
Transcriptional and Epigenetic Factors Associated with Early Thrombosis of Femoral Artery Involved in Arteriovenous Fistula



Abstract
1. Introduction
2. Materials and Methods
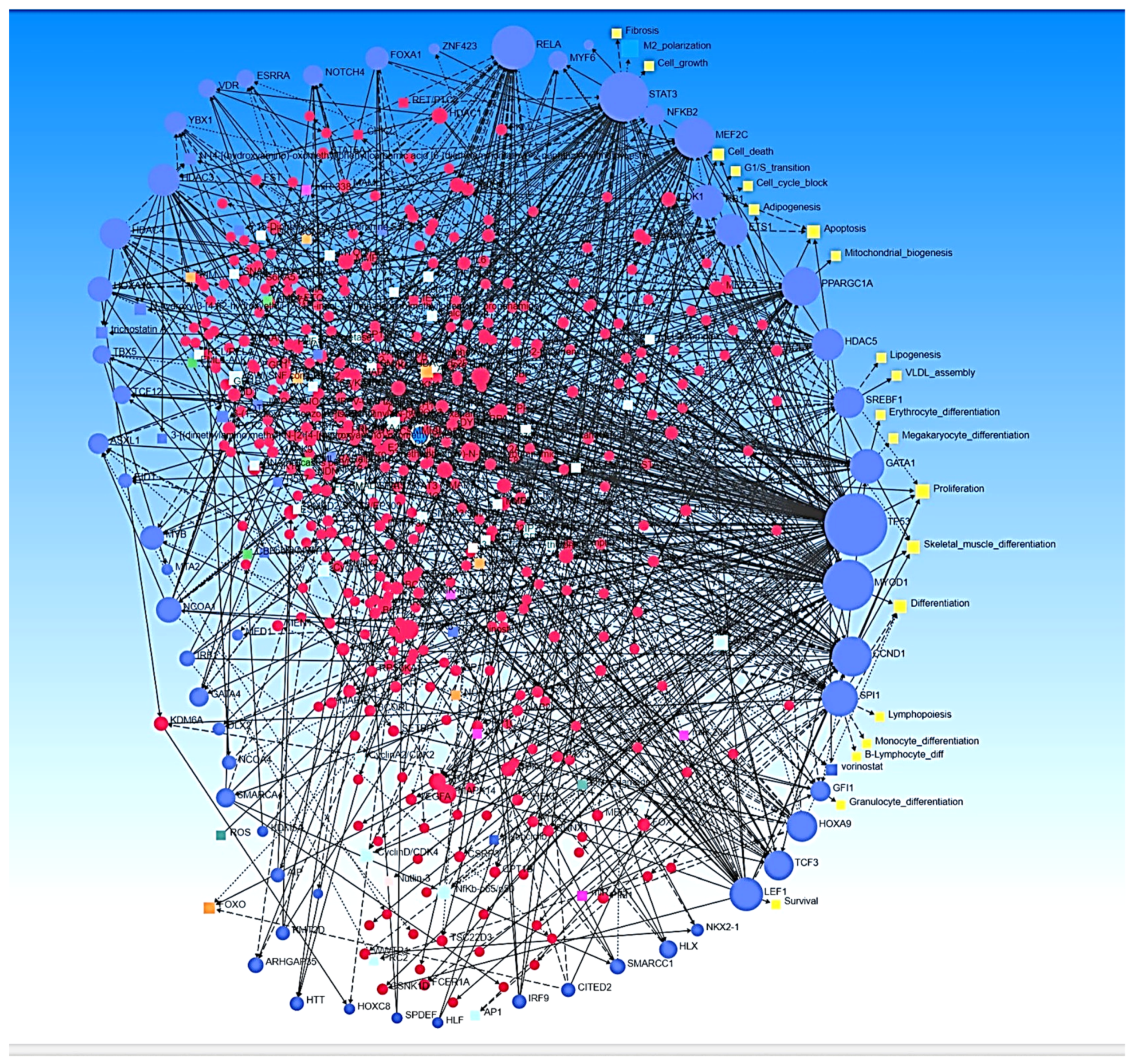
2.1. Ingenuity Pathway Analysis
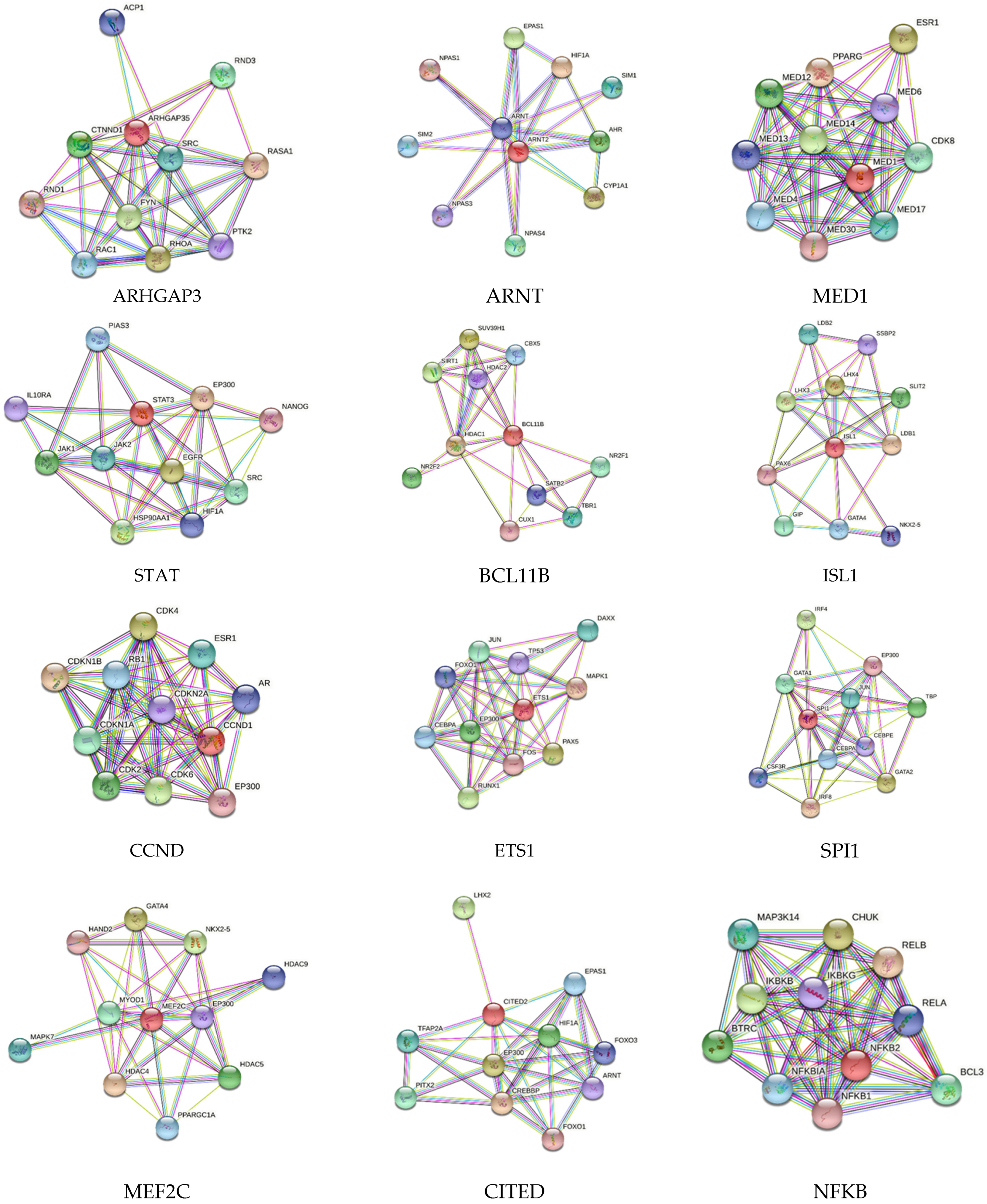
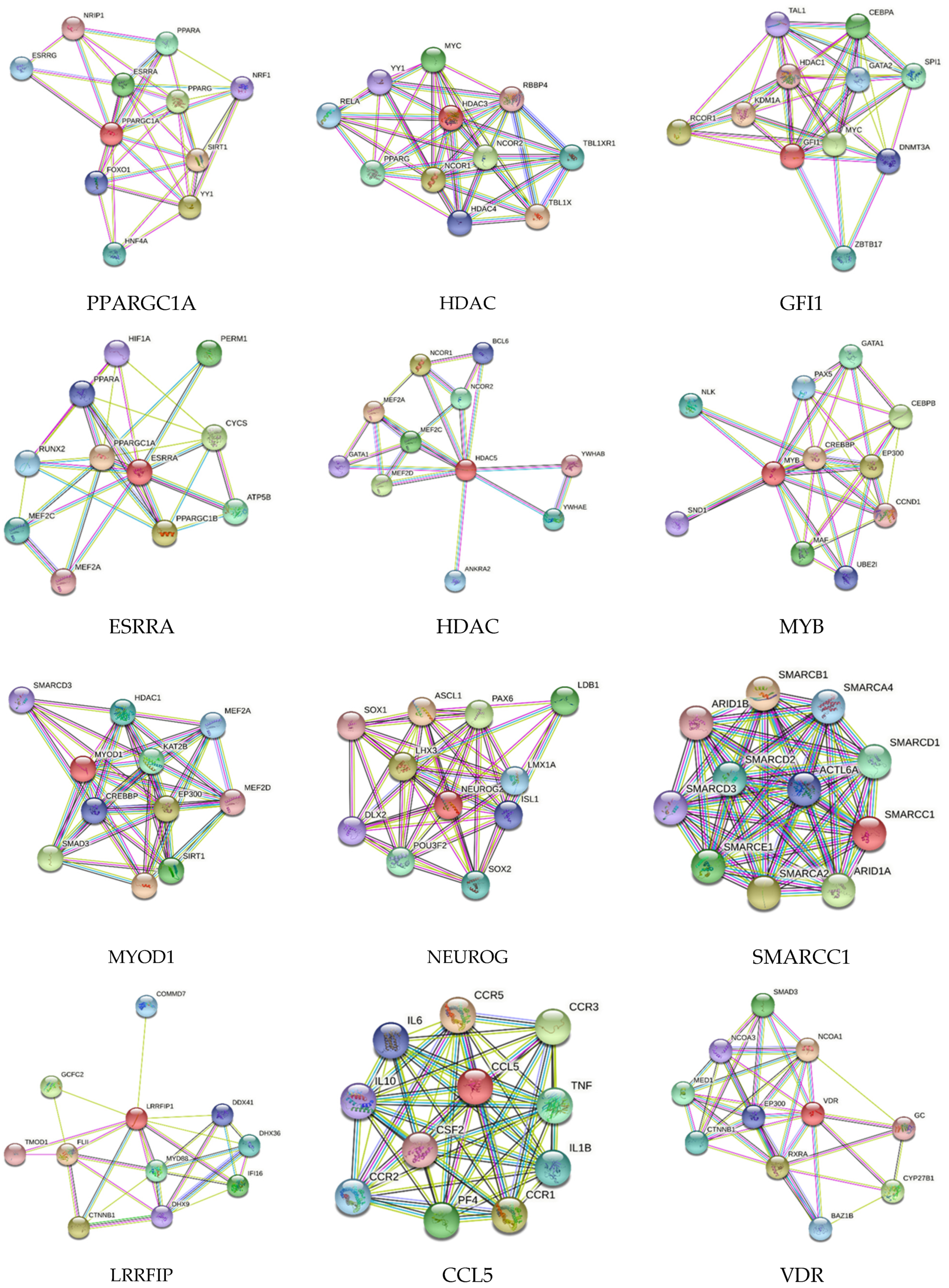
2.2. Search Tool for the Retrieval of Interacting Genes/Proteins (STRING) Network Analysis
3. Results
4. Discussion
5. Future Perspectives
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Brahmbhatt, A.; Misra, S. The Biology of Hemodialysis Vascular Access Failure. Semin. Interv. Radiol. 2016, 33, 15–20. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, M.A.; Ashraff, S.; Santos, D.; Carline, T. An overview of AVF maturation and endothelial dysfunction in an advanced renal failure. Ren. Replace. Ther. 2017, 3, 42. [Google Scholar] [CrossRef]
- Caputo, B.C.; Leong, B.; Sibona, A.; Jhajj, S.; Kohne, C.; Gabel, J.; Shih, W.; Abou Zamzam, A.; Bianchi, C.; Teruya, T. Arteriovenous fistula maturation: Physical exam versus flow study. Ann. Vasc. Surg. 2021, 77, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.; Ochoa, C.J.; Katz, S.G. Prognostic Factors for Arteriovenous Fistula Maturation. Ann. Vasc. Surg. 2018, 49, 273–276. [Google Scholar] [CrossRef]
- Beathard, G.A.; Arnold, P.; Jackson, J.; Litchfield, T.; Physician Operators Forum of, R.M.S.L. Aggressive treatment of early fistula failure. Kidney Int. 2003, 64, 1487–1494. [Google Scholar] [CrossRef]
- Vazquez-Padron, R.I.; Duque, J.C.; Tabbara, M.; Salman, L.H.; Martinez, L. Intimal Hyperplasia and Arteriovenous Fistula Failure: Looking Beyond Size Differences. Kidney360 2021, 2, 1360–1372. [Google Scholar] [CrossRef]
- Nagareddy, P.; Smyth, S.S. Inflammation and thrombosis in cardiovascular disease. Curr. Opin. Hematol. 2013, 20, 457–463. [Google Scholar] [CrossRef]
- Rai, V.; Agrawal, D.K. The role of damage- and pathogen-associated molecular patterns in inflammation-mediated vulnerability of atherosclerotic plaques. Can. J. Physiol. Pharmacol. 2017, 95, 1245–1253. [Google Scholar] [CrossRef]
- Rai, V.; Rao, V.H.; Shao, Z.; Agrawal, D.K. Dendritic Cells Expressing Triggering Receptor Expressed on Myeloid Cells-1 Correlate with Plaque Stability in Symptomatic and Asymptomatic Patients with Carotid Stenosis. PLoS ONE 2016, 11, e0154802. [Google Scholar] [CrossRef]
- Rao, V.H.; Rai, V.; Stoupa, S.; Subramanian, S.; Agrawal, D.K. Tumor necrosis factor-alpha regulates triggering receptor expressed on myeloid cells-1-dependent matrix metalloproteinases in the carotid plaques of symptomatic patients with carotid stenosis. Atherosclerosis 2016, 248, 160–169. [Google Scholar] [CrossRef]
- Roshan, M.H.; Tambo, A.; Pace, N.P. The Role of TLR2, TLR4, and TLR9 in the Pathogenesis of Atherosclerosis. Int. J. Inflam. 2016, 2016, 1532832. [Google Scholar] [CrossRef] [PubMed]
- Rai, V.; Agrawal, D.K. Transcriptomic Analysis Identifies Differentially Expressed Genes Associated with Vascular Cuffing and Chronic Inflammation Mediating Early Thrombosis in Arteriovenous Fistula. Biomedicines 2022, 10, 433. [Google Scholar] [CrossRef] [PubMed]
- Phillips, T. Regulation of transcription and gene expression in eukaryotes. Nat. Educ. 2008, 1, 199. [Google Scholar]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, Mechanisms of Actions, and Circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef]
- Ying, S.Y.; Chang, D.C.; Lin, S.L. The microRNA (miRNA): Overview of the RNA genes that modulate gene function. Mol. Biotechnol. 2008, 38, 257–268. [Google Scholar] [CrossRef]
- Valinezhad Orang, A.; Safaralizadeh, R.; Kazemzadeh-Bavili, M. Mechanisms of miRNA-Mediated Gene Regulation from Common Downregulation to mRNA-Specific Upregulation. Int. J. Genomics. 2014, 2014, 970607. [Google Scholar] [CrossRef]
- Ling, H.; Fabbri, M.; Calin, G.A. MicroRNAs and other non-coding RNAs as targets for anticancer drug development. Nat. Rev. Drug Discov. 2013, 12, 847–865. [Google Scholar] [CrossRef]
- Rai, V.; Jadhav, G.; Boosani, C. Role of transcription factors in regulating development and progression of atherosclerosis. Annals Vasc. Med. Surg. 2019, 2, 1007. [Google Scholar]
- Tsang, H.G.; Rashdan, N.A.; Whitelaw, C.B.; Corcoran, B.M.; Summers, K.M.; MacRae, V.E. Large animal models of cardiovascular disease. Cell Biochem. Funct. 2016, 34, 113–132. [Google Scholar] [CrossRef]
- Flentje, A.; Kalsi, R.; Monahan, T.S. Small GTPases and Their Role in Vascular Disease. Int. J. Mol. Sci. 2019, 20, 917. [Google Scholar] [CrossRef]
- Strassheim, D.; Gerasimovskaya, E.; Irwin, D.; Dempsey, E.C.; Stenmark, K.; Karoor, V. RhoGTPase in Vascular Disease. Cells 2019, 8, 551. [Google Scholar] [CrossRef] [PubMed]
- Sadowitz, B.; Maier, K.G.; Gahtan, V. Basic science review: Statin therapy-Part I: The pleiotropic effects of statins in cardiovascular disease. Vasc. Endovasc. Surg. 2010, 44, 241–251. [Google Scholar] [CrossRef] [PubMed]
- Sadaghianloo, N.; Contenti, J.; Dardik, A.; Mazure, N.M. Role of Hypoxia and Metabolism in the Development of Neointimal Hyperplasia in Arteriovenous Fistulas. Int. J. Mol. Sci. 2019, 20, 5387. [Google Scholar] [CrossRef] [PubMed]
- Ichihara, S.; Yamada, Y.; Ichihara, G.; Nakajima, T.; Li, P.; Kondo, T.; Gonzalez, F.J.; Murohara, T. A role for the aryl hydrocarbon receptor in regulation of ischemia-induced angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1297–1304. [Google Scholar] [CrossRef]
- Bentzon, J.F.; Otsuka, F.; Virmani, R.; Falk, E. Mechanisms of plaque formation and rupture. Circ. Res. 2014, 114, 1852–1866. [Google Scholar] [CrossRef]
- Badimon, L.; Vilahur, G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J. Intern. Med. 2014, 276, 618–632. [Google Scholar] [CrossRef]
- Huang, X.; Guan, J.; Sheng, Z.; Wang, M.; Xu, T.; Guo, G.; Wan, P.; Tian, B.; Zhou, J.; Huang, A.; et al. Effect of local anti-vascular endothelial growth factor therapy to prevent the formation of stenosis in outflow vein in arteriovenous fistula. J. Transl. Int. Med. 2021, 9, 307–317. [Google Scholar] [CrossRef]
- Mandl, M.; Depping, R. Hypoxia-inducible aryl hydrocarbon receptor nuclear translocator (ARNT) (HIF-1beta): Is it a rare exception? Mol. Med. 2014, 20, 215–220. [Google Scholar] [CrossRef]
- Yi, T.; Wang, J.; Zhu, K.; Tang, Y.; Huang, S.; Shui, X.; Ding, Y.; Chen, C.; Lei, W. Aryl Hydrocarbon Receptor: A New Player of Pathogenesis and Therapy in Cardiovascular Diseases. Biomed. Res. Int. 2018, 2018, 6058784. [Google Scholar] [CrossRef]
- Zhang, Y.; Fu, Y.; Zhang, C.; Jia, L.; Yao, N.; Lin, Y.; Dong, Y.; Fatima, N.; Alam, N.; Wang, R.; et al. MED1 Deficiency in Macrophages Accelerates Intimal Hyperplasia via ROS Generation and Inflammation. Oxid. Med. Cell Longev. 2021, 2021, 3010577. [Google Scholar] [CrossRef]
- Wincewicz, A.; Sulkowska, M.; Rutkowski, R.; Sulkowski, S.; Musiatowicz, B.; Hirnle, T.; Famulski, W.; Koda, M.; Sokol, G.; Szarejko, P. STAT1 and STAT3 as intracellular regulators of vascular remodeling. Eur. J. Intern. Med. 2007, 18, 267–271. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.; Gushiken, F.C.; Bolgiano, D.; Salsbery, B.J.; Aghakasiri, N.; Jing, N.; Wu, X.; Vijayan, K.V.; Rumbaut, R.E.; Adachi, R.; et al. Signal transducer and activator of transcription 3 (STAT3) regulates collagen-induced platelet aggregation independently of its transcription factor activity. Circulation 2013, 127, 476–485. [Google Scholar] [CrossRef] [PubMed]
- Valisno, J.A.C.; May, J.; Singh, K.; Helm, E.Y.; Venegas, L.; Budbazar, E.; Goodman, J.B.; Nicholson, C.J.; Avram, D.; Cohen, R.A.; et al. BCL11B Regulates Arterial Stiffness and Related Target Organ Damage. Circ. Res. 2021, 128, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Sporkova, A.; Ghosh, S.; Al-Hasani, J.; Hecker, M. Lin11-Isl1-Mec3 Domain Proteins as Mechanotransducers in Endothelial and Vascular Smooth Muscle Cells. Front. Physiol. 2021, 12, 769321. [Google Scholar] [CrossRef] [PubMed]
- Shikatani, E.A.; Chandy, M.; Besla, R.; Li, C.C.; Mommen, A.; El-Mounayri, O.; Robbins, C.S.; Husain, M. c-Myb Regulates Proliferation and Differentiation of Adventitial Sca1+ Vascular Smooth Muscle Cell Progenitors by Transactivation of Myocardin. Arterioscler. Thromb. Vasc. Biol. 2016, 36, 1367–1376. [Google Scholar] [CrossRef]
- You, X.M.; Mungrue, I.N.; Kalair, W.; Afroze, T.; Ravi, B.; Sadi, A.M.; Gros, R.; Husain, M. Conditional expression of a dominant-negative c-Myb in vascular smooth muscle cells inhibits arterial remodeling after injury. Circ. Res. 2003, 92, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Mehrhof, F.B.; Schmidt-Ullrich, R.; Dietz, R.; Scheidereit, C. Regulation of vascular smooth muscle cell proliferation: Role of NF-κB revisited. Circ. Res. 2005, 96, 958–964. [Google Scholar] [CrossRef]
- Wu, H.Y.; Li, J.Y.; Wen, H.; Li, Y.Q.; Li, Y.L.; Li, G.Y.; Jiang, Y.; Lv, J.Y.; Yang, D.L. Icariside II attenuates vascular remodeling via Wnt7b/CCND1 axis. J. Cardiovasc. Pharmacol. 2022. [Google Scholar] [CrossRef]
- Rao, V.H.; Rai, V.; Stoupa, S.; Agrawal, D.K. Blockade of Ets-1 attenuates epidermal growth factor-dependent collagen loss in human carotid plaque smooth muscle cells. Am. J. Physiol. Heart Circ. Physiol. 2015, 309, H1075–H1086. [Google Scholar] [CrossRef]
- Zhan, Y.; Brown, C.; Maynard, E.; Anshelevich, A.; Ni, W.; Ho, I.C.; Oettgen, P. Ets-1 is a critical regulator of Ang II-mediated vascular inflammation and remodeling. J. Clin. Investig. 2005, 115, 2508–2516. [Google Scholar] [CrossRef]
- Jeong, K.; Kim, J.H.; Murphy, J.M.; Park, H.; Kim, S.J.; Rodriguez, Y.A.R.; Kong, H.; Choi, C.; Guan, J.L.; Taylor, J.M.; et al. Nuclear Focal Adhesion Kinase Controls Vascular Smooth Muscle Cell Proliferation and Neointimal Hyperplasia Through GATA4-Mediated Cyclin D1 Transcription. Circ. Res. 2019, 125, 152–166. [Google Scholar] [CrossRef] [PubMed]
- Nakaoka, H.; Mogi, M.; Suzuki, J.; Kan-No, H.; Min, L.J.; Iwanami, J.; Horiuchi, M. Interferon regulatory factor 1 attenuates vascular remodeling; roles of angiotensin II type 2 receptor. J. Am. Soc. Hypertens. 2016, 10, 811–818. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.M.; Zhu, L.H.; Chen, H.Z.; Zhang, R.; Zhang, P.; Jiang, D.S.; Gao, L.; Tian, S.; Wang, L.; Zhang, Y.; et al. Interferon regulatory factor 9 is critical for neointima formation following vascular injury. Nat. Commun. 2014, 5, 5160. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.W.; Martino, N.; Gerlach, B.D.; Lamar, J.M.; Vincent, P.A.; Adam, A.P.; Schwarz, J.J. MEF2 (Myocyte Enhancer Factor 2) Is Essential for Endothelial Homeostasis and the Atheroprotective Gene Expression Program. Arterioscler. Thromb. Vasc. Biol. 2021, 41, 1105–1123. [Google Scholar] [CrossRef]
- Lee, J.Y.; Taub, P.J.; Wang, L.; Clark, A.; Zhu, L.L.; Maharam, E.R.; Leong, D.J.; Ramcharan, M.; Li, Z.; Liu, Z.; et al. Identification of CITED2 as a negative regulator of fracture healing. Biochem. Biophys. Res. Commun. 2009, 387, 641–645. [Google Scholar] [CrossRef]
- Yin, Z.; Haynie, J.; Yang, X.; Han, B.; Kiatchoosakun, S.; Restivo, J.; Yuan, S.; Prabhakar, N.R.; Herrup, K.; Conlon, R.A.; et al. The essential role of Cited2, a negative regulator for HIF-1alpha, in heart development and neurulation. Proc. Natl. Acad. Sci. USA 2002, 99, 10488–10493. [Google Scholar] [CrossRef]
- Pong Ng, H.; Kim, G.D.; Ricky Chan, E.; Dunwoodie, S.L.; Mahabeleshwar, G.H. CITED2 limits pathogenic inflammatory gene programs in myeloid cells. FASEB J. 2020, 34, 12100–12113. [Google Scholar] [CrossRef]
- Chou, Y.T.; Hsieh, C.H.; Chiou, S.H.; Hsu, C.F.; Kao, Y.R.; Lee, C.C.; Chung, C.H.; Wang, Y.H.; Hsu, H.S.; Pang, S.T.; et al. CITED2 functions as a molecular switch of cytokine-induced proliferation and quiescence. Cell Death Differ. 2012, 19, 2015–2028. [Google Scholar] [CrossRef]
- Castier, Y.; Ramkhelawon, B.; Riou, S.; Tedgui, A.; Lehoux, S. Role of NF-kappaB in flow-induced vascular remodeling. Antioxid. Redox Signal. 2009, 11, 1641–1649. [Google Scholar] [CrossRef]
- Zhu, M.; Goetsch, S.C.; Wang, Z.; Luo, R.; Hill, J.A.; Schneider, J.; Morris, S.M., Jr.; Liu, Z.P. FoxO4 promotes early inflammatory response upon myocardial infarction via endothelial Arg1. Circ. Res. 2015, 117, 967–977. [Google Scholar] [CrossRef]
- Chuang, P.Y.; Yu, Q.; Fang, W.; Uribarri, J.; He, J.C. Advanced glycation endproducts induce podocyte apoptosis by activation of the FOXO4 transcription factor. Kidney Int. 2007, 72, 965–976. [Google Scholar] [CrossRef]
- Rai, V.; Agrawal, D.K. Immunomodulation of IL-33 and IL-37 with Vitamin D in the Neointima of Coronary Artery: A Comparative Study between Balloon Angioplasty and Stent in Hyperlipidemic Microswine. Int. J. Mol. Sci. 2021, 22, 8824. [Google Scholar] [CrossRef]
- Rai, V.; Agrawal, D.K. Role of Vitamin D in Cardiovascular Diseases. Endocrinol. Metab. Clin. N. Am. 2017, 46, 1039–1059. [Google Scholar] [CrossRef]
- Gupta, G.K.; Agrawal, T.; Rai, V.; Del Core, M.G.; Hunter, W.J., 3rd; Agrawal, D.K. Vitamin D Supplementation Reduces Intimal Hyperplasia and Restenosis following Coronary Intervention in Atherosclerotic Swine. PLoS ONE 2016, 11, e0156857. [Google Scholar] [CrossRef]
- Krohn, R.; Raffetseder, U.; Bot, I.; Zernecke, A.; Shagdarsuren, E.; Liehn, E.A.; van Santbrink, P.J.; Nelson, P.J.; Biessen, E.A.; Mertens, P.R.; et al. Y-box binding protein-1 controls CC chemokine ligand-5 (CCL5) expression in smooth muscle cells and contributes to neointima formation in atherosclerosis-prone mice. Circulation 2007, 116, 1812–1820. [Google Scholar] [CrossRef]
- Chen, T.Q.; Hu, N.; Huo, B.; Masau, J.F.; Yi, X.; Zhong, X.X.; Chen, Y.J.; Guo, X.; Zhu, X.H.; Wei, X.; et al. EHMT2/G9a Inhibits Aortic Smooth Muscle Cell Death by Suppressing Autophagy Activation. Int. J. Biol. Sci. 2020, 16, 1252–1263. [Google Scholar] [CrossRef]
- Wei, X.; Yi, X.; Zhu, X.H.; Jiang, D.S. Histone methylation and vascular biology. Clin. Epigenetics 2020, 12, 30. [Google Scholar] [CrossRef]
- Fan, C.; Ouyang, P.; Timur, A.A.; He, P.; You, S.A.; Hu, Y.; Ke, T.; Driscoll, D.J.; Chen, Q.; Wang, Q.K. Novel roles of GATA1 in regulation of angiogenic factor AGGF1 and endothelial cell function. J. Biol. Chem. 2009, 284, 23331–23343. [Google Scholar] [CrossRef]
- Yang, D.; Xiao, C.; Long, F.; Su, Z.; Jia, W.; Qin, M.; Huang, M.; Wu, W.; Suguro, R.; Liu, X.; et al. HDAC4 regulates vascular inflammation via activation of autophagy. Cardiovasc. Res. 2018, 114, 1016–1028. [Google Scholar] [CrossRef]
- Bai, L.; Kee, H.J.; Choi, S.Y.; Seok, Y.M.; Kim, G.R.; Kee, S.J.; Kook, H.; Jeong, M.H. HDAC5 inhibition reduces angiotensin II-induced vascular contraction, hypertrophy, and oxidative stress in a mouse model. Biomed. Pharmacother. 2021, 134, 111162. [Google Scholar] [CrossRef]
- Wang, T.Y.; Chang, M.M.; Li, Y.J.; Huang, T.C.; Chien, S.; Wu, C.C. Maintenance of HDACs and H3K9me3 Prevents Arterial Flow-Induced Venous Endothelial Damage. Front. Cell Dev. Biol. 2021, 9, 642150. [Google Scholar] [CrossRef] [PubMed]
- Choe, N.; Kwon, J.S.; Kim, J.R.; Eom, G.H.; Kim, Y.; Nam, K.I.; Ahn, Y.; Kee, H.J.; Kook, H. The microRNA miR-132 targets Lrrfip1 to block vascular smooth muscle cell proliferation and neointimal hyperplasia. Atherosclerosis 2013, 229, 348–355. [Google Scholar] [CrossRef] [PubMed]
- Torella, D.; Iaconetti, C.; Catalucci, D.; Ellison, G.M.; Leone, A.; Waring, C.D.; Bochicchio, A.; Vicinanza, C.; Aquila, I.; Curcio, A.; et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ. Res. 2011, 109, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Park, M.; Choi, S.; Kim, S.; Kim, J.; Lee, D.K.; Park, W.; Kim, T.; Jung, J.; Hwang, J.Y.; Won, M.H.; et al. NF-kappaB-responsive miR-155 induces functional impairment of vascular smooth muscle cells by downregulating soluble guanylyl cyclase. Exp. Mol. Med. 2019, 51, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.; Ye, C.; Zheng, F.; Wan, G.W.; Wu, L.L.; Chen, Q.; Li, Y.H.; Kang, Y.M.; Zhu, G.Q. MiR155-5p Inhibits Cell Migration and Oxidative Stress in Vascular Smooth Muscle Cells of Spontaneously Hypertensive Rats. Antioxidants 2020, 9, 204. [Google Scholar] [CrossRef]
- Ren, X.S.; Tong, Y.; Qiu, Y.; Ye, C.; Wu, N.; Xiong, X.Q.; Wang, J.J.; Han, Y.; Zhou, Y.B.; Zhang, F.; et al. MiR155-5p in adventitial fibroblasts-derived extracellular vesicles inhibits vascular smooth muscle cell proliferation via suppressing angiotensin-converting enzyme expression. J. Extracell. Vesicles 2020, 9, 1698795. [Google Scholar] [CrossRef]
- Raucci, A.; Macri, F.; Castiglione, S.; Badi, I.; Vinci, M.C.; Zuccolo, E. MicroRNA-34a: The bad guy in age-related vascular diseases. Cell Mol. Life Sci. 2021, 78, 7355–7378. [Google Scholar] [CrossRef]
- Hua, C.C.; Liu, X.M.; Liang, L.R.; Wang, L.F.; Zhong, J.C. Targeting the microRNA-34a as a Novel Therapeutic Strategy for Cardiovascular Diseases. Front. Cardiovasc. Med. 2021, 8, 784044. [Google Scholar] [CrossRef]
- Weng, C.F.; Wu, C.F.; Kao, S.H.; Chen, J.C.; Lin, H.H. Down-Regulation of miR-34a-5p Potentiates Protective Effect of Adipose-Derived Mesenchymal Stem Cells Against Ischemic Myocardial Infarction by Stimulating the Expression of C1q/Tumor Necrosis Factor-Related Protein-9. Front. Physiol. 2019, 10, 1445. [Google Scholar] [CrossRef]
- Tao, W.; Sun, W.; Zhu, H.; Zhang, J. miR-205-5p suppresses pulmonary vascular smooth muscle cell proliferation by targeting MICAL2-mediated Erk1/2 signaling. Microvasc. Res. 2019, 124, 43–50. [Google Scholar] [CrossRef]
- Zhao, M.; Wang, W.; Lu, Y.; Wang, N.; Kong, D.; Shan, L. MicroRNA153 attenuates hypoxiainduced excessive proliferation and migration of pulmonary arterial smooth muscle cells by targeting ROCK1 and NFATc3. Mol. Med. Rep. 2021, 23, 194. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Sun, Y.; Ma, X.; Wang, C.; Zhang, L.; Wang, J.; Wang, G.; Li, Z.; Tian, W.; Zhao, Z.; et al. MicroRNA-22 Inhibits the Apoptosis of Vascular Smooth Muscle Cell by Targeting p38MAPKalpha in Vascular Remodeling of Aortic Dissection. Mol. Ther. Nucleic Acids 2020, 22, 1051–1062. [Google Scholar] [CrossRef] [PubMed]
- Cirino, M.; Porsani, L.; Neto, F.L.; Tazima, M.; Zimak, R.; Carlotti Jr, C.; Colli, B.; Tirapelli, L.; Tirapelli, D. Expression of miR-15b, miR-29b, miR-219 and miR-222 microRNAs in rats with focal cerebral ischemia submitted to physical exercise. Genet. Mol. Res. 2019, 18, gmr18213. [Google Scholar] [CrossRef]
- Fang, Y.C.; Yeh, C.H. Role of microRNAs in Vascular Remodeling. Curr. Mol. Med. 2015, 15, 684–696. [Google Scholar] [CrossRef]
- Henn, D.; Abu-Halima, M.; Wermke, D.; Falkner, F.; Thomas, B.; Kopple, C.; Ludwig, N.; Schulte, M.; Brockmann, M.A.; Kim, Y.J.; et al. MicroRNA-regulated pathways of flow-stimulated angiogenesis and vascular remodeling in vivo. J. Transl. Med. 2019, 17, 22. [Google Scholar] [CrossRef]
- McDonald, R.A.; Hata, A.; MacLean, M.R.; Morrell, N.W.; Baker, A.H. MicroRNA and vascular remodelling in acute vascular injury and pulmonary vascular remodelling. Cardiovasc. Res. 2012, 93, 594–604. [Google Scholar] [CrossRef]
- Welten, S.M.; Goossens, E.A.; Quax, P.H.; Nossent, A.Y. The multifactorial nature of microRNAs in vascular remodelling. Cardiovasc. Res. 2016, 110, 6–22. [Google Scholar] [CrossRef]
- Pujol-López, M.; Ortega-Paz, L.; Garabito, M.; Brugaletta, S.; Sabaté, M.; Dantas, A.P. miRNA update: A review focus on clinical implications of miRNA in vascular remodeling. AIMS Med. Sci. 2017, 4, 99–112. [Google Scholar] [CrossRef]
- Cunnane, E.M.; Weinbaum, J.S.; O’Brien, F.J.; Vorp, D.A. Future Perspectives on the Role of Stem Cells and Extracellular Vesicles in Vascular Tissue Regeneration. Front. Cardiovasc. Med. 2018, 5, 86. [Google Scholar] [CrossRef]
- Pedroza, A.J.; Tashima, Y.; Shad, R.; Cheng, P.; Wirka, R.; Churovich, S.; Nakamura, K.; Yokoyama, N.; Cui, J.Z.; Iosef, C.; et al. Single-Cell Transcriptomic Profiling of Vascular Smooth Muscle Cell Phenotype Modulation in Marfan Syndrome Aortic Aneurysm. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 2195–2211. [Google Scholar] [CrossRef]
- Scapoli, C.; Ziliotto, N.; Lunghi, B.; Menegatti, E.; Salvi, F.; Zamboni, P.; Baroni, M.; Mascoli, F.; Bernardi, F.; Marchetti, G. Combination of Genomic and Transcriptomic Approaches Highlights Vascular and Circadian Clock Components in Multiple Sclerosis. Int. J. Mol. Sci. 2021, 23, 310. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liu, D.; Bu, D.; Chen, S.; Wu, J.; Tang, C.; Du, J.; Jin, H. Brg1-dependent epigenetic control of vascular smooth muscle cell proliferation by hydrogen sulfide. Biochim. Biophys. Acta 2013, 1833, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Xin, W.; Zhang, M.; Yu, Y.; Li, S.; Ma, C.; Zhang, J.; Jiang, Y.; Li, Y.; Zheng, X.; Zhang, L.; et al. BCAT1 binds the RNA-binding protein ZNF423 to activate autophagy via the IRE1-XBP-1-RIDD axis in hypoxic PASMCs. Cell Death Dis. 2020, 11, 764. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Associated with Inhibited Network | DEGs (log2 > 2, p < 0.05) | DEGs (log2 < −2, p < 0.05) |
|---|---|---|
| mir-133 | VCAN | PPARG, TNFSF10 |
| miR-155-5p | BACH1, CD69, CXCL8, PMAIP1, PPL, RIPK1 |
| Associated with Activated Network | DEGs (log2 > 2, p < 0.05) | DEGs (log2 < −2, p < 0.05) | DEGs (log2 < 2 or >−2, p < 0.05) |
|---|---|---|---|
| miR-205-5p | ADAM8, ADAMTS4, ARG1, CD27, CD3D, CD5, CSF2RB, CXCL8, DNAH11, DUOX2, GP1BA, GP1BB, HPSE, IL1R1, IL2RB, IL7R, KDM6B, LAG3, MMP25, NCOR2, PCDH8, PHACTR1, PPRC1, PTX3, RELT, SEMA4A, SERPINB2, STAT4, STEAP4 | ADAMTS8, ADIPOR2, AQP11, BCAM, CAT, CEBPA, CIDEC, DHH, EIF4EBP1, HCAR1, LMO2, MYLPF, PLIN1, PLIN5, PNPLA2, PPARG, RETSAT, SEMA3G, SERPINI1, TEK, WNT11 | CD6, CXCR5, EGR2 |
| Associated with inhibited networks | |||
| mir-515 | ADAM8, ADAMTS4, ALAS2, BCL3, CCR7, CD27, CD3D, CD69, COL6A3, CXCL8, DUOX2, ECM1, EDIL3, EGR1, FCMR, FERMT3, GP1BA, GPR84, HPSE, ICOS, IL2RB, IL7R, KDM6B, LAG3, MAP3K14, MUC13, MYC, NCOR2, PCDH8, PHACTR1, PLAGL1, PPRC1, PRDM1, PTX3, RCAN1, RNF213, SBNO2, SELP, SEMA4A, SERPINB2, SLAMF7, ST18, STAT4, STC1, TUBB1, VWF | AGT, ALDH9A1, APOD, APOE, ASS1, BCAM, BMX, CAVIN2, CIDEC, CLEC14A, CYP4B1, DDIT4L, ECH1, EIF4EBP1, ENHO, EPHX2, HCAR1, MMD, MYH4, MYL1, OSM, OSMR, PPARG, PLIN5, PNPLA2, PON3, S100A1, SERPINI1, TCAP, TEK, TMOD4, TNNC2, TSPAN5 | CD6, CXCR5 |
| mir-153 | ADAMTS1, CD5, CD69, CSF2RB, CXCL8, DUOX2, EGR3, FCMR, GP1BA, HAS1, ICOS, IL2RB, ITGA2B, ITGB7, KDM6B, NCOR2, NFATC2, PDE4B, PHACTR1, PRDM1, RNF213, SERPINB2, ST18, STAT4, STEAP4, TEAD3, VDR | AGT, ALDH1A1, APOD, APOE, BCAM, DHH, MYH8, MYL1, MYOT, OSM, OSMR, PLIN1, RAMP2, RETSAT, S100A1, SCD, TCAP, TNNC2, WNT11 | |
| mir-219 | ALAS2, BCL3, BLK, CCL22, CSF2RB, CXCL8, DUOX2, FCMR, ICOS, IL2RB, IL7R, ITGA2B, NFATC2, PDE4B, PHACTR1, PLAGL1, PRDM1, PTX3, RNF213, SBNO2, STEAP4, TUBB1, VWF | ALDH1A1, APOD, APOE, ASS1, BCAM, CAT, CAVIN2, LDHB, PPARG, MMD, MYL1, MYLPF, MYOT, OSM, PEX11A, RAMP1, TEK | EGR2, IL9R |
| mir-22 | CCL22, CCR7, DUOX2, HAS1, IL1R1, IL2RB, IL7R, ITGA2B, ITGB7, NFATC2,PTX3, RCAN1, SERPINB2, STEAP4, VDR | ALDH1A2, ASS1, BCAM, MYLPF, TNNC2 | EGR2 |
| Associated with Activated Networks | DEGs (log2 > 2, p < 0.05) | DEGs (log2 < −2, p < 0.05) | DEGs (log2 < 2 or >−2, p < 0.05) |
|---|---|---|---|
| MYOD1 | CXCL14, MYH4, MYL1, MYLPF, MYOD1, TNNC2 | IGF1 | |
| SMARCA4 | IL7R, ITGA3, MYL1, MYLPF, TNNC2 | CNTN1, MAP1B, MFGE8, | IGF1 |
| MEF2C | MYLPF, MYOD1, MYOT | FRZB, KCNA5, | IGF1 |
| MYF6 | EGF, IL15, IL7, MYOD1, | ||
| HOXC8 | IL1R2, SLC16A3 | ||
| SPDEF | ITGA3 | TNC | COL4A2 |
| SPI1 | CD1D, CXCL14, IL18, IL1R2, IL7R | CD79B | |
| IRF1 | IL15, IL18, IL7, MMP9 | ||
| RB1 | HOMER1, MYH4, MYH8, MYL1, TCAP, TNNC2 | IGF1, COL4A2 | |
| ARNT2 | APOE, EGF, HOMER1 | ||
| SIM1 | APOE, EGF, HOMER1 | ||
| Associated with inhibited network | |||
| HDAC4 | HOMER1, MMP9, MYLK2, MYOT | CNTN1 | |
| KDM5A | HOMER1, MYH4, MYH8, MYL1, TCAP, TNNC2 | ||
| HTT | APOE, HOMER1, IL15, MYL1, MYOD1, | PRELP | COL4A2 |
| VDR | IL18, MYH8, S100A8 | COL4A2 | |
| Associated with Activated Networks | DEGs (log2 > 2, p < 0.05) | DEGs (log2 < −2, p < 0.05) | DEGs (log2 < 2 or >−2, p < 0.05) |
|---|---|---|---|
| MYF6 | ANK1, APOE, CASQ1, EGF, HOMER1, IL15, IL18, IL7R, MYL1, MYLPF, MYOD1, MYOT, TCAP, TNNC2, TRIM63 | AQP5, NOTCH3, TNC | IGF1 |
| MYOD1 | ANK1, APOE, CASQ1, EGF, HOMER1, IL18, IL7R, MYL1, MYLPF, MYOT, TCAP, TNNC2, TRIM63 | AQP5, NOTCH3, TNC | IGF1 |
| NCOA4 | AMPD1, APOE, CD1D, CD247, CD3G, CYGB, DDIT4L, EGF, FGR, HABP2, ICAM3, IL15, IL1R2, IL7R, MMP9, MMRN1, MYH8, MYLPF, MYOT, MYPN, PRKCQ, S100A1, SLN, TFPI, TNNC2, XIRP2 | ADAMTSL4, AEBP1, COL8A1, ELN, FBLN5, FRZB, KCNA5, MAP1B, MFGE8, NOTCH3, SLIT3 | IGF1, COL4A2 |
| NCOA1 | ANK1, CNKSR1, CXCL14, DOK5, EGF, HOMER1, ICAM3, IFIT1, IL15, IL18, IL1R2, IL7, IL7R, MYH4, MYH8, MYL1, MYOT, MYPN, PLIN5, STK17B, TCAP, TNNC2, TRIM63, XIRP2 | AEBP1, AQP5, COL8A1, ELN, FBLN5, FMOD, ITGA3, MAP1B, NOTCH3, SLIT3 | COL4A2 |
| FOXA1 | CD1D, CD247, CNKSR1, CXCL14, CYGB, DDIT4L, FGR, HOMER1, ICAM3, IFIT1, IL15, IL18, IL7, MMP9, MYH4, MYH8, MYL1, MYLPF, MYOD1, MYPN, PLIN5, PRKCQ, S100A1, STK17B, TCAP, TNNC2, XIRP2 | ADAMTSL4, BGN, ELN, COL8A1, FBLN5, FMOD, FRZB, ITGA3, MAP1B, TNC, NOTCH3, PRELP, SLIT3 | IGF1, COL4A2, CD79B |
| SMARCC1 | CNKSR1, ICAM3, IL7R, MMP9, MYOT | ITGA3, MAP1B, NOTCH3, TNC | IGF1, CD79B COL4A2 |
| SMARCA4 | IFIT1, MYL1, MYLPF, TNNC2 | CNTN1 | IGF1 |
| HOXC8 | IL1R2, SLC16A3 | ||
| SPDEF | ITGA3, TNC | COL4A2 | |
| IRF9 | IFIT1, IL18 | ||
| Associated with inhibited networks | |||
| AHRR | CD1D, CD247, CD3G, CXCL14, HABP2, ICAM3, IFIT1, IL15, IL18, IL1R2, IL7R, MYH4, MYH8, MYL1, MYLK2, MYOT, MYPN, PLIN5, PRKCQ, STK17B, TCAP, TRIM63, XIRP2 | AEBP1, FBLN5, FMOD, FRZB, MFGE8, NOTCH3, SLIT3 | COL4A2 |
| HDAC3 | ANK1, CXCL14, DOK5, HABP2, IFIT1, IL15, IL18, IL7, IL7R, MYH8, MYLPF, PLIN5, S100A8, SLN, TFPI, TRIM63 | AQP5, BGN, COL8A1, ELN, FBLN5, ITGA3, KCNA5, NOTCH3, RASL11B | IGF1, COL4A2 |
| EID1 | CNKSR1, CXCL14, CYGB, DDIT4L, DOK5, EGF, FGR, HOMER1, ICAM3, IL15, IL18, IL7, MMP9, MYH4, MYH8, MYL1, MYOT, MYPN, PLIN5, S100A8, STK17B, TMOD4, TNNC2, XIRP2 | ADAMTSL4, ADRA1D, AEBP1, AQP5, BGN, FBLN5, FRZB, KCNA5, MAP1B, NOTCH3, SLIT3, TNC | IGF1 |
| HOXA10 | APOE, CNKSR1, CXCL14, CYGB, DDIT4L, DOK5, EGF, FGR, HOMER1, ICAM3, IL15, IL18, IL7, MMP9, MYH4, MYH8, MYL1, MYOT, MYPN, PLIN5, S100A8, TK17B, TMOD4, TNNC2, XIRP2 | ADAMTSL4, ADRA1D, AEBP1, FBLN5, FRZB, KCNA5, MAP1B, NOTCH3, SLIT3, TNC | CD79B |
| MTA2 | CD1D, COL8A1, CXCL14, DOK5, ICAM3, IFIT1, IL15, IL18, IL1R2, IL7R, MMP9, MYH4, MYH8, MYL1, MYLPF, MYOD1, MYOT, PLIN5, S100A8, SLC16A3, TNNC2, TRIM63 | AQP5, ELN, FBLN5, MAP1B, | |
| HOXA9 | ADAMDEC1, C5AR2, CNKSR1, CXCL14, CYGB, DOK5, FGR, ICAM3, IL18, IL7R, MYH4, MYH8, MYL1, MYOD1, MYOT, MYPN, PLIN5, PRKCQ, S100A8, STK17B, TNNC2, TRIM63, XIRP2 | ADRA1D, AEBP1, FBLN5, FRZB, ITGA8, KCNA5, MAP1B, NOTCH3, SLIT3, TNC | CD79B |
| TCF3 | CD3G, IL7R, MMP9, MYL1, MYLPF, MYOD1, STK17B, TNNC2 | NOTCH3 | IGF1, CD79B |
| AHRR | CD247, CD3G, IL15, IL1R2, IL7R, MYLK2, MYOT, PRKCQ | FBLN5, NOTCH3 | COL4A2 |
| AIP | ADRA1D, APOE, CD247, CD3G, IL15, IL1R2, IL7R, PLIN5, PRKCQ, S100A8 | FBLN5, NOTCH3 | COL4A2 |
| KDM5A | HOMER1, MYH4, MYH8, MYL1, TCAP, TNNC2 | ||
| DNMT3L | CASQ1, IFIT1, S100A1, SLIT3, SLN | CD79B | |
| EHF | HOMER1, MMP9, MYH4, MYH8, MYL1, S100A8, TCAP, TFPI, TNC, TNNC2 | NOTCH3 | IGF1 |
| HDAC4 | MYLK2, MYOT | ||
| SOX15 | MMP9, MYOD1 | ||
| Associated with Activated Networks | DEGs (log2 < −2, p < 0.05) | DEGs (log2 > 2, p < 0.05) | DEGs (log2 < 2 or >−2, p < 0.05) |
|---|---|---|---|
| STAT3 | AGT, ALDH1A1, CAT, CEBPA, LDHB | ALAS2, ARG1, BCL3, SELP COL5A1, CXCL8, EGR1,EGR3, ICOS, IL1R1,IL2RB, MYC, SBNO2, NFATC2, PLAGL1, PRDM1 | CXCR5, EGR2, IL9R |
| TP53 | ALDH1A1, ALDH1A2, APOE, ALDH9A1, ASS1, BMX, CAT, CEBPA, DDIT4L, ECH1, LDHB, PPARG, RAMP2, SCD, TCAP, TMOD4 | ADAM8, ARG1, BCL3, CXCL8, EGR1, EGR3, MYC, DMRT1, ECM1, EDIL3, ITGA2B, ITGB7, NCOR2, PDE4B, PRDM1, SELP, SERPINB2, VDR | EGR2 |
| HTT | AGT, APOE, CEBPA, LDHB, MYL1, PPARG | ADAMTS4, BCL3, EGR1, PDE4B, COL6A3, PPRC1, PTPN22, RGS14 | CXCR5, EGR2 |
| ETS1 | HPSE | CD27, CD69, CRTAM, EGR1, IL2RB, MYC, ITGA2B, PRDM1 | |
| BCL11B | CXCL8 | ||
| RELA | AGT, APOE, PPARG | BCL3, CCL22, CCR7, CD69, CXCL8, EGR1, GP1BB, MYC, NFATC2, PDE4B, PRDM1, PTX3, SELP | |
| VDR | AGT, MYH8, PNPLA2, PPARG | CXCL8, EGR1, MYC, ITGB7, STAT4, VDR | |
| HDAC4 | MYLK2, MYOT | SERPINB2 | EGR2 |
| RELB | CXCL8, MYC, PRDM1, STAT4 | ||
| KDM5A | MYH4, MYH8, MYL1, TCAP TNNC2 | ||
| ASXL1 | PLIN1, PPARG, SCD | ||
| LEF1 | ECM1, MYC, PRDM1 | ||
| NFKB2 | CCR7, CXCL8, MYC | CXCR5 | |
| NUPR1 | MMD | ABL2, CXCL8, MYC | |
| Associated with inhibited networks | |||
| GFI1 | CEBPA, | BCL3, CCR7,CXCL8, IL1R1, ITGB7, MYC, STAT4, VDR | EGR2 |
| PPARGC1A | AGT, APOD, CAT, LDHB, MYC, PLIN5, PNPLA2, SCD, SEMA3G | ADAMTS1, IL1R2, COL6A3, OSMR, STC1 | |
| MYOD1 | AGT, ASS1, MYH4, MYL1, MYLPF, TNNC2 | ||
| HLX | SEMA3G | CXCL8, EGR1, MYC, PRDM1, | |
| SPDEF | COL5A1, COL6A3, CXCL8 | ||
| RB1 | CEBPA, MYH4, MYH8, MYL1, PPARG, RAMP2, TCAP, TNNC2 | COL5A1, CXCL8, EGR1, EGR3, MYC, OSMR, PTX3 | |
| IKZF2 | CD69, ICOS, IL1R1, LAG3, STAT4 | ||
| NCOA1 | CEBPA, PPARG | EGR1, MYC | |
| NKX2-3 | CAVIN2 | CXCL8, RNF213 | |
| NEUROG1 | ASS1, CAVIN2 | ||
| KMT2D | IGSF1, PPARG | ||
| Associated with Activated Networks | DEGs (log2 > 2, p < 0.05) | DEGs (log2 < −2, p < 0.05) | DEGs (log2 < 2 or >−2, p < 0.05) |
|---|---|---|---|
| MEF2C | ADIPOR2, AGT, AQP11, ASS1, BACH1, BCAM, BST1, CAVIN2, CEBPA, DDIT4L, ECH1, EPHX2, LDHB, MDK, MYL1, MYLPF, MYOT, PEX11A, PLIN5, PNPLA2, RAMP1, RAMP2, RETSAT, SCD, SLC9A3R2, TEK, TMOD4, TNNC2, TSPAN5 | ABL2, ALAS2, ARG1, BLK, CD27, CD3D, CHST2, COL5A1, CXCL8, EGR3, DMRT1, ECM1, FCMR, GP1BA, GP1BB, GPR84, IL1R2, IL2RB, IL7R, ITGA2B, ITGB7, LAG3, LMO2, NFATC2, PLIN1, PLAGL1, PPRC1, PRDM1, PTPN22, RNF213, SEMA4A, SELP, ST18, STEAP4, TUBB1, VDR, VWF | EGR2, IL9R |
| ISL1 | AGT, ALDH1A1, ALDH1A2, AQP11, BCAM, CAT, CAVIN2, CEBPA, CIDEC, DHH, EIF4EBP1, EPHX2, KLHL31, LDHB, LGALS12, MMD, MYH4, MYL1, MYOT, PEX11A, PNPLA2, RAMP1, RAMP2, RETSAT, SERPINI1, SFRP5, TCAP, TEK, TNNC2,TSPAN5, WNT11 | ADAMTS1, ALAS2, BCL3, CD27, COL5A1, COL6A3, CXCL8, DMRT1, EGR1, EGR3, GPR84, HAS1, HPSE, ICOS, IL1R2, IL1RL1, KDM6B, LAG3, LMO2, MYC, MMP25, NFATC2, OSMR, PCDH8, PRDM1, PLAGL1, PLIN1, SBNO2, SEMA4A, SERPINB2, ST18, STEAP4, TCF7, TEAD3, TUBB1 | CXCR5, EGR2, IL9R |
| EHMT2 | ALDH1A1, APOE, ASS1, CAT, CAVIN2, CIDEC, CYP4B1, EPHX2, HCAR1, LDHB, MMD, MYL1, MYLPF, PEX11A, PLIN5, PPARG, SCD, TCAP | COL5A1, COL6A3, EGR1, FCMR, GP1BA, HAS1, IL1R1, IL1RL1, IL2RB, ITGA2B, ITGB7, MUC13, NFATC2, PDE4B, PRDM1, PLIN1, PTX3, SERPINB2, TUBB1, VDR, VWF | EGR2 |
| ASXL1 | ADRB1, ALDH1A1, ALDH1A2, ALDH9A1, APOD, APOE, BCAM, BMX, CAT, CAVIN2, CEBPA, CIDEC, CYP4B1, DDIT4L, ECH1, EPHX2, HCAR1, MYLPF, PEX11A, PLIN5, PNPLA2, PPARG, RAMP2, RETSAT, SCD, SEMA3G, SERPINI1, TCAP, TMOD4, TNNC2, WNT11 | ABL2, ARG1, ADAM8, ADAMTS1, ADAMTS4, BCL3, CCL22, CCR7, CD3D, CD5, CD27, COL6A3, CXCL8, DUOX2, EGR3, ECM1, EDIL3, GP1BA, GPR84, HAS1, HPSE, IL1RL1, IL2RB, ITGB7, ITGA11, KDM6B, LAG3, LMO2, MMP25, MUC13, NAV1, NFATC2, NCOR2, OSM, OSMR, PHACTR1, PLAGL1, PLIN1, PRDM1, RCAN1, RNF213, SBNO2, SELP, SLAMF7, ST18, STAT4, STC1, STEAP4, TUBB1, VDR | CXCR5, IL9R |
| BCL11B | AGT, ALDH1A2, ALDH9A1, APOD, APOE, ASS1, BMX, CAVIN2, CIDEC, CLEC14A, CYP4B1, DDIT4L, ECH1, EIF4EBP1, ENHO, EPHX2, HCAR1, HPSE, LDHB, MMD, MYLPF, PEX11A, PPARG, RAMP1, SERPINI1, TCAP, TEK, TMOD4, TSPAN5, WNT11 | ADAM8, ADAMTS4, ALAS2, BCL3, CCL22, CCR7, CD3D, CD27, CD69, COL6A3, CXCL8, DUOX2, ECM1, EGR1, FERMT3, GP1BA, GPR84, MYC, EDIL3, ICOS, IL1R2, IL2RB, ITGB7, IL7R, KDM6B, LAG3, MUC13, NCOR2, OSM, OSMR, PHACTR1, PCDH8, PLAGL1, PLIN5, PRDM1, PPRC1, PTX3, RCAN1, RNF213, SBNO2, SEMA4A, SERPINB2, SLAMF7, ST18, STAT4, TCF7, TUBB1,VWF | CD6, CXCR5, EGR2 |
| ANKRD42 | AGT, ALDH1A2, ALDH9A1, APOD, APOE, ASS1, BMX, CAVIN2, CIDEC, CLEC14A, CYP4B1, DDIT4L, ECH1, EIF4EBP1, ENHO, EPHX2, HCAR1, HPSE, LDHB, MMD, MYLPF, PEX11A, PPARG, RAMP1, SERPINI1, TCAP, TEK, TMOD4, TSPAN5, WNT11 | ADAM8, ADAMTS4, ALAS2, BCL3, CCL22, CCR7, CD3D, CD27, CD69, COL6A3, CXCL8, DUOX2, ECM1, EGR1, FERMT3, ICOS, EDIL3, GP1BA, GPR84, IL1R2, IL2RB, ITGB7, IL7R, KDM6B, LAG3, MUC13, MYC, NCOR2, OSM, OSMR, PCDH8, PHACTR1, PLAGL1, PLIN5, PPRC1, PRDM1, PTX3, RCAN1, RNF213, SBNO2, SEMA4A, SERPINB2, SLAMF7, ST18, STAT4, TCF7, TUBB1, VWF | CD6, CXCR5, EGR2 |
| YBX1 | AGT, ALDH1A2, ALDH9A1, APOD, APOE, ASS1, BMX, CAVIN2, CIDEC, CLEC14A, CYP4B1, DDIT4L, ECH1, EIF4EBP1, ENHO, EPHX2, HCAR1, HPSE, LDHB, MMD, MYLPF, PEX11A, PPARG, RAMP1, SERPINI1, TCAP, TEK, TMOD4, TSPAN5, WNT11 | ADAM8, ADAMTS4, ALAS2, BCL3, CCL22, CCR7, CD3D, CD27, CD69, COL6A3, CXCL8, DUOX2, EGR1, FERMT3, GP1BA, GPR84, ICOS, MYC, ECM1, EDIL3, IL1R2, IL2RB, ITGB7, IL7R, KDM6B, LAG3, MUC13, NCOR2, OSM, OSMR, PCDH8, PHACTR1, PRDM1, PPRC1, PLAGL1, PLIN5, PTX3, RCAN1, RNF213, SBNO2, SEMA4A, SERPINB2, SLAMF7, ST18, STAT4, TCF7, TUBB1, VWF | CD6, CXCR5, EGR2 |
| GATA4 | AGT, APOE, ASS1, CAT, CEBPA, CLEC14A, EIF4EBP1, MYL1, MYLPF, MYOT, PPARG, RAMP1, TCAP | ADAMTS4, ALAS2, BCL3, BLK, CCL22,CCR7, CD69, CSF2RB, CXCL8, DMRT1, FERMT3, GP1BA, HAS1, ICOS, IL1R1, IL1R2, IL1RL1, IL7R, ITGA2B, KDM6B, LMO2, MYC, NFATC2, OSM, OSMR, PDE4B, PLAGL1, PRDM1, PTX3, RCAN1, SELP, SERPINB2, ST18, TUBB1, VDR | CXCR5 |
| CCND1 | AGT, APOE, BCAM, CAT, CIDEC, CYP4B1, DHH, HCAR1, MMD, MYH4, MYH8, MYL1, MYLPF, MYOT, PEX11A, PLIN5, PNPLA2, PPARG, RAMP2, RBP7, SCD, TCAP, TNNC2, TSPAN5, WNT11 | ALAS2, ARG1, CCL22, CCR7, CD5, CD27, COL5A1, EGR3, GP1BA, GP1BB, HAS1, ICOS, IL2RB, ITGA2B, IL7R, LMO2, MYC, MAP3K14, MUC13, NCOR2, NFATC2, PCDH8, PLAGL1, PLIN1, PPRC1, PTX3, SBNO2, STEAP4, TEAD3, TUBB1, VDR | CD6, IL9R |
| MYB | AGT, APOE, BCAM, CAVIN2, MMD, RCAN1 | ALAS2, ARG1, BCL3, CCL22, CCR7, CD69, CXCL8, EGR1, FCMR, GP1BA, HPSE, ICOS, IL1R1, IL1RL1, IL2RB, IL7R, KDM6B, LMO2, MYC, ITGA2B, PDE4B, PRDM1, PTPN22, PTX3, SERPINB2, ST18, STEAP4, TUBB1, VDR, VWF | EGR2 |
| LRRFIP1 | AGT, ALDH1A1, ALDH1A2, APOE, CEBPA, PPARG, WNT11 | BCL3, CCL22, CCR7, CD69, CXCL8, EGR1, HAS1, ICOS, IL7R, KDM6B, MYC, ITGA2B, PDE4B, PLIN1, PRDM1, PTX3, SERPINB2, ST18, STAT4, TCF7 | EGR2 |
| ESRRA | AGT, ALDH1A1, ALDH1A2, APOD, CAT, CEBPA, EGFL7, LDHB, MYH8, PLIN5, PNPLA2, PPARG, SEMA3G, STC1, WNT11 | ADAM8, ADAMTS1, CCL22, CCR7, CXCL8, COL6A3, EGR3, IL1R1, IL1R2, IL2RB, MYC, OSMR, PLIN1, PDE4B, PRDM1, PTX3, STEAP4, TCF7 | |
| NOTCH4 | AGT, CEBPA, LDHB, MYL1, PPARG, SCD, TCAP, TEK | ALAS2, BCL3, CCL22, CCR7, CD27, CD69, CXCL8, EGR1, ICOS, IL7R, IL2RB, KDM6B, LMO2, OSM, PRDM1, PTX3, SERPINB2, STAT4, ST18, VWF | |
| GATA1 | AGT, ALDH1A1, CAT, CAVIN2, LDHB, MMD, PPARG | ALAS2, BCL3, CCL22, CXCL8, CD69, DUOX2, FCMR, GP1BA, GPR84, IL7R, ICOS, IL1R1, IL1R2, IL1RL1, ITGA2B, LMO2, MYC, NFATC2, PHACTR1, PLAGL1, PDE4B, PTPN22, RNF213, SBNO2, STAT4, ST18, TUBB1, VDR, VWF | IL9R |
| TBX5 | AGT, APOE, ASS1, MYL1, MYLPF, MYOT, PPARG | BCL3, BLK, CCL22, CCR7, IL7R, KDM6B, CD69, CXCL8, DMRT1, EGR1, ICOS, MYC, NFATC2, PLAGL1, PTX3, SERPINB2, ST18, STAT4 | EGR2 |
| NRIP1 | AGT, APOD, BCAM, CAT, CYP4B1, LDHB, PLIN5, PNPLA2, PPARG, SCD, SEMA3G, STC1 | ADAMTS1, ARG1, COL6A3, IL7R, CXCL8, EGR1, IL1R1, IL2RB, OSMR, PLIN1, PTX3, STEAP4 | EGR2 |
| TRIM32 | AGT, APOE, CEBPA, PPARG | ARG1, BCL3, CCL22, CCR7, CD69, EGR1, ICOS, IL7R, KDM6B, PRDM1, PTX3, SELP, SERPINB2, STAT4, ST18 | CXCR5 |
| BCL11B | CXCL8, IL7R | ||
| HDAC4 | MYLK2, MYOT | EGR2 | |
| Associated with inhibited networks | |||
| HDAC5 | AGT, ALDH1A1, ALDH1A2, APOE, ASS1, CASQ1, CAT, CEBPA, CIDEC, CYP4B1, DDIT4L, ECH1, EIF4EBP1, EPHX2, HCAR1, LDHB, MDK, MMD, MYC, MYH4, MYH8, MYL1, MYLPF, MYOT, PEX11A, PLIN5, PNPLA2, PPARG, RAMP2, RETSAT, SCD, SERPINI1, TEK, TNNC2 | ALAS2, ARG1, BLK, CD3D, CD27, CHST2, COL5A1, CSF2RB, DUOX2, EGR3, FCMR, GP1BA, GP1BB, IL1R2, IL1RL1, DMRT1, ITGA2B, KDM6B, LMO2, MUC13, NFATC2, OSM, OSMR, PHACTR1, PLAGL1, PLIN1, PTPN22, RCAN1, RNF213, SBNO2, SELP, STC1, ST18, STAT4, TUBB1, VDR | CD6, EGR2, IL9R |
| SREBF1 | ADRB1, ALDH1A1, ALDH1A2, ALDH9A1, APOD, APOE, BCAM, BMX, CAT, CAVIN2, CEBPA, CIDEC, CYP4B1, DDIT4L, ECH1, EPHX2, HCAR1, MDK, MYL1, MYLPF, PEX11A, PLIN5, PNPLA2, PPARG, RAMP2, RETSAT, SCD, SEMA3G, SERPINI1, TCAP, TMOD4, TNNC2, WNT11 | ABL2, ADAMTS1, ADAM8, ADAMTS4, ARG1, BCL3, CCL22, CCR7, CD3D, CD5, CD27, COL6A3, DUOX2, EGR3, ECM1, EDIL3, GP1BA, GPR84, HAS1, HPSE, IL1R2, IL1RL1, ITGA11, ITGB7, KDM6B, LAG3, LMO2, MMP25, MUC13, MYC, NAV1, NFATC2, OSM, OSMR, PHACTR1, NCOR2, PLAGL1, PLIN1, PRDM1, RCAN1, RNF213, SBNO2, SELP, SLAMF7, STC1, ST18, STAT4, TUBB1, VDR | CXCR5, IL9R |
| MED24 | ADRB1, AGT, ALDH1A2, ALDH9A1, APOD, APOE, BMX, CAT, CAVIN2, CEBPA, CIDEC, CYP4B1, DDIT4L, DHH, ECH1, EPHX2, HCAR1, MYH4, MYH8, MYLPF, MYOT, PEX11A, PLIN5, PNPLA2, PPARG, RAMP2, RETSAT, SCD, SEMA3G, SERPINI1, SGK2, STC1, TCAP, TMOD4, TNNC2, WNT11 | ABL2, ADAM8, ADAMTS4, ADIPOR2, ARG1, BCL3, CCL22, CCR7, CD3D, CD5, CD27, CXCL8, DUOX2, EGR1, EGR3, HAS1, HPSE, IL1RL1, ECM1, EDIL3, GP1BA, GPR84, ITGB7, ITGA11, KDM6B, LAG3, LMO2, MAP3K14, MUC13, MYC, MMP25, NCOR2, NFATC2, NAV1, OSM, PHACTR1, PLAGL1, PLIN1, PRDM1, RCAN1, RNF213, SBNO2, SEMA4A, SELP, ST18, STAT4, SLAMF7, TEAD3, TUBB1, VDR | CXCR5, IL9R |
| Ncoa6 | ADRB1, ALDH1A1, ALDH1A2, ALDH9A1, APOD, APOE, BCAM, BMX, CAT, CAVIN2, CEBPA, CIDEC, CYP4B1, DDIT4L, ECH1, EPHX2, HCAR1, MYLPF, PEX11A, PLIN5, PNPLA2, RAMP2, RETSAT, SCD, SEMA3G, SERPINI1, TCAP, TMOD4, TNNC2, WNT11 | ABL2, ADAMTS1, ADAMTS4, ADAM8, ARG1, BCL3, CCL22, CCR7, CD3D, CD5, CD27, COL6A3, CXCL8, DUOX2, EGR3, EGR1, GP1BA, GPR84, IL1RL1, IL2RB, ECM1, EDIL3, HAS1, HPSE, ITGB7, ITGA11, KDM6B, LAG3, LMO2, MMP25, MUC13, NAV1, NCOR2, NFATC2, OSM, OSMR, PHACTR1, PLAGL1, PLIN1, PRDM1, RCAN1, RNF213, SBNO2, SELP, SLAMF7, STC1, ST18, STEAP4, STAT4, TUBB1, VDR | CXCR5, IL9R |
| ZBTB32 | AGT, ALDH1A2, ALDH9A1, APOD, APOE, ASS1, BMX, CAVIN2, CIDEC, CLEC14A, CYP4B1, DDIT4L, ECH1, EIF4EBP1, ENHO, EPHX2, HCAR1, LDHB, MMD, MYLPF, PEX11A, PPARG, RAMP1, SERPINI1, TCAP, TEK, TMOD4, TSPAN5, WNT11 | ADAM8, ADAMTS4, ALAS2, BCL3, CCL22, CCR7, CD3D, CD27, CD69, COL6A3, CSF2RB, CXCL8, DUOX2, ECM1, EDIL3, FERMT3, GP1BA, GPR84, HPSE, ITGB7, ICOS, IL1R2, IL2RB, IL7R, KDM6B, LAG3, MYC, MUC13, NCOR2, OSM, OSMR, PHACTR1, PCDH8, PLAGL1, PLIN5, PPRC1, PRDM1, PTX3, RCAN1, RNF213, SBNO2, SEMA4A, SERPINB2, SLAMF7, ST18, STAT4, TCF7, TUBB1, VWF | CD6, CXCR5, EGR2 |
| NKX2-1 | AGT, ALDH1A2, APOE, ASS1, BACH1, CAT, CAVIN2, CEBPA, DHH, EPHX2, LDHB, LGALS12, MDK, MMD, MYH4, MYH8, MYL1, MYLPF, PEX11A, RAMP1, SCD, TCAP, WNT11 | ADAM8, ALAS2, CD3D, COL5A1, COL6A3, CSF2RB, EGR3, FCMR, HAS1, ICOS, IL1R2, IL1RL1, IL2RB, IL7R, LAG3, MYC, ITGA2B, MMP25, NAV1, NCOR2, NFATC2, OSM, OSMR, PLAGL1, PLIN1, PRDM1, PTX3, RCAN1, SBNO2, SELP, SERPINB2, TCF7, TUBB1, VDR, VWF | CD6, CXCR5, IL9R |
| NEUROG2 | ADRB1, AGT, ALDH1A1, ALDH1A2, ALDH9A1, APOD, ASS1, BACH1, BCAM, BMX, CAVIN2, CIDEC, CYP4B1, DDIT4L, DHH, ECH1, EIF4EBP1, EPHX2, HCAR1, LDHB, MDK, MMD, MYH4, MYL1, MYLPF, PLIN5, RAMP2, TCAP, TEK, TMOD4, WNT11 | ARG1, BCL3, CCL22, CCR7, COL5A1, EGR1, EGR3, HAS1, HPSE, IL1R1, IL2RB, ECM1, EDIL3, FCMR, GP1BA, ITGB7, KDM6B, MMP25, MUC13, NFATC2, OSM, OSMR, PLAGL1, PLIN1, RCAN1, SBNO2, ST18, STAT4, STEAP4, TCF7, TUBB1, VWF | CXCR5, EGR2, IL9R |
| DLX2 | ALDH1A2, APOE, AQP11, ASS1, BACH1, BST1, CEBPA, EIF4EBP1, ENHO, FFAR4, MMD, TSPAN5, WNT11 | ADAM8, ARG1, BCL3, CD69, CD3D, CSF2RB, CXCL8, DUOX2, EGR1, EGR3, FCMR, GPR84, HSH2D, IL1R2, IL1RL1, IL2RB, ITGB7, KDM6B, LAG3, MYC, NFATC2, OSM, MMP25, NAV1, PHACTR1, PRDM1, PTX3, RNF213, SBNO2, SELP. SEMA4A, SLAMF7, SERPINB2, ST18, STAT4, TCF7, VDR | EGR2 |
| NCOA6 | ADRB1, AGT, APOD, APOE, AQP11, ASS1, CAVIN2, CEBPA, DNAH11, EIF4EBP1, ENHO, EPHX2, HCAR1, MMD, MYH4, MYLPF, PEX11A, PON3, PPARG, RETSAT, S100A1, SEMA3B, SERPINI1, SLC9A3R2, TNNC2, WNT11 | ADAMTS1, ADAMTS4, ADIPOR2, CD27, CD69, CD3D, CD5, COL5A1, CSF2RB, COL6A3, CXCL8, EGR1, FCMR, GP1BA, HAS1, ICOS, IL1R2, IL1RL1, ITGA2B, KDM6B, LMO2, MAP3K14, MYC, OSM, PHACTR1, PCDH8, PLAGL1, PLIN1, PTX3, RELT, RNF213, SBNO2, SEMA4A, STAT4, STC1, TCF7, TUBB1, VWF, VDR | IL9R |
| FOXJ1 | ALDH1A2, APOE, AQP11, ASS1, BACH1, BST1, CEBPA, EIF4EBP1, ENHO, FFAR4, MMD, TSPAN5, WNT11 | ADAM8, ARG1, BCL3, CD3D, CD69, CSF2RB, CXCL8, DUOX2, EGR1, EGR3, FCMR, GPR84, HSH2D, IL1R2, IL1RL1, IL2RB, ITGB7, KDM6B, LAG3, MMP25, NAV1, NFATC2, OSM, PHACTR1, PRDM1, PTX3, RNF213, SBNO2, SELP, SEMA4A, SLAMF7, SERPINB2, ST18, STAT4, TCF7, VDR | EGR2 |
| FOXD1 | ALDH1A2, APOE, AQP11, ASS1, BACH1, BST1, CEBPA, ENHO, FFAR4, MMD, TSPAN5, WNT11 | ADAM8, ARG1, BCL3, CD3D, CD69, CSF2RB, CXCL8, DUOX2, EGR1, EGR3, FCMR, GPR84, HSH2D, ITGB7, IL1R2, IL1RL1, IL2RB, KDM6B, LAG3, MYC, MMP25, NAV1, PRDM1, PTX3, NFATC2, OSM, PHACTR1, RNF213, SBNO2, SELP, SERPINB2, SEMA4A, SLAMF7, ST18, STAT4, TCF7, VDR | EGR2 |
| ARHGAP35 | ADRB1, AGT, ALDH1A1, ALDH9A1, APOD, ASS1, BACH1, BCAM, BMX, CIDEC, CYP4B1, DDIT4L, DHH, ECH1, EIF4EBP1, HCAR1, KDM6B, LDHB, MMD, MYH4, MYLPF, PEX11A, PLIN5, RAMP1, RAMP2, SCD, TCAP, TEK, TMOD4, TNNC2 | ALAS2, BCL3, COL5A1, CSF2RB, CXCL8, DUOX2, EGR3, FCMR, GP1BA, IL1R1, IL1RL1, IL2RB, ECM1, EDIL3, ITGB7, MUC13, NFATC2, OSM, OSMR, PLAGL1, PLIN1, PTX3, RCAN1, SBNO2, STEAP4, ST18, STAT4, TUBB1, VWF | CXCR5, IL9R |
| MED1 | ADRB1, ALDH1A2, APOE, CAT, CAVIN2, CEBPA, CIDEC, CYP4B1, HCAR1, MUC13, PEX11A, PLIN5, PNPLA2, PPARG, SCD, SEMA3B | ARG1, CCL22, CCR7, CD69, COL5A1, COL6A3, EGR1, IL1R2, IL1RL1, IL2RB, MYC, PDE4B, PLIN1, PRDM1, STC1, VDR | |
| Ncoa6 | ALDH1A2, APOE, CAT, CAVIN2, CEBPA, CIDEC, CYP4B1, HCAR1, MUC13, PEX11A, PLIN5, SCD | ARG1, CCL22, CCR7, CXCL8, PLIN1, PRDM1, VDR | |
| ZNF423 | ALDH1A2, APOE, CAT, CAVIN2, CEBPA, CIDEC, CYP4B1, HCAR1, MUC13, PEX11A, PLIN5, PPARG, SCD | ARG1, CCL22, CCR7, CXCL8, MYC, PLIN1, PRDM1, VDR | |
| CITED2 | AGT, ALDH1A2, APOE, CAT, CAVIN2, CIDEC, CYP4B1, HCAR1, PEX11A, PLIN5, PPARG, SCD, SEMA3B, TEK | ALAS2, ARG1, BCL3, CCL22, CCR7, CD69, CXCL8, EGR1, EGR3, ICOS, IL7R, KDM6B, MUC13, MYC, PLIN1, PTX3, SERPINB2, ST18, STAT4, VDR | |
| CBFA2T3 | AGT, ALDH1A1, CAT, CAVIN2, LDHB, MMD, PPARG | ALAS2, BCL3, CD69, CXCL8, CCL22, DUOX2, FCMR, GP1BA, GPR84, IL7R, ICOS, IL1R1, IL1R2, IL1RL1, ITGA2B, LMO2, MYC, NFATC2, PHACTR1, PLAGL1, PDE4B, PTPN22, RCAN1, RNF213,SBNO2, STAT4, ST18, TUBB1, VDR, VWF | IL9R |
| TCF12 | AGT, ASS1, EIF4EBP1, MYL1, MYLPF, PPARG, TNNC2 | CD3D, CD5, CXCL8, ICOS, IL1RL1, IL2RB, ITGB7, IL7R, LMO2, PRDM1, SEMA4A, SLAMF7 | IL9R |
| HLF | APOE, ASS1, CAT, CEBPA, CIDEC, ECH1, MMD, PEX11A, PLIN5, PNPLA2, PPARG, RETSAT, SCD | ARG1, LMO2, MYC, PLIN1, | |
| IKZF2 | CD69, ICOS, IL1R1, LAG3, STAT4 | ||
| MYOD1 | AGT, ASS1, MYL1, MYLPF, TNNC2 | ||
| GFI1 | CEBPA | BCL3, CCR7, IL1R1, IL7R, ITGB7, STAT4 | |
| HLX | RAMP1, RAMP2, SEMA3G, TSPAN5 | BCL3, CXCL8, EGR1, KDM6B, MYC, PRDM1 | |
| SPDEF | COL5A1, COL6A3, CXCL8 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rai, V.; Agrawal, D.K. Transcriptional and Epigenetic Factors Associated with Early Thrombosis of Femoral Artery Involved in Arteriovenous Fistula. Proteomes 2022, 10, 14. https://doi.org/10.3390/proteomes10020014
Rai V, Agrawal DK. Transcriptional and Epigenetic Factors Associated with Early Thrombosis of Femoral Artery Involved in Arteriovenous Fistula. Proteomes. 2022; 10(2):14. https://doi.org/10.3390/proteomes10020014
Chicago/Turabian StyleRai, Vikrant, and Devendra K. Agrawal. 2022. "Transcriptional and Epigenetic Factors Associated with Early Thrombosis of Femoral Artery Involved in Arteriovenous Fistula" Proteomes 10, no. 2: 14. https://doi.org/10.3390/proteomes10020014
APA StyleRai, V., & Agrawal, D. K. (2022). Transcriptional and Epigenetic Factors Associated with Early Thrombosis of Femoral Artery Involved in Arteriovenous Fistula. Proteomes, 10(2), 14. https://doi.org/10.3390/proteomes10020014