
Comparison of the Proteomes and Phosphoproteomes of S. cerevisiae Cells Harvested with Different Strategies



{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Material and Methods
2.1. Materials
2.2. Yeast Growth and Protein Extraction
2.3. Protein Digestion, TMT Labeling and Sample Processing
2.4. Spin Column-Based Phosphopeptide Enrichment
2.5. Mass Spectrometry Data Acquisition and Processing
3. Results and Discussion
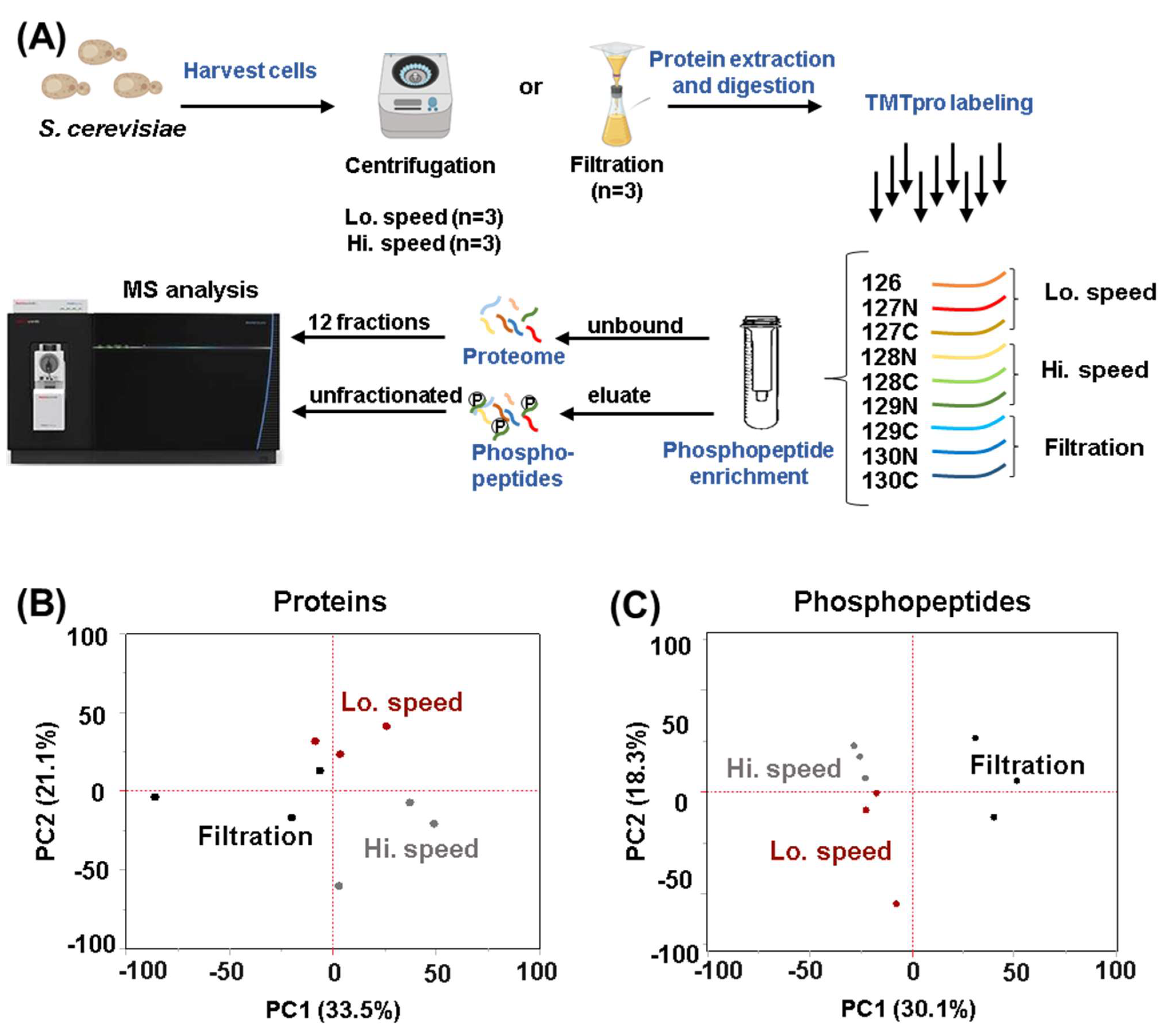
3.1. Proteome-Wide Abundance Profiling Revealed Minimal Changes among the Different Harvesting Methods
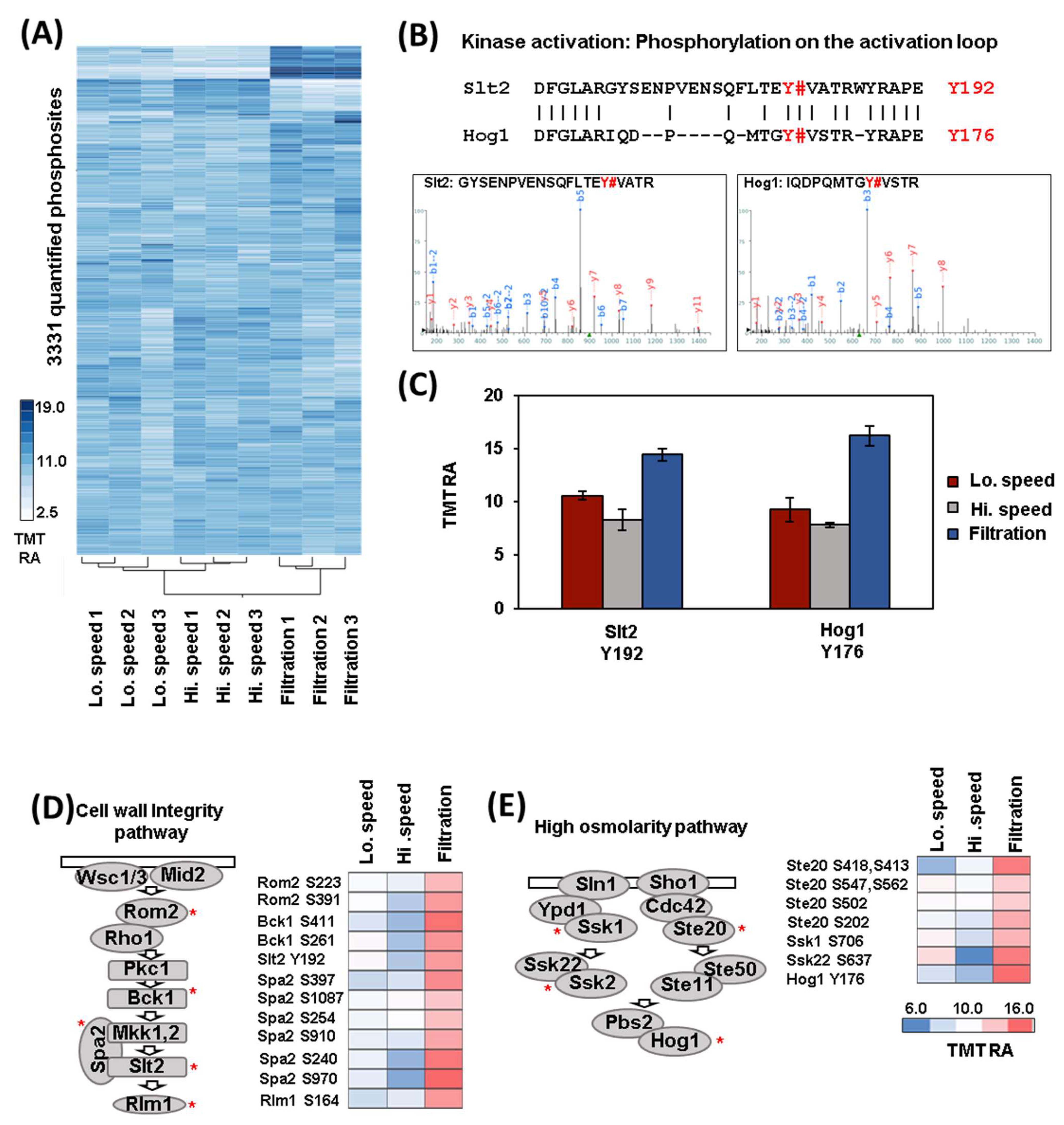
3.2. The Quantification of Hundreds of Phosphorylation Events Differed between Cells Harvested by Filtration and Centrifugation
3.3. Analysis of Differentially Regulated Kinases through Phosphorylation
4. Conclusions and Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Vanderwaeren, L.; Dok, R.; Voordeckers, K.; Nuyts, S.; Verstrepen, K.J. Saccharomyces cerevisiae as a Model System for Eukaryotic Cell Biology, from Cell Cycle Control to DNA Damage Response. Int. J. Mol. Sci. 2022, 23, 11665. [Google Scholar] [CrossRef] [PubMed]
- Rossio, V.; Kazatskaya, A.; Hirabayashi, M.; Yoshida, S. Comparative genetic analysis of PP2A-Cdc55 regulators in budding yeast. Cell Cycle 2014, 13, 2073–2083. [Google Scholar] [CrossRef] [PubMed]
- Navarrete-Perea, J.; Yu, Q.; Gygi, S.P.; Paulo, J.A. Streamlined Tandem Mass Tag (SL-TMT) Protocol: An Efficient Strategy for Quantitative (Phospho)proteome Profiling Using Tandem Mass Tag-Synchronous Precursor Selection-MS3. J. Proteome Res. 2018, 17, 2226–2236. [Google Scholar] [CrossRef] [PubMed]
- Rappsilber, J.; Ishihama, Y.; Mann, M. Stop and go extraction tips for matrix-assisted laser desorption/ionization, nanoelectrospray, and LC/MS sample pretreatment in proteomics. Anal. Chem. 2003, 75, 663–670. [Google Scholar] [CrossRef] [PubMed]
- Paulo, J.A.; O’Connell, J.D.; Everley, R.A.; O’Brien, J.; Gygi, M.A.; Gygi, S.P. Quantitative mass spectrometry-based multiplexing compares the abundance of 5000 S. cerevisiae proteins across 10 carbon sources. J. Proteom. 2016, 148, 85–93. [Google Scholar] [CrossRef]
- Paulo, J.A.; Navarrete-Perea, J.; Erickson, A.R.; Knott, J.; Gygi, S.P. An Internal Standard for Assessing Phosphopeptide Recovery from Metal Ion/Oxide Enrichment Strategies. J. Am. Soc. Mass Spectrom. 2018, 29, 1505–1511. [Google Scholar] [CrossRef]
- Saba, J.; Bonneil, E.; Pomies, C.; Eng, K.; Thibault, P. Enhanced sensitivity in proteomics experiments using FAIMS coupled with a hybrid linear ion trap/Orbitrap mass spectrometer. J. Proteome Res. 2009, 8, 3355–3366. [Google Scholar] [CrossRef]
- Chambers, M.C.; Maclean, B.; Burke, R.; Amodei, D.; Ruderman, D.L.; Neumann, S.; Gatto, L.; Fischer, B.; Pratt, B.; Egertson, J.; et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat. Biotechnol. 2012, 30, 918–920. [Google Scholar] [CrossRef]
- Beausoleil, S.A.; Villen, J.; Gerber, S.A.; Rush, J.; Gygi, S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006, 24, 1285–1292. [Google Scholar] [CrossRef]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villen, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010, 604, 55–71. [Google Scholar] [CrossRef] [PubMed]
- Elias, J.E.; Gygi, S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods 2007, 4, 207–214. [Google Scholar] [CrossRef] [PubMed]
- McAlister, G.C.; Huttlin, E.L.; Haas, W.; Ting, L.; Jedrychowski, M.P.; Rogers, J.C.; Kuhn, K.; Pike, I.; Grothe, R.A.; Blethrow, J.D.; et al. Increasing the Multiplexing Capacity of TMTs Using Reporter Ion Isotopologues with Isobaric Masses. Anal. Chem. 2012, 84, 7469–7478. [Google Scholar] [CrossRef] [PubMed]
- Hornbeck, P.V.; Kornhauser, J.M.; Tkachev, S.; Zhang, B.; Skrzypek, E.; Murray, B.; Latham, V.; Sullivan, M. PhosphoSitePlus: A comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012, 40, D261–D270. [Google Scholar] [CrossRef]
- Rubenstein, E.M.; Schmidt, M.C. Mechanisms regulating the protein kinases of Saccharomyces cerevisiae. Eukaryot Cell 2007, 6, 571–583. [Google Scholar] [CrossRef]
- Levin, D.E. Regulation of cell wall biogenesis in Saccharomyces cerevisiae: The cell wall integrity signaling pathway. Genetics 2011, 189, 1145–1175. [Google Scholar] [CrossRef]
- Harvey, S.L.; Charlet, A.; Haas, W.; Gygi, S.P.; Kellogg, D.R. Cdk1-dependent regulation of the mitotic inhibitor Wee1. Cell 2005, 122, 407–420. [Google Scholar] [CrossRef]
- Lew, D.J.; Reed, S.I. A cell cycle checkpoint monitors cell morphogenesis in budding yeast. J. Cell Biol. 1995, 129, 739–749. [Google Scholar] [CrossRef]
- Alexander, M.R.; Tyers, M.; Perret, M.; Craig, B.M.; Fang, K.S.; Gustin, M.C. Regulation of cell cycle progression by Swe1p and Hog1p following hypertonic stress. Mol. Biol. Cell 2001, 12, 53–62. [Google Scholar] [CrossRef]
- Harrison, J.C.; Bardes, E.S.; Ohya, Y.; Lew, D.J. A role for the Pkc1p/Mpk1p kinase cascade in the morphogenesis checkpoint. Nat. Cell Biol. 2001, 3, 417–420. [Google Scholar] [CrossRef]
- Perez-Riverol, Y.; Csordas, A.; Bai, J.; Bernal-Llinares, M.; Hewapathirana, S.; Kundu, D.J.; Inuganti, A.; Griss, J.; Mayer, G.; Eisenacher, M.; et al. The PRIDE database and related tools and resources in 2019: Improving support for quantification data. Nucleic Acids Res. 2019, 47, D442–D450. [Google Scholar] [CrossRef] [PubMed]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rossio, V.; Paulo, J.A. Comparison of the Proteomes and Phosphoproteomes of S. cerevisiae Cells Harvested with Different Strategies. Proteomes 2023, 11, 28. https://doi.org/10.3390/proteomes11040028
Rossio V, Paulo JA. Comparison of the Proteomes and Phosphoproteomes of S. cerevisiae Cells Harvested with Different Strategies. Proteomes. 2023; 11(4):28. https://doi.org/10.3390/proteomes11040028
Chicago/Turabian StyleRossio, Valentina, and Joao A. Paulo. 2023. "Comparison of the Proteomes and Phosphoproteomes of S. cerevisiae Cells Harvested with Different Strategies" Proteomes 11, no. 4: 28. https://doi.org/10.3390/proteomes11040028
APA StyleRossio, V., & Paulo, J. A. (2023). Comparison of the Proteomes and Phosphoproteomes of S. cerevisiae Cells Harvested with Different Strategies. Proteomes, 11(4), 28. https://doi.org/10.3390/proteomes11040028