
Comparative Proteomic Analysis of Two Commonly Used Laboratory Yeast Strains: W303 and BY4742




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
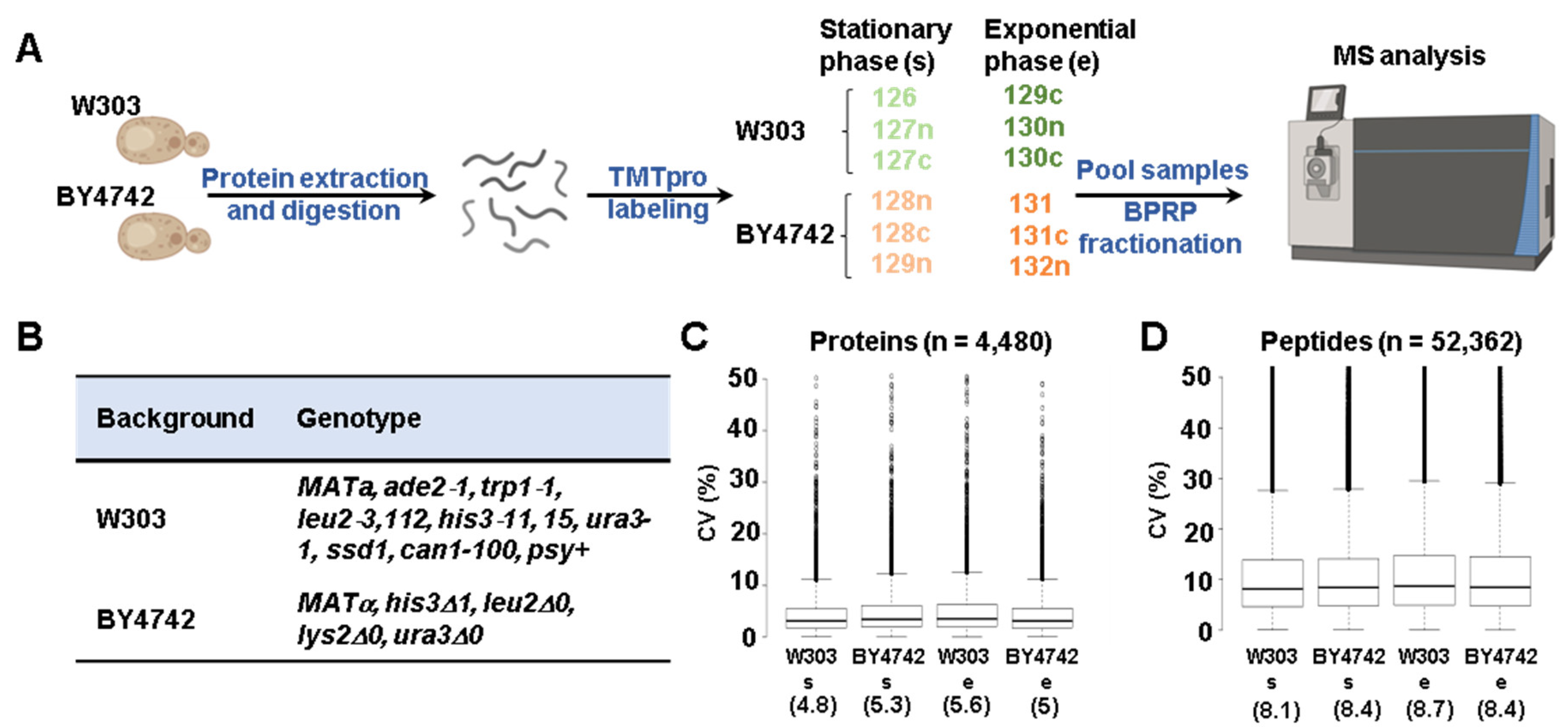
2.1. Materials
2.2. Yeast Growth and Protein Extraction
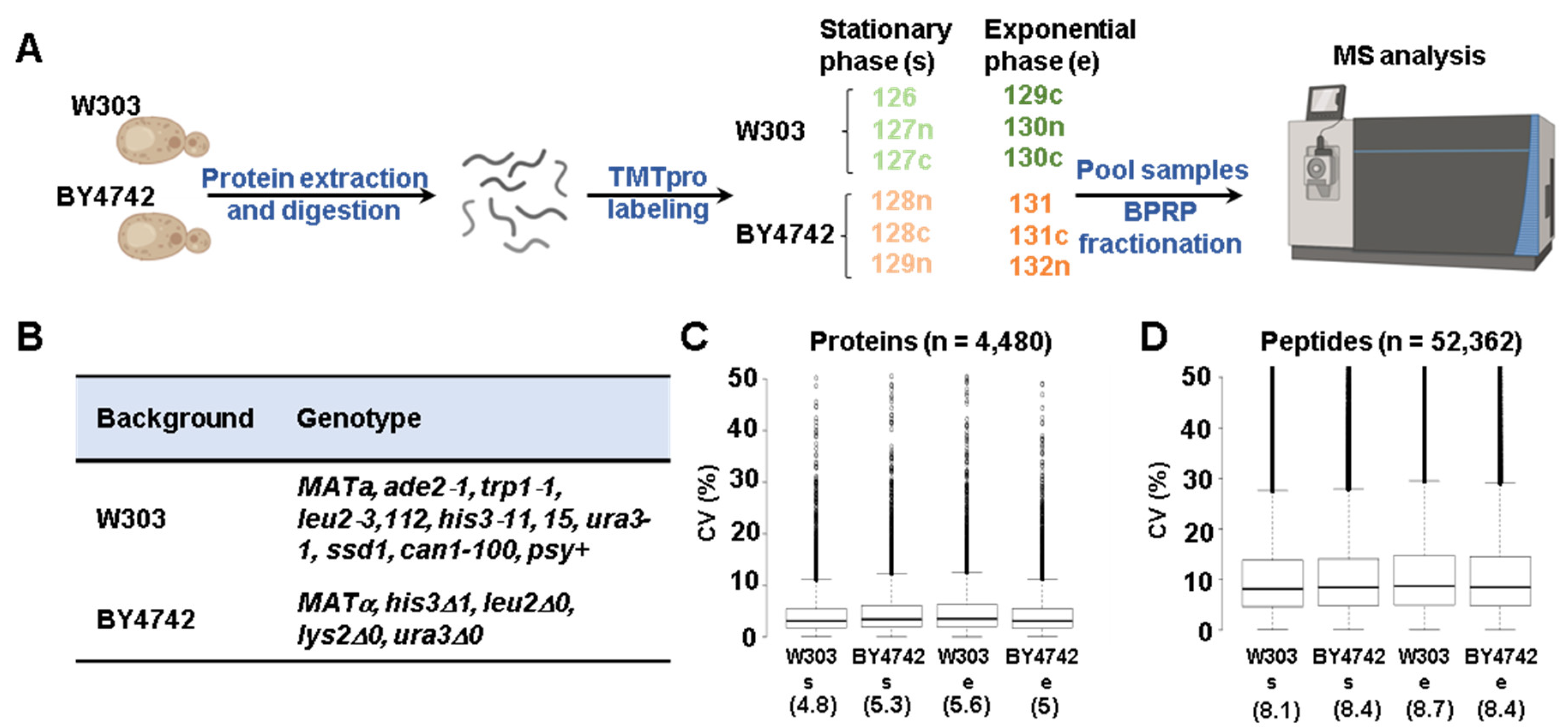
2.3. Protein Digestion, TMT Labeling, and Sample Processing
2.4. Mass Spectrometry Data Acquisition and Processing
3. Results and Discussion
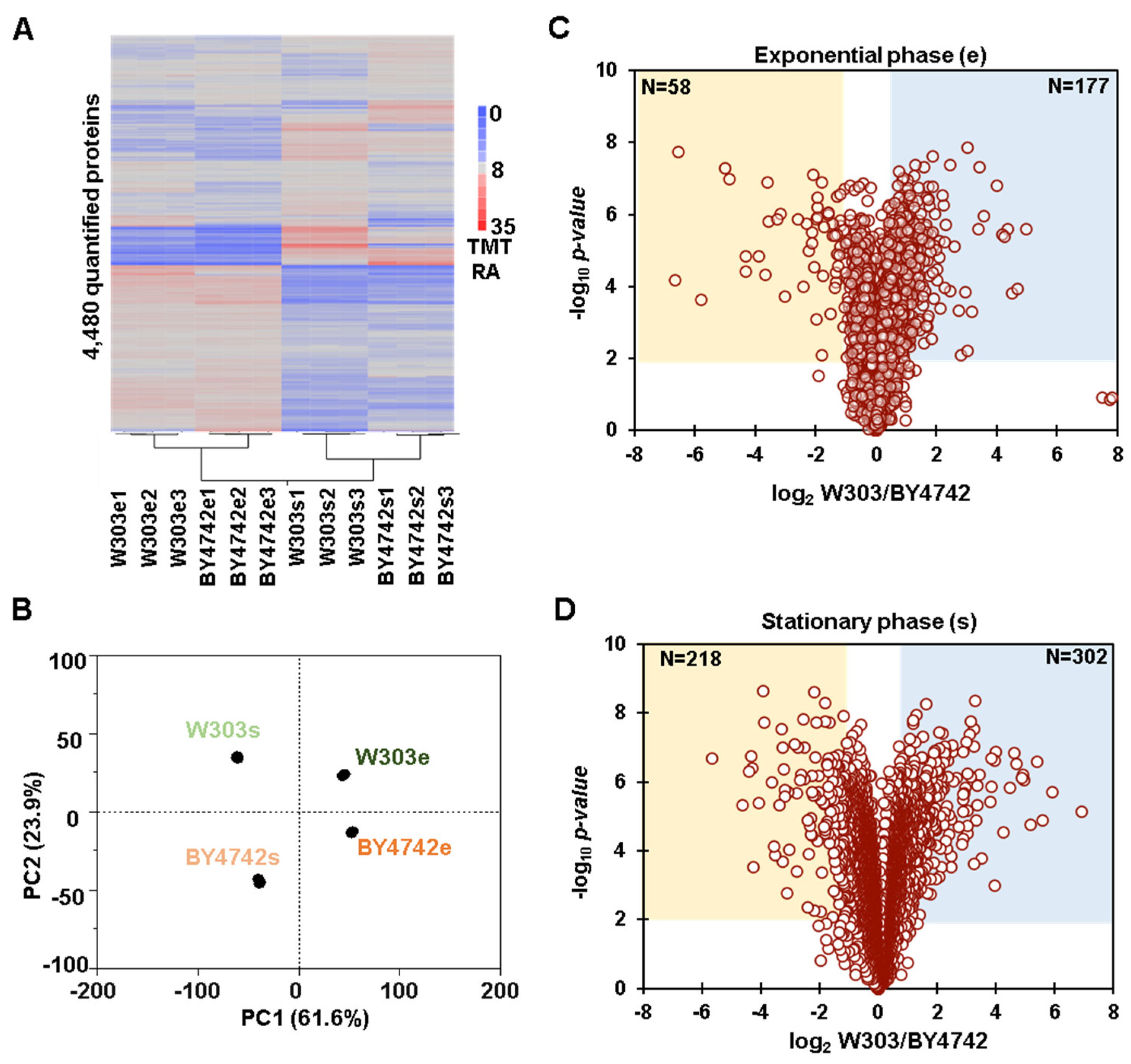
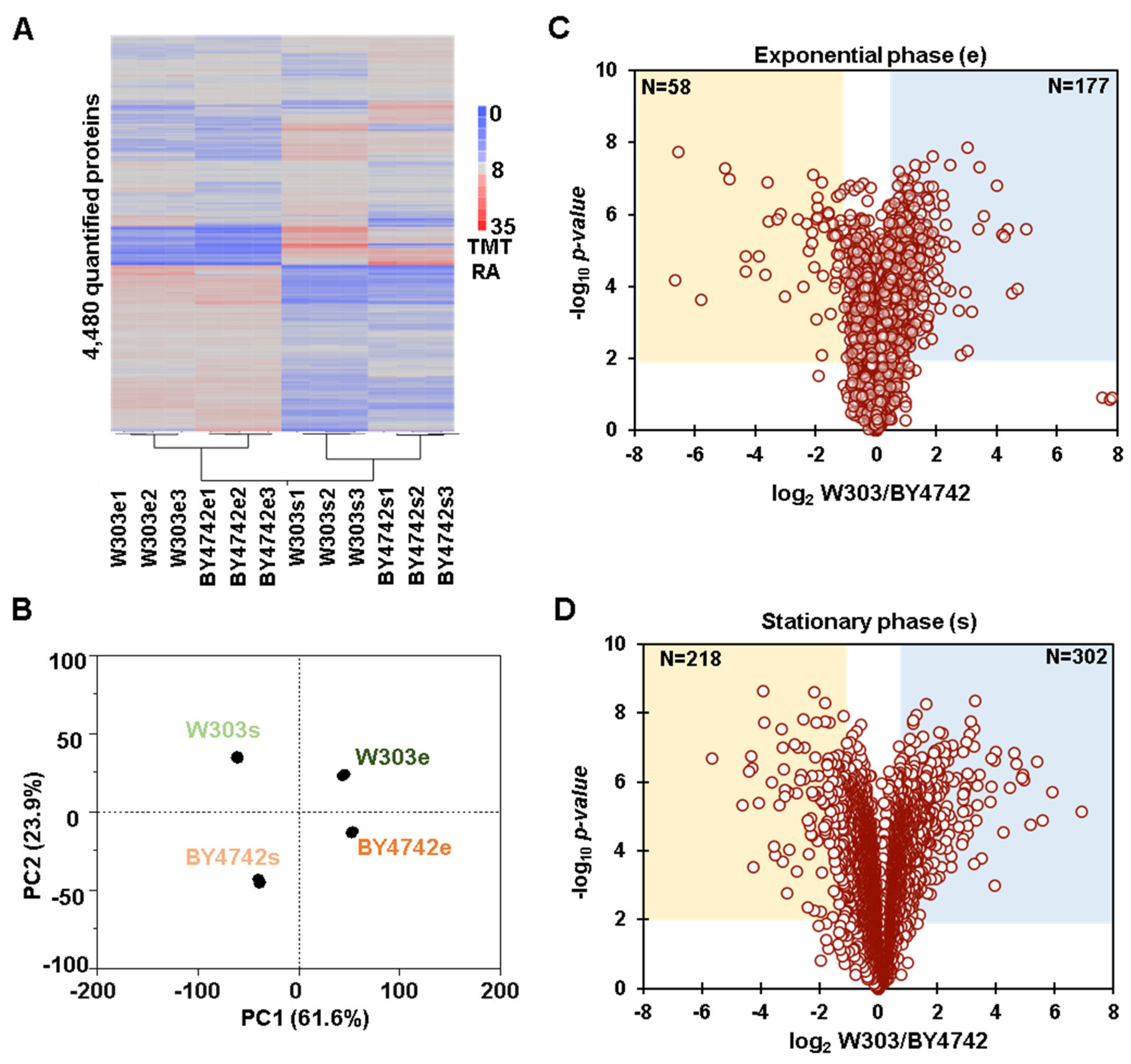
3.1. Whole Proteome Abundance Profiling Revealed Hundreds of Proteins That Differed in Abundance between BY4742 and W303 Yeast Genetic Backgrounds
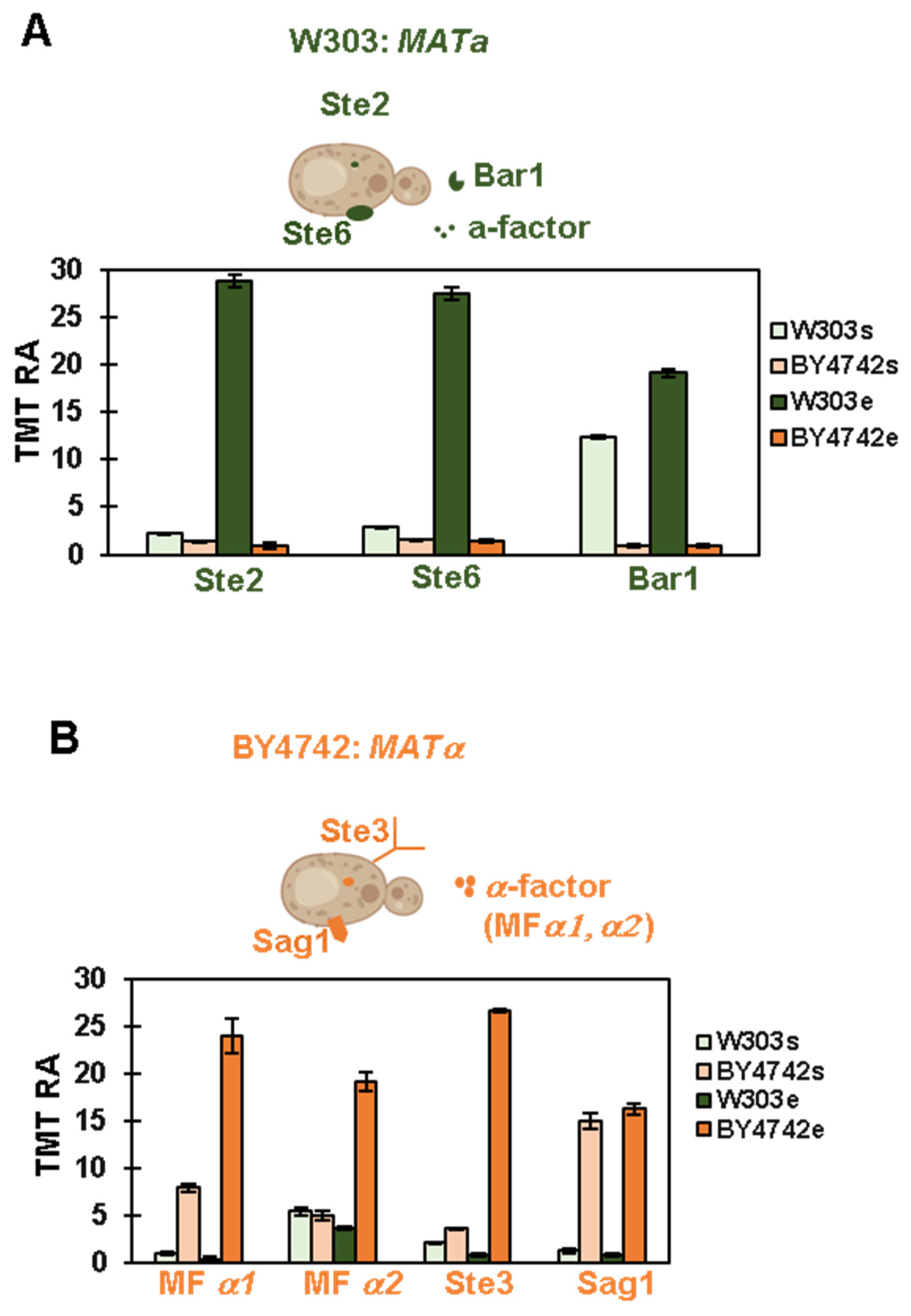
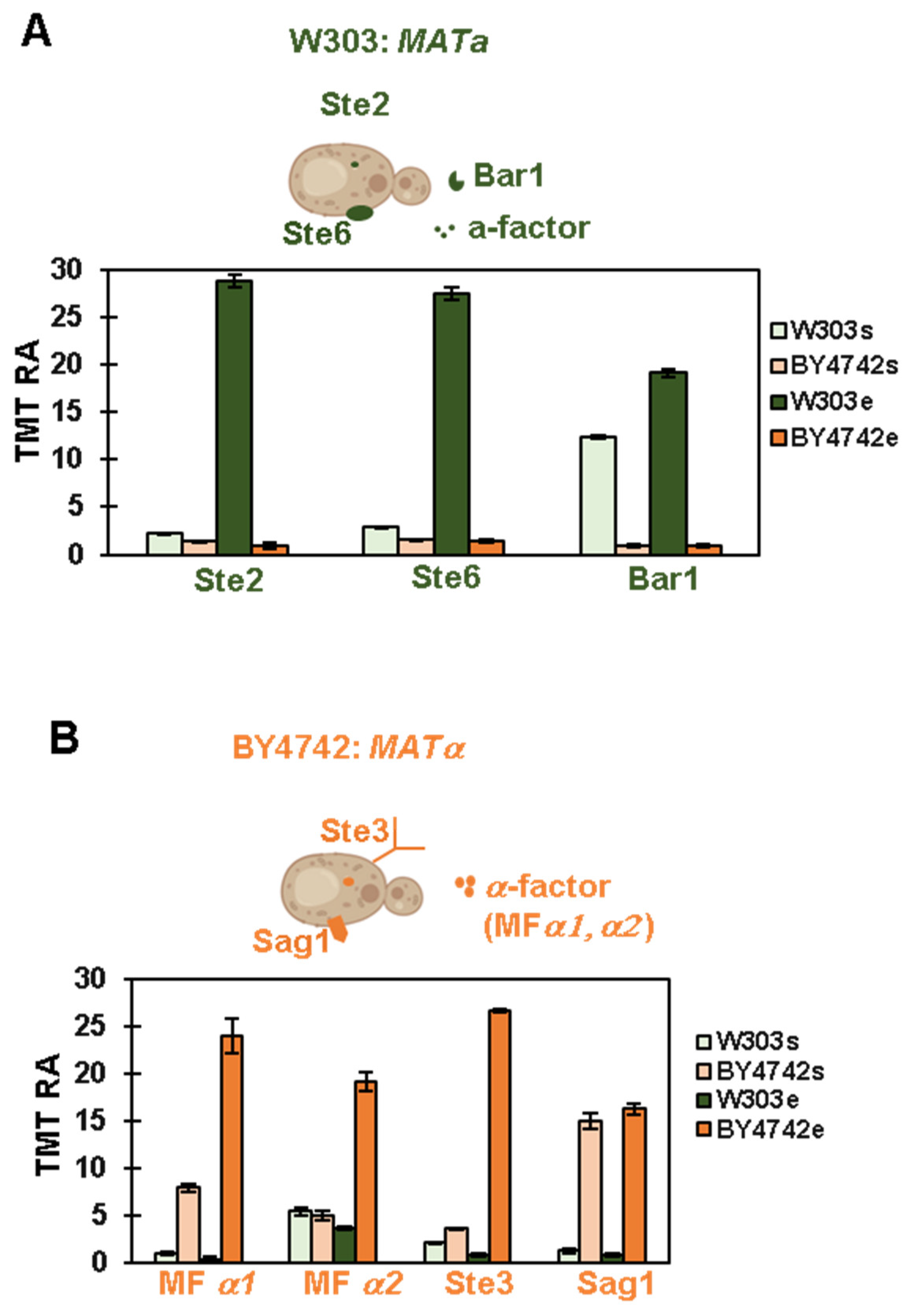
3.2. Proteomic Analysis Confirms the Mating Type of the Strains Used in This Experiment
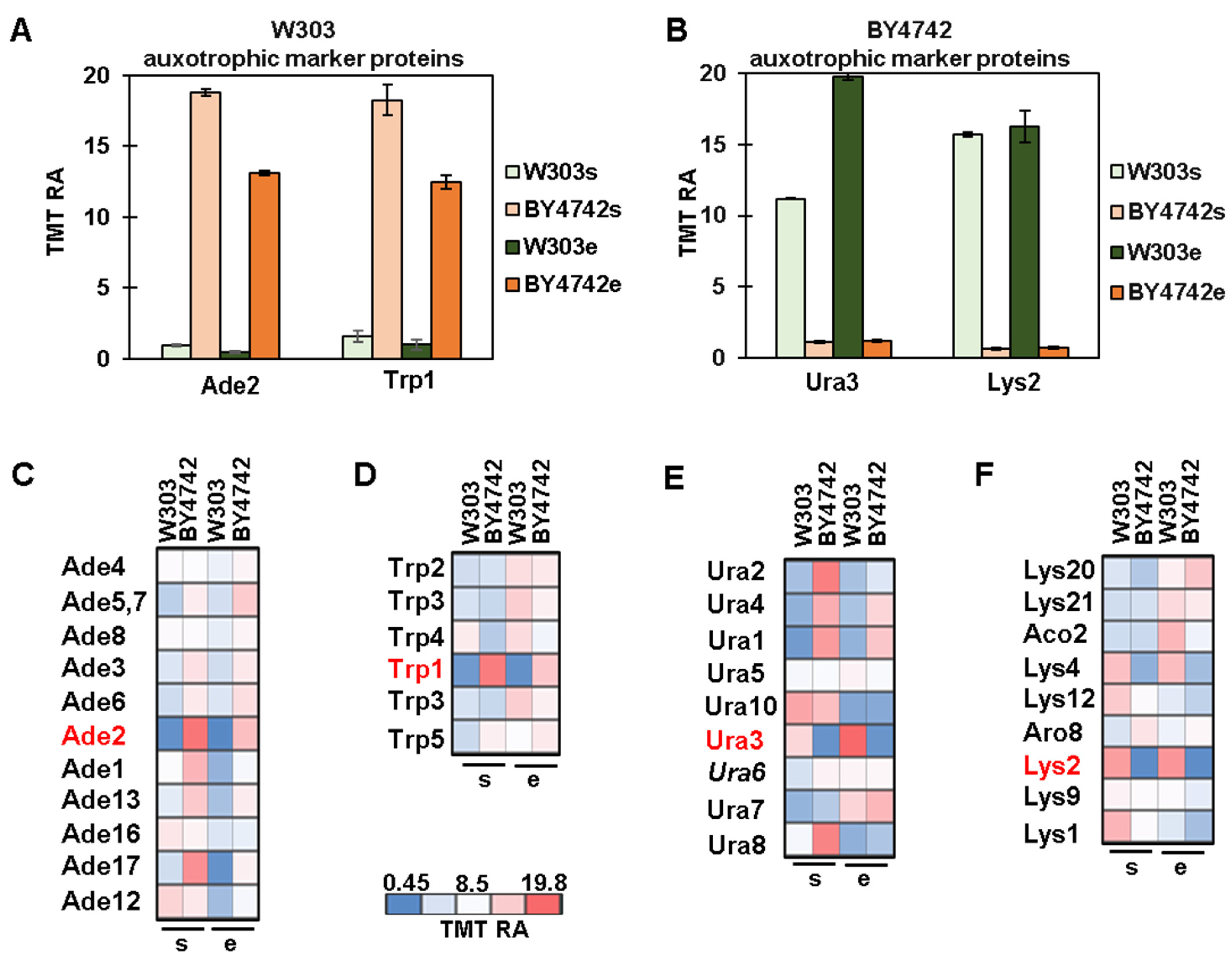
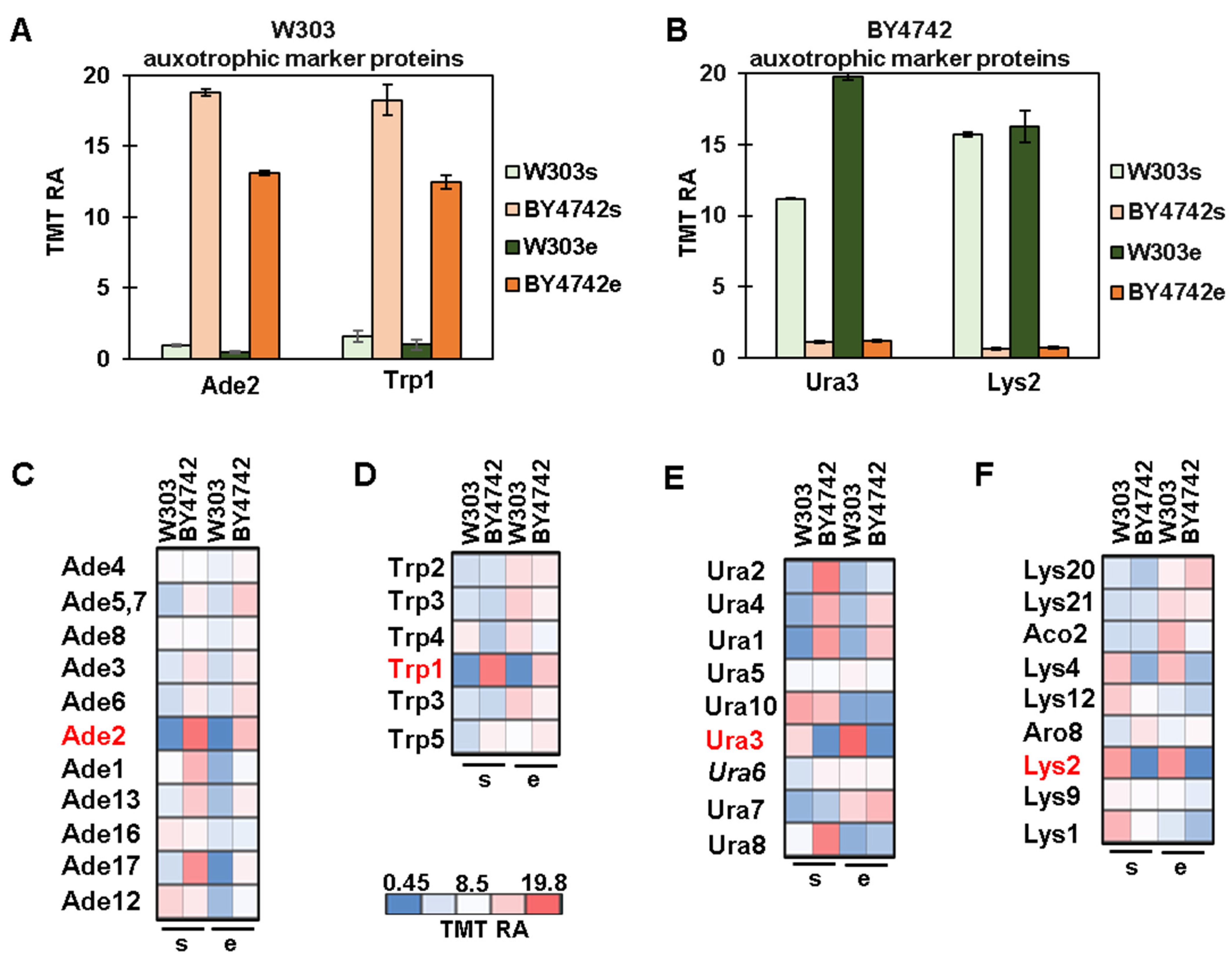
3.3. Protein Profiles of Auxotrophic Markers and Their Metabolic Pathways
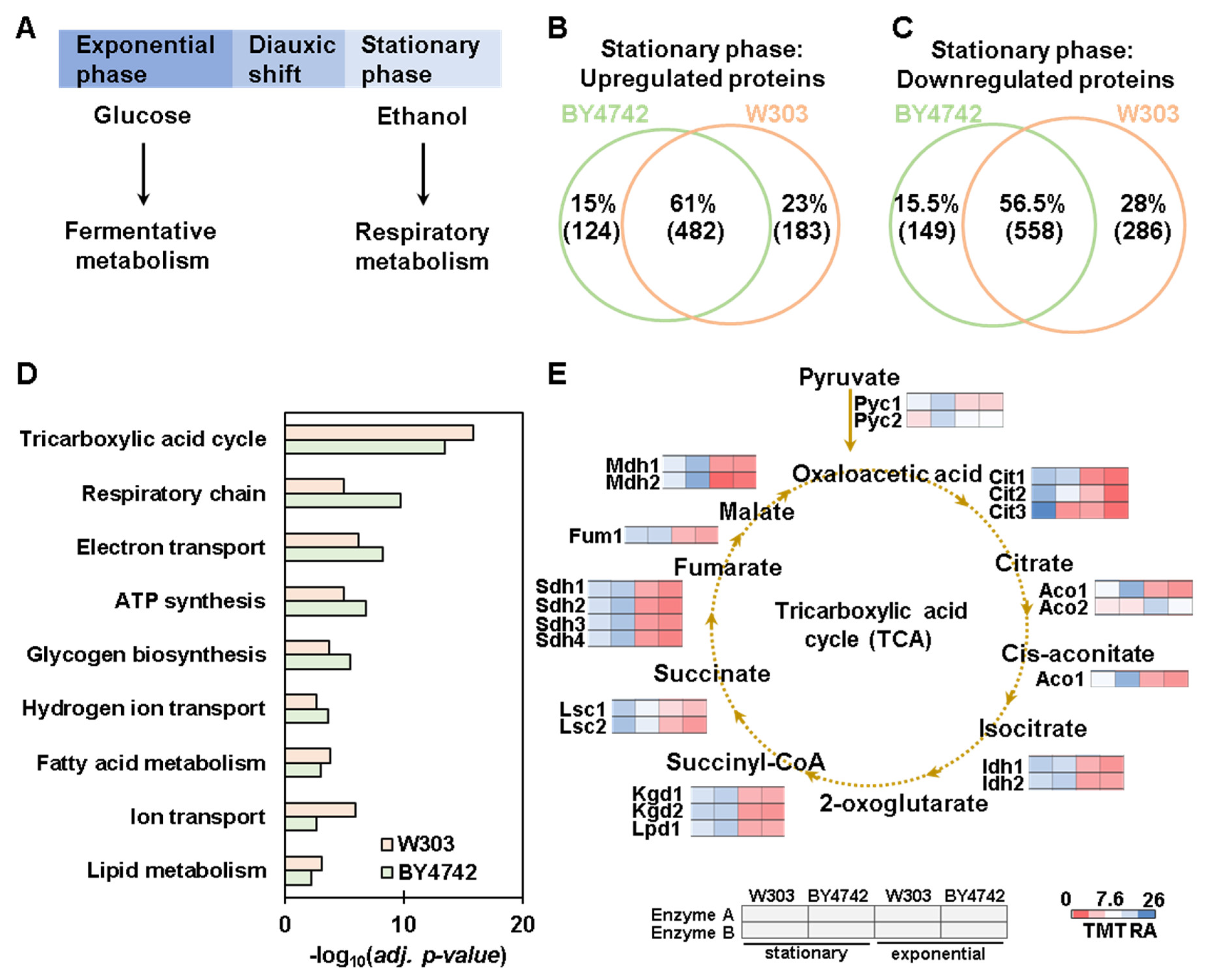
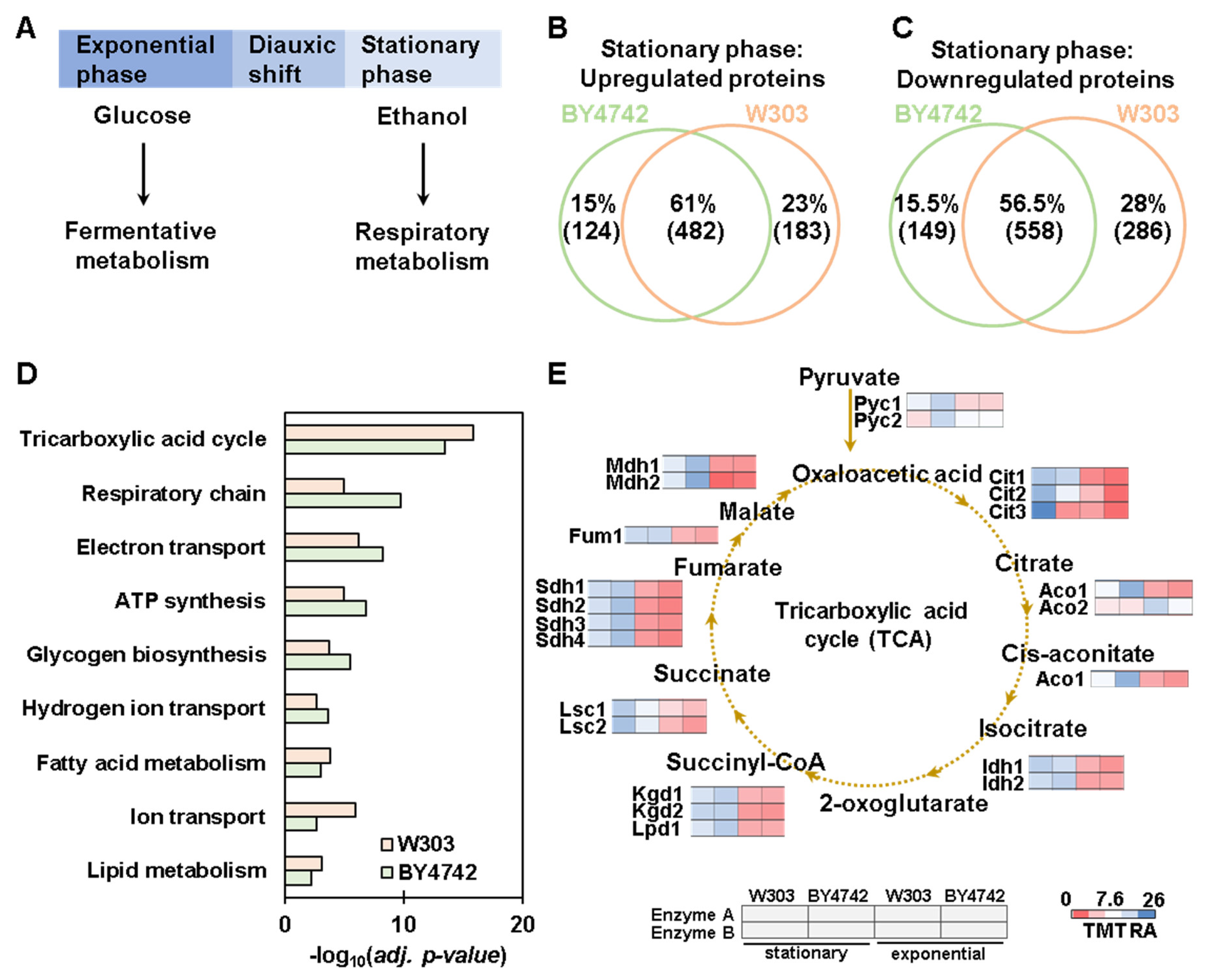
3.4. Proteome-Wide Abundance Profiling of the Transition from Exponential to Stationary Growth Phase
4. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kachroo, A.H.; Laurent, J.M.; Yellman, C.M.; Meyer, A.G.; Wilke, C.O.; Marcotte, E.M. Evolution. Systematic humanization of yeast genes reveals conserved functions and genetic modularity. Science 2015, 348, 921–925. [Google Scholar] [CrossRef] [PubMed]
- Laurent, J.M.; Garge, R.K.; Teufel, A.I.; Wilke, C.O.; Kachroo, A.H.; Marcotte, E.M. Humanization of yeast genes with multiple human orthologs reveals functional divergence between paralogs. PLoS Biol. 2020, 18, e3000627. [Google Scholar] [CrossRef] [PubMed]
- Louis, E.J. Historical Evolution of Laboratory Strains of Saccharomyces cerevisiae. Cold Spring Harb. Protoc. 2016, 7, 2016. [Google Scholar]
- Elion, E.A.; Brill, J.A.; Fink, G.R. FUS3 represses CLN1 and CLN2 and in concert with KSS1 promotes signal transduction. Proc. Natl. Acad. Sci. USA 1991, 88, 9392–9396. [Google Scholar] [CrossRef]
- Rossio, V.; Kazatskaya, A.; Hirabayashi, M.; Yoshida, S. Comparative genetic analysis of PP2A-Cdc55 regulators in budding yeast. Cell Cycle 2014, 13, 2073–2083. [Google Scholar] [CrossRef]
- Liti, G.; Haricharan, S.; Cubillos, F.A.; Tierney, A.L.; Sharp, S.; Bertuch, A.A.; Parts, L.; Bailes, E.; Louis, E.J. Segregating YKU80 and TLC1 alleles underlying natural variation in telomere properties in wild yeast. PLoS Genet. 2009, 5, e1000659. [Google Scholar] [CrossRef]
- Ralser, M.; Kuhl, H.; Ralser, M.; Werber, M.; Lehrach, H.; Breitenbach, M.; Timmermann, B. The Saccharomyces cerevisiae W303-K6001 cross-platform genome sequence: Insights into ancestry and physiology of a laboratory mutt. Open Biol. 2012, 2, 120093. [Google Scholar] [CrossRef]
- Baker Brachmann, C.; Davies, A.; Cost, G.J.; Caputo, E.; Li, J.; Hieter, P.; Boeke, J.D. Designer deletion strains derived from Saccharomyces cerevisiae S288C: A useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast 1998, 14, 115–132. [Google Scholar] [CrossRef]
- Rogowska-Wrzesinska, A.; Larsen, P.M.; Blomberg, A.; Gorg, A.; Roepstorff, P.; Norbeck, J.; Fey, S.J. Comparison of the proteomes of three yeast wild type strains: CEN.PK2, FY1679 and W303. Comp. Funct. Genomics 2001, 2, 207–225. [Google Scholar] [CrossRef]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef]
- Paulo, J.A.; O’Connell, J.D.; Everley, R.A.; O’Brien, J.; Gygi, M.A.; Gygi, S.P. Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J. Proteom. 2016, 148, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Saba, J.; Bonneil, E.; Pomiès, C.; Eng, K.; Thibault, P. Enhanced sensitivity in proteomics experiments using FAIMS coupled with a hybrid linear ion trap/Orbitrap mass spectrometer. J. Proteome Res. 2009, 8, 3355–3366. [Google Scholar] [CrossRef] [PubMed]
- Gygi, J.P.; Yu, Q.; Navarrete-Perea, J.; Rad, R.; Gygi, S.P.; Paulo, J.A. Web-Based Search Tool for Visualizing Instrument Performance Using the Triple Knockout (TKO) Proteome Standard. J. Proteome Res. 2019, 18, 687–693. [Google Scholar] [CrossRef]
- Paulo, J.A.; O’Connell, J.D.; Gygi, S.P. A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass. Spectrom. 2016, 27, 1620–1625. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. [Google Scholar]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the Multiplexing Capacity of TMTs Using Reporter Ion Isotopologues with Isobaric Masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef]
- Balzi, E.; Goffeau, A. Yeast multidrug resistance: The PDR network. J. Bioenerg. Biomembr. 1995, 27, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Roberts, C.A.; Miller, J.H.; Atkinson, P.H. The genetic architecture in Saccharomyces cerevisiae that contributes to variation in drug response to the antifungals benomyl and ketoconazole. FEMS Yeast Res. 2017, 17, fox027. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, K.C.; Gorman, O.; Lew, D.J. Mechanism of commitment to a mating partner in Saccharomyces cerevisiae. Mol. Biol. Cell 2022, 33, ar112. [Google Scholar] [CrossRef] [PubMed]
- Erickson, B.K.; Rose, C.M.; Braun, C.R.; Erickson, A.R.; Knott, J.; McAlister, G.C.; Wühr, M.; Paulo, J.A.; Everley, R.A.; Gygi, S.P. A Strategy to Combine Sample Multiplexing with Targeted Proteomics Assays for High-Throughput Protein Signature Characterization. Mol. Cell 2017, 65, 361–370. [Google Scholar] [CrossRef] [PubMed]
- Pronk, J.T. Auxotrophic yeast strains in fundamental and applied research. Appl. Environ. Microbiol. 2002, 68, 2095–2100. [Google Scholar] [CrossRef]
- Murphy, J.P.; Stepanova, E.; Everley, R.A.; Paulo, J.A.; Gygi, S.P. Comprehensive Temporal Protein Dynamics during the Diauxic Shift in Saccharomyces cerevisiae. Mol. Cell. Proteom. 2015, 14, 2454–2465. [Google Scholar] [CrossRef] [PubMed]
- Talarek, N.; Cameroni, E.; Jaquenoud, M.; Luo, X.; Bontron, S.; Lippman, S.; Devgan, G.; Snyder, M.; Broach, J.R.; De Virgilio, C. Initiation of the TORC1-regulated G0 program requires Igo1/2, which license specific mRNAs to evade degradation via the 5′-3′ mRNA decay pathway. Mol. Cell 2010, 38, 345–355. [Google Scholar] [CrossRef]
- Sherman, B.T.; Hao, M.; Qiu, J.; Jiao, X.; Baseler, M.W.; Lane, H.C.; Imamichi, T.; Chang, W. DAVID: A web server for functional enrichment analysis and functional annotation of gene lists (2021 update). Nucleic Acids Res. 2022, 50, W216–W221. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Kirsch, R.; Koutrouli, M.; Nastou, K.; Mehryary, F.; Hachilif, R.; Gable, A.L.; Fang, T.; Doncheva, N.T.; Pyysalo, S.; et al. The STRING database in 2023: Protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic Acids Res. 2023, 51, D638–D646. [Google Scholar] [CrossRef]
- Key, M. A tutorial in displaying mass spectrometry-based proteomic data using heat maps. BMC Bioinform. 2012, 13 (Suppl. S16), S10. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossio, V.; Liu, X.; Paulo, J.A. Comparative Proteomic Analysis of Two Commonly Used Laboratory Yeast Strains: W303 and BY4742. Proteomes 2023, 11, 30. https://doi.org/10.3390/proteomes11040030
Rossio V, Liu X, Paulo JA. Comparative Proteomic Analysis of Two Commonly Used Laboratory Yeast Strains: W303 and BY4742. Proteomes. 2023; 11(4):30. https://doi.org/10.3390/proteomes11040030
Chicago/Turabian StyleRossio, Valentina, Xinyue Liu, and Joao A. Paulo. 2023. "Comparative Proteomic Analysis of Two Commonly Used Laboratory Yeast Strains: W303 and BY4742" Proteomes 11, no. 4: 30. https://doi.org/10.3390/proteomes11040030
APA StyleRossio, V., Liu, X., & Paulo, J. A. (2023). Comparative Proteomic Analysis of Two Commonly Used Laboratory Yeast Strains: W303 and BY4742. Proteomes, 11(4), 30. https://doi.org/10.3390/proteomes11040030