [CrossRef]
- Heise, D.; Rentsch, K.; Braeuer, A.; Friedrich, M.; Quintel, M. Comparison of Urinary Neutrophil Glucosaminidase-Associated Lipocalin, Cystatin C, and Alpha1-Microglobulin for Early Detection of Acute Renal Injury after Cardiac Surgery. Eur. J. Cardiothorac Surg. 2011, 39, 38–43. [Google Scholar] [CrossRef]
- Du, Y.; Zappitelli, M.; Mian, A.; Bennett, M.; Ma, Q.; Devarajan, P.; Mehta, R.; Goldstein, S.L. Urinary Biomarkers to Detect Acute Kidney Injury in the Pediatric Emergency Center. Pediatr. Nephrol. 2011, 26, 267–274. [Google Scholar] [CrossRef]
- Endre, Z.H.; Pickering, J.W.; Walker, R.J.; Devarajan, P.; Edelstein, C.L.; Bonventre, J.V.; Frampton, C.M.; Bennett, M.R.; Ma, Q.; Sabbisetti, V.S.; et al. Improved Performance of Urinary Biomarkers of Acute Kidney Injury in the Critically Ill by Stratification for Injury Duration and Baseline Renal Function. Kidney Int. 2011, 79, 1119–1130. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Li, Q.; Wang, J.; Xu, Z.; Song, C.; Zhuang, R.; Yang, K.; Yang, A.; Jin, B. Cystatin C, a Novel Urinary Biomarker for Sensitive Detection of Acute Kidney Injury during Haemorrhagic Fever with Renal Syndrome. Biomarkers 2010, 15, 410–417. [Google Scholar] [CrossRef] [PubMed]
- Iorember, F.M.; Vehaskari, V.M. Uromodulin: Old Friend with New Roles in Health and Disease. Pediatr. Nephrol. 2014, 29, 1151–1158. [Google Scholar] [CrossRef] [PubMed]
- Devuyst, O.; Bochud, M. Uromodulin, Kidney Function, Cardiovascular Disease, and Mortality. Kidney Int. 2015, 88, 944–946. [Google Scholar] [CrossRef]
- Youhanna, S.; Weber, J.; Beaujean, V.; Glaudemans, B.; Sobek, J.; Devuyst, O. Determination of Uromodulin in Human Urine: Influence of Storage and Processing. Nephrol. Dial. Transplant. 2014, 29, 136–145. [Google Scholar] [CrossRef]
- Prajczer, S.; Heidenreich, U.; Pfaller, W.; Kotanko, P.; Lhotta, K.; Jennings, P. Evidence for a Role of Uromodulin in Chronic Kidney Disease Progression. Nephrol. Dial. Transplant. 2010, 25, 1896–1903. [Google Scholar] [CrossRef]
- Casado, J.L.; Bañón, S.; Santiuste, C.; Serna, J.; Guzman, P.; Tenorio, M.; Liaño, F.; Rey, J.M. del Prevalence and Significance of Proximal Renal Tubular Abnormalities in HIV-Infected Patients Receiving Tenofovir. AIDS 2016, 30, 231–239. [Google Scholar] [CrossRef]
- Waheed, S.; Attia, D.; Estrella, M.M.; Zafar, Y.; Atta, M.G.; Lucas, G.M.; Fine, D.M. Proximal Tubular Dysfunction and Kidney Injury Associated with Tenofovir in HIV Patients: A Case Series. Clin. Kidney J. 2015, 8, 420–425. [Google Scholar] [CrossRef]
- Vidal, F.; Domingo, J.C.; Guallar, J.; Saumoy, M.; Cordobilla, B.; de la Rosa, R.; Giralt, M.; Alvarez, M.L.; Lopez-Dupla, M.; Torres, F.; et al. In Vitro Cytotoxicity and Mitochondrial Toxicity of Tenofovir Alone and in Combination with Other Antiretrovirals in Human Renal Proximal Tubule Cells. Antimicrob. Agents Chemother. 2006, 50, 3824–3832. [Google Scholar] [CrossRef]
- Quintana, L.F.; Campistol, J.M.; Alcolea, M.P.; Banon-Maneus, E.; Sol-Gonzalez, A.; Cutillas, P.R. Application of Label-Free Quantitative Peptidomics for the Identification of Urinary Biomarkers of Kidney Chronic Allograft Dysfunction. Mol. Cell Proteom. 2009, 8, 1658–1673. [Google Scholar] [CrossRef]
- El-Hefnawy, S.M.; Kasemy, Z.A.; Eid, H.A.; Elmadbouh, I.; Mostafa, R.G.; Omar, T.A.; Kasem, H.E.; Ghonaim, E.M.; Ghonaim, M.M.; Saleh, A.A. Potential Impact of Serpin Peptidase Inhibitor Clade (A) Member 4 SERPINA4 (Rs2093266) and SERPINA5 (Rs1955656) Genetic Variants on COVID-19 Induced Acute Kidney Injury. Human Gene 2022, 32, 101023. [Google Scholar] [CrossRef] [PubMed]
- Perez-Riverol, Y.; Bai, J.; Bandla, C.; García-Seisdedos, D.; Hewapathirana, S.; Kamatchinathan, S.; Kundu, D.J.; Prakash, A.; Frericks-Zipper, A.; Eisenacher, M.; et al. The PRIDE Database Resources in 2022: A Hub for Mass Spectrometry-Based Proteomics Evidences. Nucleic Acids Res. 2022, 50, D543–D552. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristic | Normal (n = 5) | AKI (n = 5) | p Value |
|---|---|---|---|
| Age (years) | 35.4 ± 6.6 | 42.4 ± 12.5 | ns |
| Serum CreatinineAdmission (µmol/L) | 53.6 ± 4.17 | 563 ± 213.9 | 0.03 |
| Estimated glomerular filtration rate(mL/min/1.73 m2) | 108.6 ± 25.97 | 14.5 ± 11.8 | 0.03 |
| Urine Phosphate (mmol/L) | 1.62 ± 0.89 | 1.68 ± 0.93 | ns |
| Urine protein:creatinine ratio (g/mmol creat) | 0.025 ± 0.008 | 0.322 ± 0.18 | 0.03 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Govender, I.S.; Mokoena, R.; Stoychev, S.; Naicker, P. Urine-HILIC: Automated Sample Preparation for Bottom-Up Urinary Proteome Profiling in Clinical Proteomics. Proteomes 2023, 11, 29. https://doi.org/10.3390/proteomes11040029
Govender IS, Mokoena R, Stoychev S, Naicker P. Urine-HILIC: Automated Sample Preparation for Bottom-Up Urinary Proteome Profiling in Clinical Proteomics. Proteomes. 2023; 11(4):29. https://doi.org/10.3390/proteomes11040029
Chicago/Turabian StyleGovender, Ireshyn Selvan, Rethabile Mokoena, Stoyan Stoychev, and Previn Naicker. 2023. "Urine-HILIC: Automated Sample Preparation for Bottom-Up Urinary Proteome Profiling in Clinical Proteomics" Proteomes 11, no. 4: 29. https://doi.org/10.3390/proteomes11040029
APA StyleGovender, I. S., Mokoena, R., Stoychev, S., & Naicker, P. (2023). Urine-HILIC: Automated Sample Preparation for Bottom-Up Urinary Proteome Profiling in Clinical Proteomics. Proteomes, 11(4), 29. https://doi.org/10.3390/proteomes11040029