The Treasure Vault Can be Opened: Large-Scale Genome Skimming Works Well Using Herbarium and Silica Gel Dried Material

 ,
,  , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results
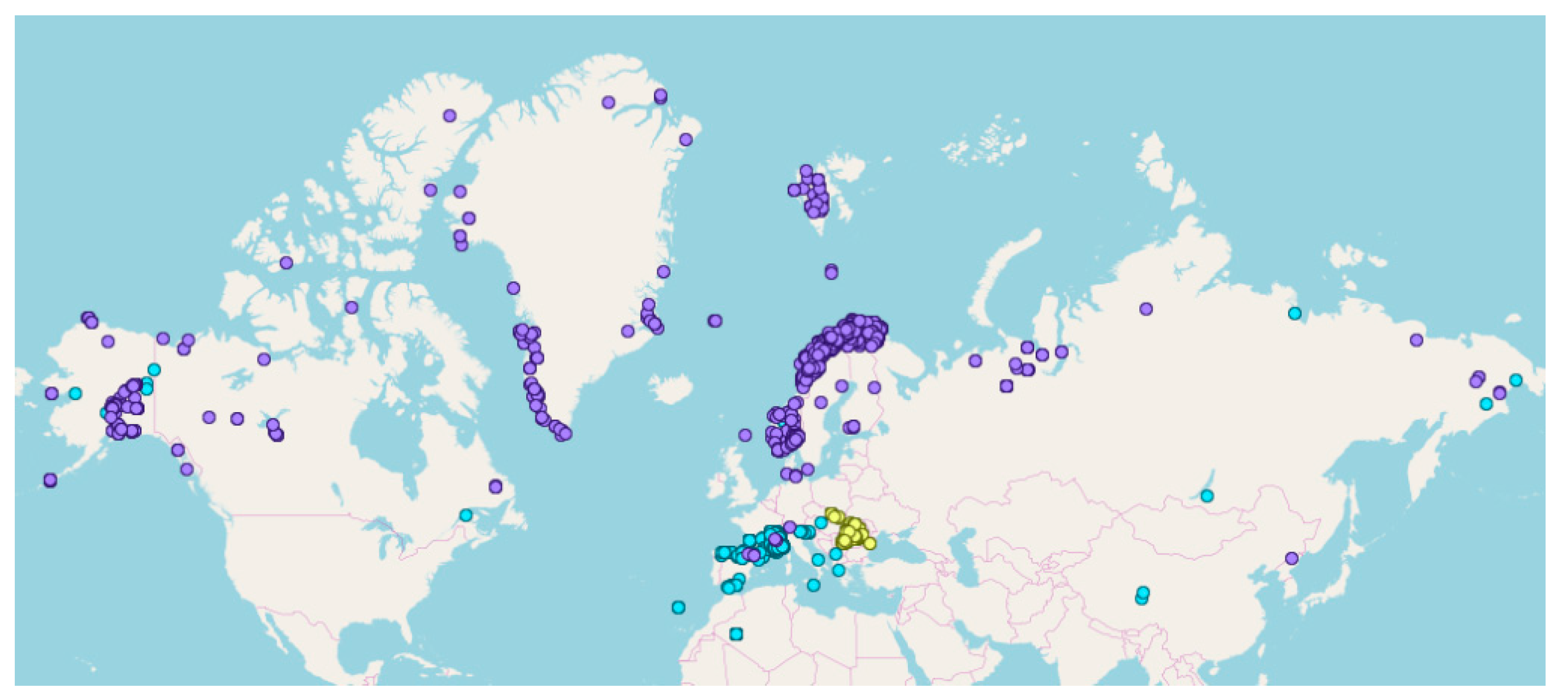
2.1. The Total Dataset
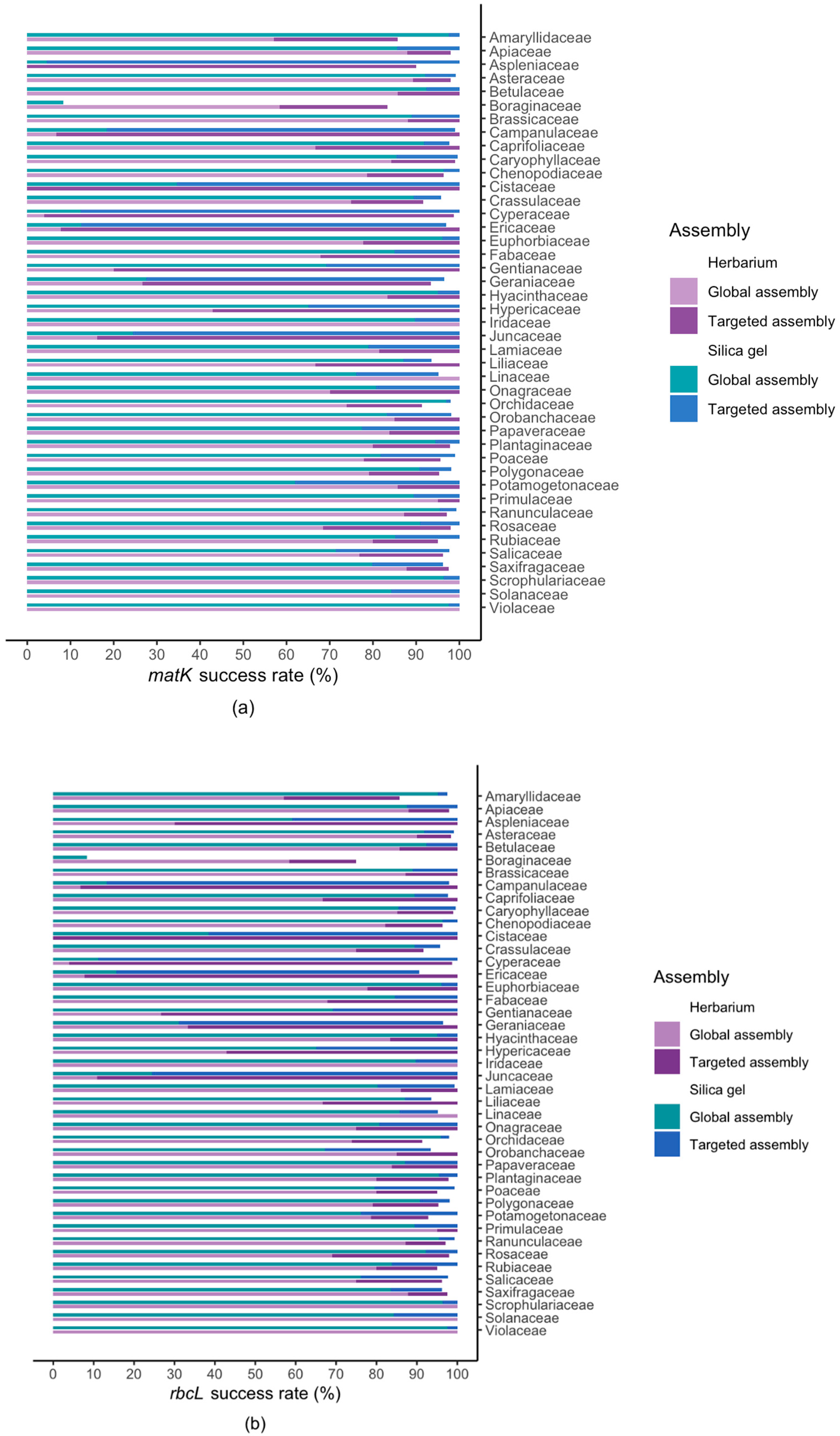
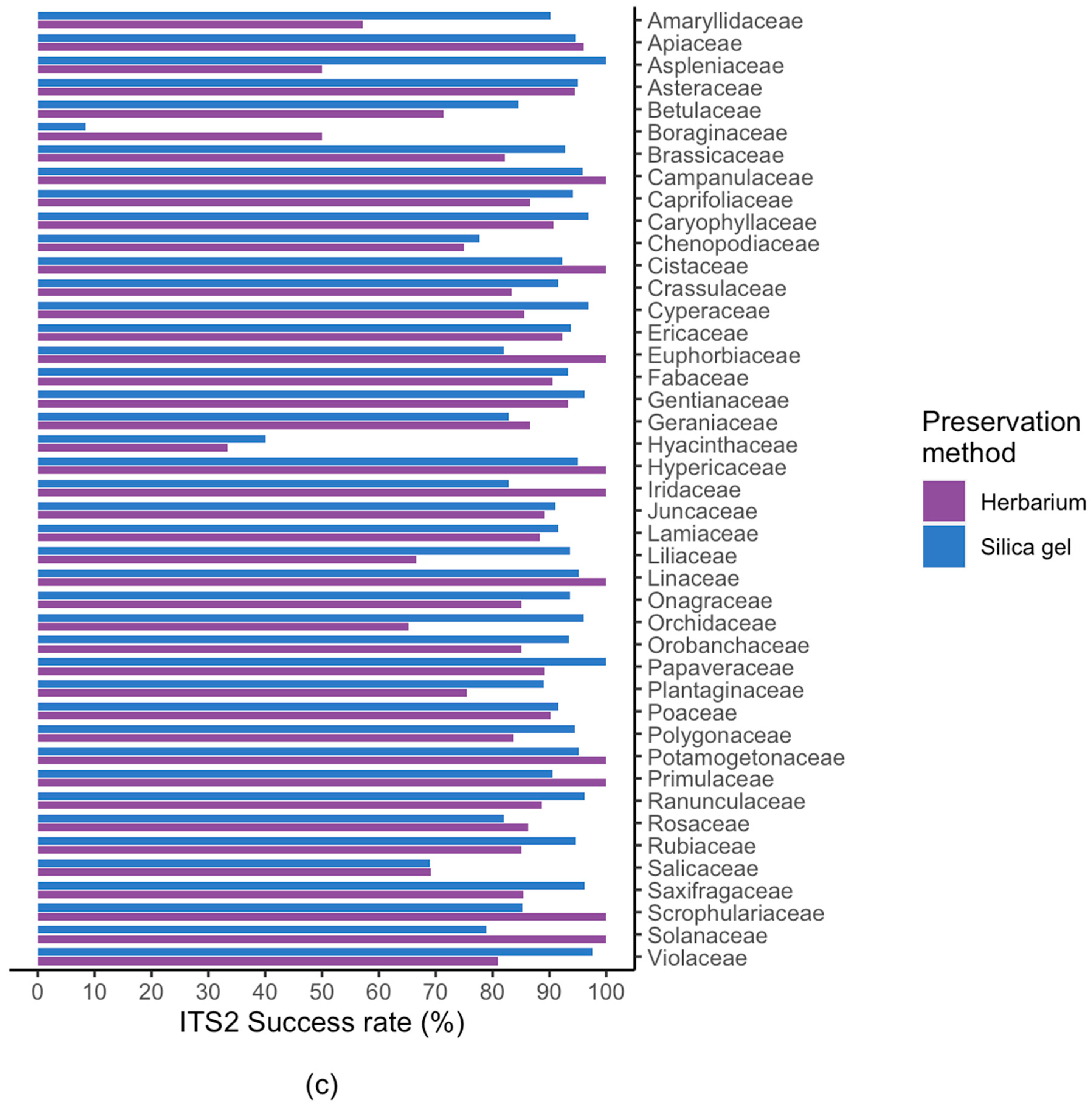
2.2. Results For Recovery of Standard Barcode Loci From Large Families
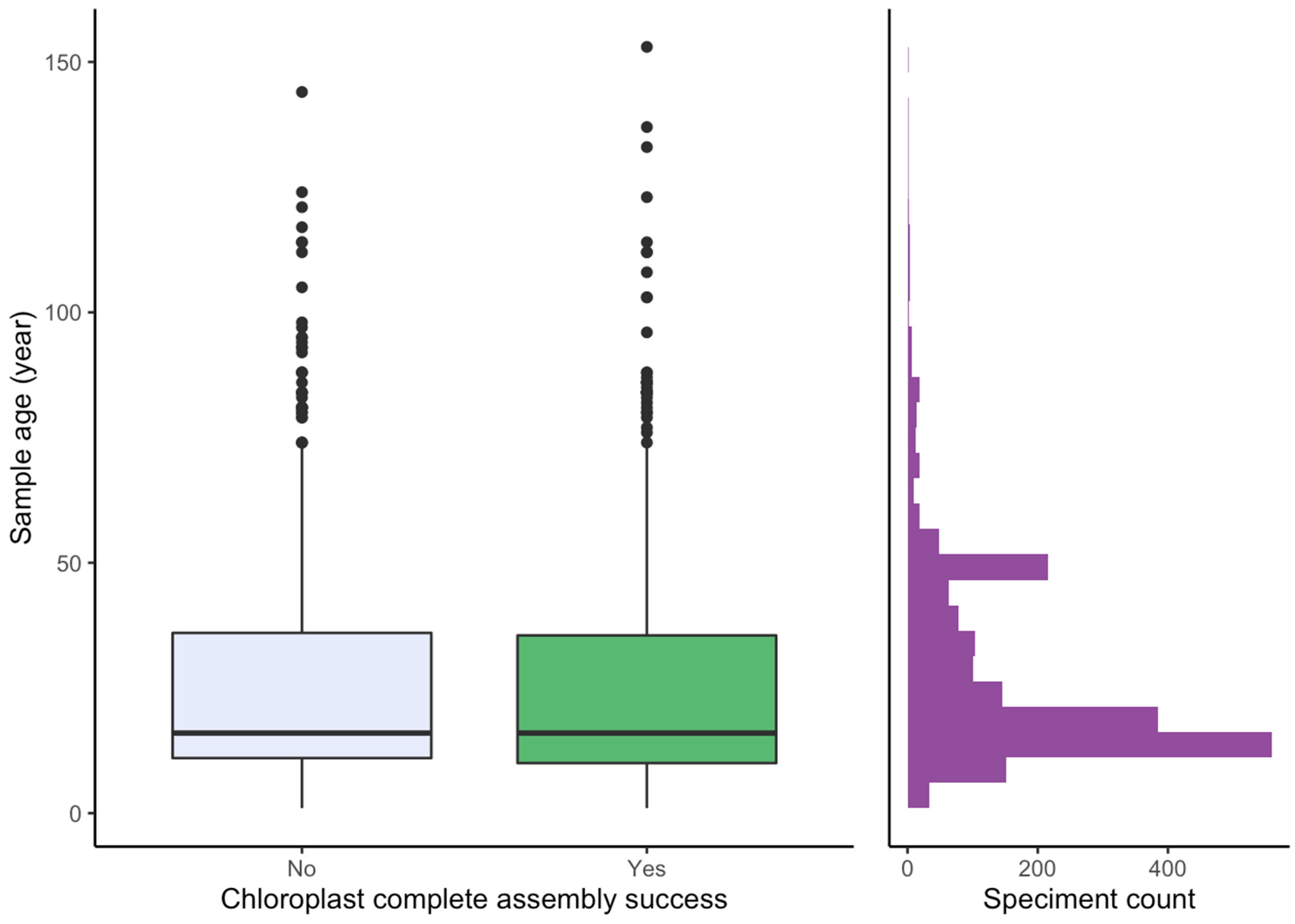
2.3. Effect of Sample Age and Time of Season
3. Discussion
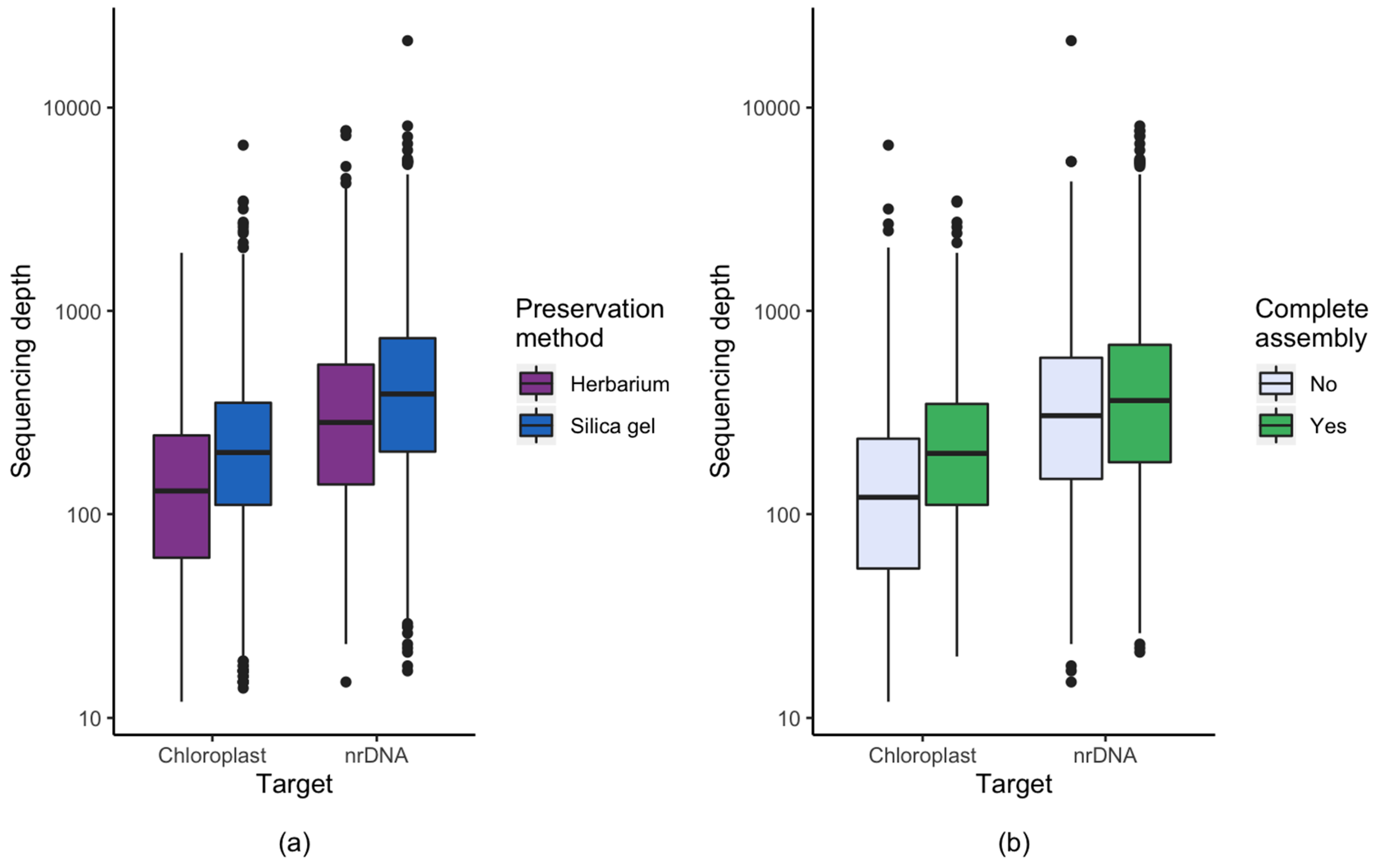
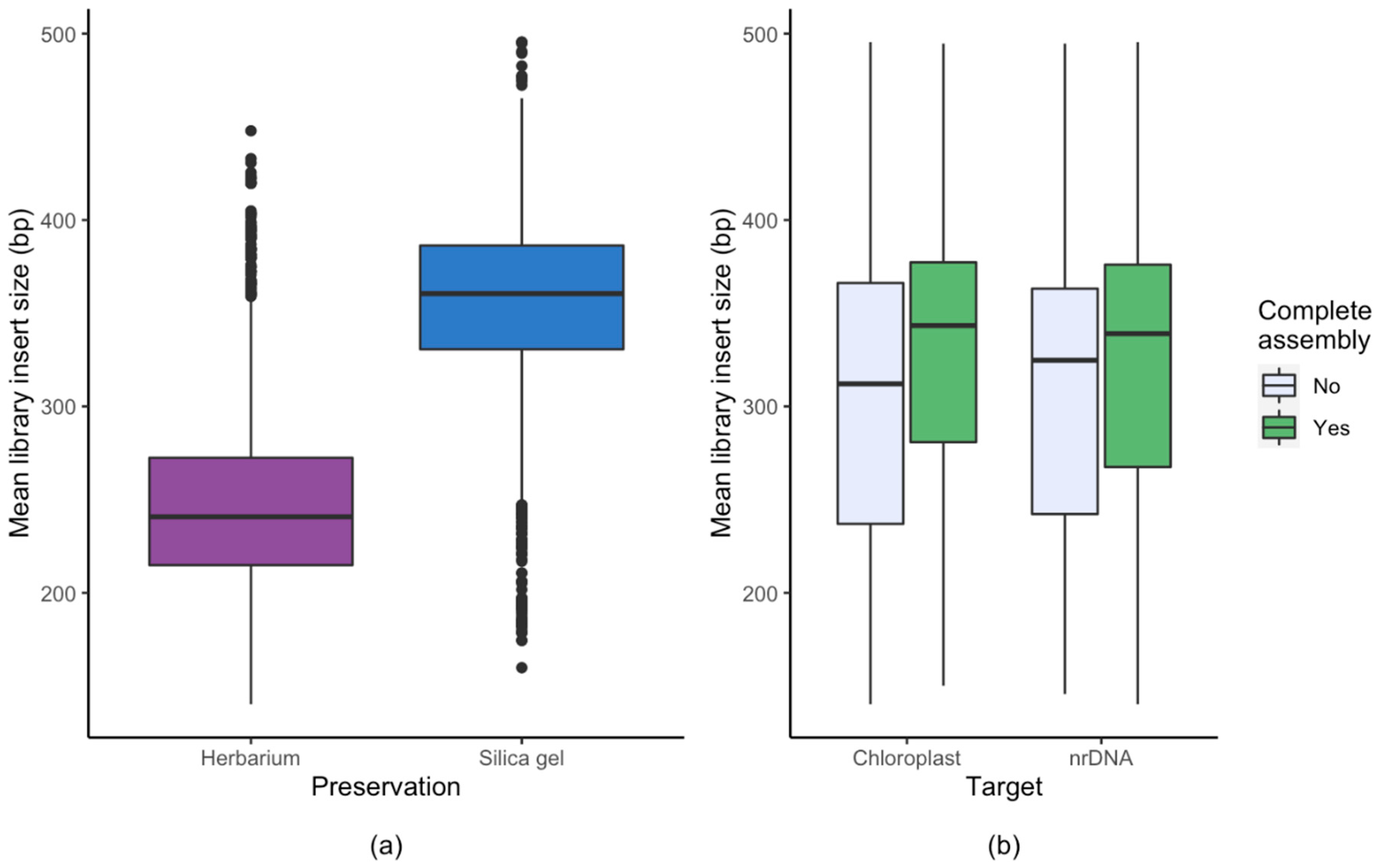
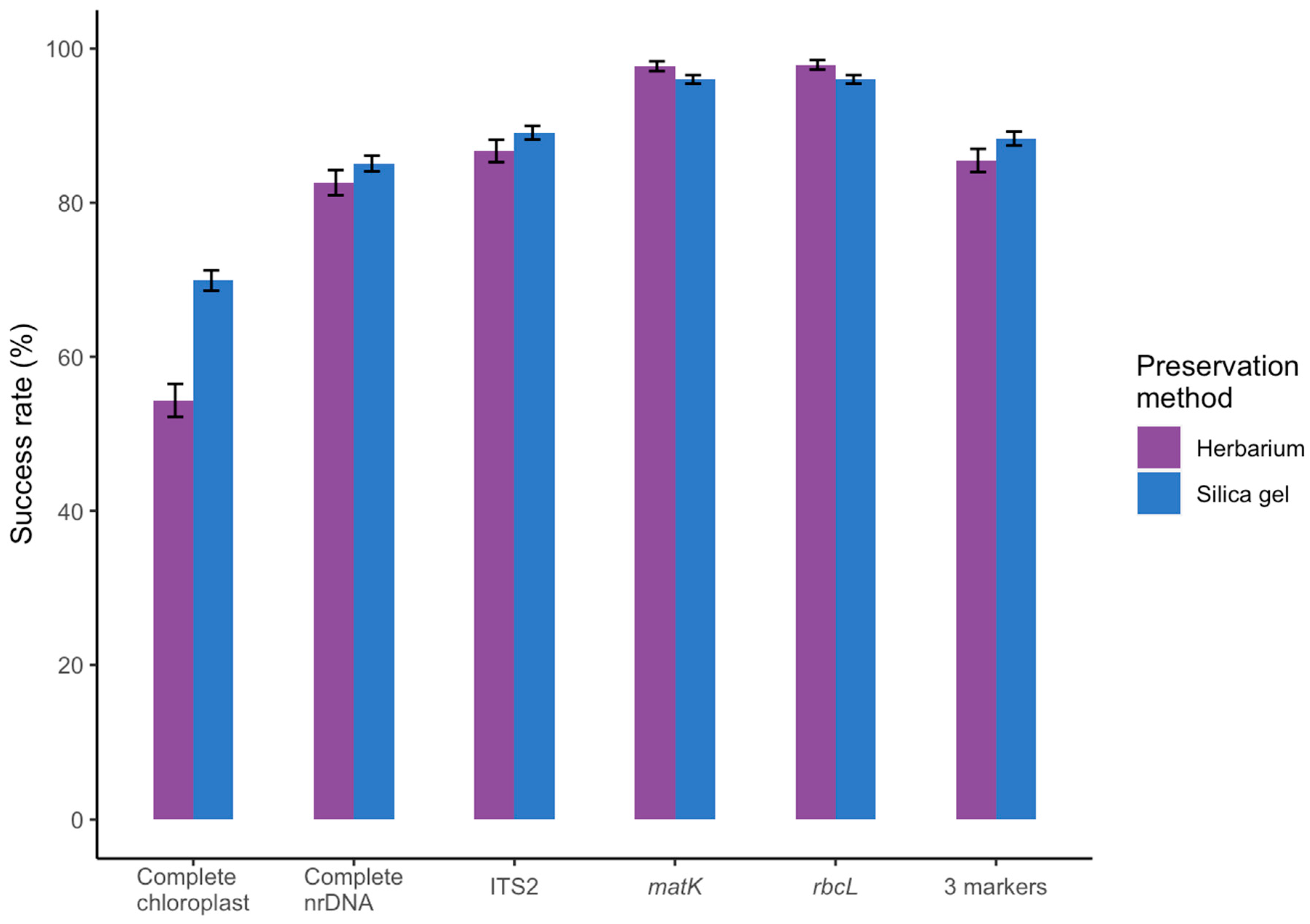
3.1. Effect of Starting Material
3.2. Success Rate Compared to Amplicon-based DNA Barcoding
3.3. Success Rate Among Families
3.4. Effect of Age and Time of Season
3.5. Utilisation of the Genome Skimming Data
4. Materials and Methods
4.1. Sampling and DNA Extraction
4.2. Library Preparation and Sequencing
4.3. Global Assembly and Annotation
4.4. Targeted Assembly for matK and rbcL
4.5. Quality Control
4.6. Statistical Analyses
4.7. Data Availability
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Plant Working Group; Hollingsworth, P.M.; Forrest, L.L.; Spouge, J.L.; Hajibabaei, M.; Ratnasingham, S.; van der Bank, M.; Chase, M.W.; Cowan, R.S.; Erickson, D.L.; et al. A DNA barcode for land plants. Proc. Natl. Acad. Sci. USA 2009, 106, 12794–12797. [Google Scholar] [CrossRef] [PubMed]
- Hollingsworth, P.M.; Graham, S.W.; Little, D.P. Choosing and using a plant DNA barcode. PLoS ONE 2011, 6, e19254. [Google Scholar] [CrossRef] [PubMed]
- Braukmann, T.W.A.; Kuzmina, M.L.; Sills, J.; Zakharov, E.V.; Hebert, P.D.N. Testing the efficacy of DNA barcodes for identifying the vascular plants of Canada. PLoS ONE 2017, 12, e0169515. [Google Scholar] [CrossRef] [PubMed]
- Li, H.-T.; Yi, T.-S.; Gao, L.-M.; Ma, P.-F.; Zhang, T.; Yang, J.-B.; Gitzendanner, M.A.; Fritsch, P.W.; Cai, J.; Luo, Y.; et al. Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 2019, 5, 461–470. [Google Scholar] [CrossRef] [PubMed]
- Gitzendanner, M.A.; Soltis, P.S.; Wong, G.K.-S.; Ruhfel, B.R.; Soltis, D.E. Plastid phylogenomic analysis of green plants: A billion years of evolutionary history. Am. J. Bot. 2018, 105, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Zhai, W.; Duan, X.; Zhang, R.; Guo, C.; Li, L.; Xu, G.; Shan, H.; Kong, H.; Ren, Y. Chloroplast genomic data provide new and robust insights into the phylogeny and evolution of the Ranunculaceae. Mol. Phylogenet. Evol. 2019, 135, 12–21. [Google Scholar] [CrossRef]
- Fahner, N.A.; Shokralla, S.; Baird, D.J.; Hajibabaei, M. Large-scale monitoring of plants through environmental DNA metabarcoding of soil: Recovery, resolution, and annotation of four DNA markers. PLoS ONE 2016, 11, e0157505. [Google Scholar] [CrossRef]
- Liu, J.; Yan, H.-F.; Newmaster, S.G.; Pei, N.; Ragupathy, S.; Ge, X.-J. The use of DNA barcoding as a tool for the conservation biogeography of subtropical forests in China. Divers. Distrib. 2015, 21, 188–199. [Google Scholar] [CrossRef]
- Willerslev, E.; Davison, J.; Moora, M.; Zobel, M.; Coissac, E.; Edwards, M.E.; Lorenzen, E.D.; Vestergård, M.; Gussarova, G.; Haile, J.; et al. Fifty thousand years of Arctic vegetation and megafaunal diet. Nature 2014, 506, 47–51. [Google Scholar] [CrossRef]
- Parducci, L.; Bennett, K.D.; Ficetola, G.F.; Alsos, I.G.; Suyama, Y.; Wood, J.R.; Pedersen, M.W. Ancient plant DNA in lake sediments. New Phytol. 2017, 214, 924–942. [Google Scholar] [CrossRef]
- Clarke, C.L.; Edwards, M.E.; Gielly, L.; Ehrich, D.; Hughes, P.D.M.; Morozova, L.M.; Haflidason, H.; Mangerud, J.; Svendsen, J.I.; Alsos, I.G. Persistence of arctic-alpine flora during 24,000 years of environmental change in the Polar Urals. Sci. Rep. 2019, 9, 19613. [Google Scholar] [CrossRef] [PubMed]
- Kool, A.; de Boer, H.J.; Krüger, A.; Rydberg, A.; Abbad, A.; Björk, L.; Martin, G. Molecular identification of commercialized medicinal plants in southern Morocco. PLoS ONE 2012, 7, e39459. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Zhang, M.-F.; Xue, J.; Dong, R.; Du, Y.-P.; Zhang, X.-H. Chloroplast genomic resources for phylogeny and DNA barcoding: A case study on Fritillaria. Sci. Rep. 2018, 8, 1184. [Google Scholar] [CrossRef] [PubMed]
- Soininen, E.M.; Gauthier, G.; Bilodeau, F.; Berteaux, D.; Gielly, L.; Taberlet, P.; Gussarova, G.; Bellemain, E.; Hassel, K.; Stenøien, H.K.; et al. Highly overlapping winter diet in two sympatric lemming species revealed by DNA metabarcoding. PLoS ONE 2015, 10, e0115335. [Google Scholar] [CrossRef] [PubMed]
- Bell, K.L.; Burgess, K.S.; Okamoto, K.C.; Aranda, R.; Brosi, B.J. Review and future prospects for DNA barcoding methods in forensic palynology. Forensic Sci. Int. Genet. 2016, 21, 110–116. [Google Scholar] [CrossRef] [PubMed]
- Lang, D.; Tang, M.; Hu, J.; Zhou, X. Genome-skimming provides accurate quantification for pollen mixtures. Mol. Ecol. Resour. 2019, 19, 1433–1446. [Google Scholar] [CrossRef]
- Xu, S.-Z.; Li, Z.-Y.; Jin, X.-H. DNA barcoding of invasive plants in China: A resource for identifying invasive plants. Mol. Ecol. Resour. 2018, 18, 128–136. [Google Scholar] [CrossRef]
- Fazekas, A.J.; Kuzmina, M.L.; Newmaster, S.G.; Hollingsworth, P.M. DNA barcoding methods for land plants. In DNA Barcodes: Methods and Protocols; Kress, W.J., Erickson, D.L., Eds.; Humana Press: Totowa, NJ, USA, 2012; pp. 223–252. ISBN 9781617795916. [Google Scholar]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Gielly, L.; Miquel, C.; Valentini, A.; Vermat, T.; Corthier, G.; Brochmann, C.; Willerslev, E. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucleic Acids Res. 2007, 35, e14. [Google Scholar] [CrossRef]
- Li, X.; Yang, Y.; Henry, R.J.; Rossetto, M.; Wang, Y.; Chen, S. Plant DNA barcoding: From gene to genome. Biol. Rev. Camb. Philos. Soc. 2015, 90, 157–166. [Google Scholar] [CrossRef]
- Kress, W.J. Plant DNA barcodes: Applications today and in the future. J. Syst. Evol. 2017, 55, 291–307. [Google Scholar] [CrossRef]
- Hollingsworth, P.M.; Li, D.-Z.; van der Bank, M.; Twyford, A.D. Telling plant species apart with DNA: From barcodes to genomes. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2016, 371. [Google Scholar] [CrossRef] [PubMed]
- Coissac, E.; Hollingsworth, P.M.; Lavergne, S.; Taberlet, P. From barcodes to genomes: Extending the concept of DNA barcoding. Mol. Ecol. 2016, 25, 1423–1428. [Google Scholar] [CrossRef] [PubMed]
- Tonti-Filippini, J.; Nevill, P.G.; Dixon, K.; Small, I. What can we do with 1000 plastid genomes? Plant J. 2017, 90, 808–818. [Google Scholar] [CrossRef] [PubMed]
- McKain, M.R.; Johnson, M.G.; Uribe-Convers, S.; Eaton, D.; Yang, Y. Practical considerations for plant phylogenomics. Appl. Plant. Sci. 2018, 6, e1038. [Google Scholar] [CrossRef] [PubMed]
- Staats, M.; Erkens, R.H.J.; van de Vossenberg, B.; Wieringa, J.J.; Kraaijeveld, K.; Stielow, B.; Geml, J.; Richardson, J.E.; Bakker, F.T. Genomic treasure troves: Complete genome sequencing of herbarium and insect museum specimens. PLoS ONE 2013, 8, e69189. [Google Scholar] [CrossRef] [PubMed]
- Straub, S.C.K.; Fishbein, M.; Livshultz, T.; Foster, Z.; Parks, M.; Weitemier, K.; Cronn, R.C.; Liston, A. Building a model: Developing genomic resources for common milkweed (Asclepias syriaca) with low coverage genome sequencing. BMC Genomics 2011, 12, 211. [Google Scholar] [CrossRef]
- Kane, N.C.; Cronk, Q. Botany without borders: Barcoding in focus. Mol. Ecol. 2008, 17, 5175–5176. [Google Scholar] [CrossRef]
- Parks, M.; Cronn, R.; Liston, A. Increasing phylogenetic resolution at low taxonomic levels using massively parallel sequencing of chloroplast genomes. BMC Biol. 2009, 7, 84. [Google Scholar] [CrossRef]
- Nock, C.J.; Waters, D.L.E.; Edwards, M.A.; Bowen, S.G.; Rice, N.; Cordeiro, G.M.; Henry, R.J. Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 2011, 9, 328–333. [Google Scholar] [CrossRef]
- Chen, J.-H.; Huang, Y.; Brachi, B.; Yun, Q.-Z.; Zhang, W.; Lu, W.; Li, H.-N.; Li, W.-Q.; Sun, X.-D.; Wang, G.-Y.; et al. Genome-wide analysis of Cushion willow provides insights into alpine plant divergence in a biodiversity hotspot. Nat. Commun. 2019, 10, 5230. [Google Scholar] [CrossRef]
- Bendich, A.J. Circular chloroplast chromosomes: The grand illusion. Plant. Cell 2004, 16, 1661–1666. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Lee, S.-C.; Lee, J.; Yu, Y.; Yang, K.; Choi, B.-S.; Koh, H.-J.; Waminal, N.E.; Choi, H.-I.; Kim, N.-H.; et al. Complete chloroplast and ribosomal sequences for 30 accessions elucidate evolution of Oryza AA genome species. Sci. Rep. 2015, 5, 15655. [Google Scholar] [CrossRef] [PubMed]
- Steele, P.R.; Hertweck, K.L.; Mayfield, D.; McKain, M.R.; Leebens-Mack, J.; Pires, J.C. Quality and quantity of data recovered from massively parallel sequencing: Examples in Asparagales and Poaceae. Am. J. Bot. 2012, 99, 330–348. [Google Scholar] [CrossRef] [PubMed]
- Kuzmina, M.L.; Braukmann, T.W.A.; Fazekas, A.J.; Graham, S.W.; Dewaard, S.L.; Rodrigues, A.; Bennett, B.A.; Dickinson, T.A.; Saarela, J.M.; Catling, P.M.; et al. Using herbarium-derived DNAs to assemble a large-scale DNA barcode library for the vascular plants of Canada. Appl. Plant Sci. 2017, 5. [Google Scholar] [CrossRef]
- De Vere, N.; Rich, T.C.G.; Ford, C.R.; Trinder, S.A.; Long, C.; Moore, C.W.; Satterthwaite, D.; Davies, H.; Allainguillaume, J.; Ronca, S.; et al. DNA barcoding the native flowering plants and conifers of Wales. PLoS ONE 2012, 7, e37945. [Google Scholar] [CrossRef]
- Nevill, P.G.; Zhong, X.; Tonti-Filippini, J.; Byrne, M.; Hislop, M.; Thiele, K.; van Leeuwen, S.; Boykin, L.M.; Small, I. Large scale genome skimming from herbarium material for accurate plant identification and phylogenomics. Plant. Methods 2020, 16, 1. [Google Scholar] [CrossRef]
- Pyke, G.H.; Ehrlich, P.R. Biological collections and ecological/environmental research: A review, some observations and a look to the future. Biol. Rev. Camb. Philos. Soc. 2010, 85, 247–266. [Google Scholar] [CrossRef]
- Suarez, A.V.; Tsutsui, N.D. The value of museum collections for research and society. Bioscience 2004, 54, 66–74. [Google Scholar] [CrossRef]
- Särkinen, T.; Staats, M.; Richardson, J.E.; Cowan, R.S.; Bakker, F.T. How to open the treasure chest? Optimising DNA extraction from herbarium specimens. PLoS ONE 2012, 7, e43808. [Google Scholar] [CrossRef]
- Zeng, C.-X.; Hollingsworth, P.M.; Yang, J.; He, Z.-S.; Zhang, Z.-R.; Li, D.-Z.; Yang, J.-B. Genome skimming herbarium specimens for DNA barcoding and phylogenomics. Plant Methods 2018, 14, 43. [Google Scholar] [CrossRef]
- Dormontt, E.E.; van Dijk, K.-J.; Bell, K.L.; Biffin, E.; Breed, M.F.; Byrne, M.; Caddy-Retalic, S.; Encinas-Viso, F.; Nevill, P.G.; Shapcott, A.; et al. Advancing DNA barcoding and metabarcoding applications for plants requires systematic analysis of herbarium collections—An Australian perspective. Front. Ecol. Evol. 2018, 6, 134. [Google Scholar] [CrossRef]
- Lang, P.L.M.; Willems, F.M.; Scheepens, J.F.; Burbano, H.A.; Bossdorf, O. Using herbaria to study global environmental change. New Phytol. 2019, 221, 110–122. [Google Scholar] [CrossRef] [PubMed]
- Besnard, G.; Christin, P.-A.; Malé, P.-J.G.; Lhuillier, E.; Lauzeral, C.; Coissac, E.; Vorontsova, M.S. From museums to genomics: Old herbarium specimens shed light on a C3 to C4 transition. J. Exp. Bot. 2014, 65, 6711–6721. [Google Scholar] [CrossRef]
- Zedane, L.; Hong-Wa, C.; Murienne, J.; Jeziorski, C.; Baldwin, B.G.; Besnard, G. Museomics illuminate the history of an extinct, paleoendemic plant lineage (Hesperelaea, Oleaceae) known from an 1875 collection from Guadalupe Island, Mexico. Biol. J. Linn. Soc. Lond. 2016, 117, 44–57. [Google Scholar] [CrossRef]
- Bakker, F.T.; Lei, D.; Yu, J.; Mohammadin, S.; Wei, Z.; van de Kerke, S.; Gravendeel, B.; Nieuwenhuis, M.; Staats, M.; Alquezar-Planas, D.E.; et al. Herbarium genomics: Plastome sequence assembly from a range of herbarium specimens using an Iterative Organelle Genome Assembly pipeline. Biol. J. Linn. Soc. Lond. 2016, 117, 33–43. [Google Scholar] [CrossRef]
- Elliott, T.L.; Jonathan Davies, T. Challenges to barcoding an entire flora. Mol. Ecol. Resour. 2014, 14, 883–891. [Google Scholar] [CrossRef]
- Johnson, B.M.; Kemp, B.M. Rescue PCR: Reagent-rich PCR recipe improves amplification of degraded DNA extracts. J. Archaeol. Sci. Rep. 2017, 11, 683–694. [Google Scholar] [CrossRef]
- Carøe, C.; Gopalakrishnan, S.; Vinner, L.; Mak, S.S.T.; Sinding, M.H.S.; Samaniego, J.A.; Wales, N.; Sicheritz-Pontén, T.; Gilbert, M.T.P. Single-tube library preparation for degraded DNA. Methods Ecol. Evol. 2018, 9, 410–419. [Google Scholar] [CrossRef]
- Gansauge, M.-T.; Gerber, T.; Glocke, I.; Korlevic, P.; Lippik, L.; Nagel, S.; Riehl, L.M.; Schmidt, A.; Meyer, M. Single-stranded DNA library preparation from highly degraded DNA using T4 DNA ligase. Nucleic Acids Res. 2017, 45, e79. [Google Scholar]
- El-Shazly, A.; Wink, M. Diversity of pyrrolizidine alkaloids in the Boraginaceae structures, distribution, and biological properties. Diversity 2014, 6, 188–282. [Google Scholar] [CrossRef]
- Sønstebø, J.H.; Gielly, L.; Brysting, A.K.; Elven, R.; Edwards, M.; Haile, J.; Willerslev, E.; Coissac, E.; Rioux, D.; Sannier, J.; et al. Using next-generation sequencing for molecular reconstruction of past Arctic vegetation and climate. Mol. Ecol. Resour. 2010, 10, 1009–1018. [Google Scholar] [CrossRef] [PubMed]
- Brewer, G.E.; Clarkson, J.J.; Maurin, O.; Zuntini, A.R.; Barber, V.; Bellot, S.; Biggs, N.; Cowan, R.S.; Davies, N.M.J.; Dodsworth, S.; et al. Factors affecting targeted sequencing of 353 nuclear genes from herbarium specimens spanning the diversity of Angiosperms. Front. Plant. Sci. 2019, 10, 1102. [Google Scholar] [CrossRef] [PubMed]
- Korpelainen, H.; Pietiläinen, M. The effects of sample age and taxonomic origin on the success rate of DNA barcoding when using herbarium material. Plant. Syst. Evol. 2019, 305, 319–324. [Google Scholar] [CrossRef]
- Malé, P.-J.G.; Bardon, L.; Besnard, G.; Coissac, E.; Delsuc, F.; Engel, J.; Lhuillier, E.; Scotti-Saintagne, C.; Tinaut, A.; Chave, J. Genome skimming by shotgun sequencing helps resolve the phylogeny of a pantropical tree family. Mol. Ecol. Resour. 2014, 14, 966–975. [Google Scholar] [CrossRef] [PubMed]
- Gryta, H.; Van de Paer, C.; Manzi, S.; Holota, H.; Roy, M.; Besnard, G. Genome skimming and plastid microsatellite profiling of alder trees (Alnus spp., Betulaceae): Phylogenetic and phylogeographical prospects. Tree Genet. Genomes 2017, 13, 118. [Google Scholar] [CrossRef]
- De La Harpe, M.; Hess, J.; Loiseau, O.; Salamin, N.; Lexer, C.; Paris, M. A dedicated target capture approach reveals variable genetic markers across micro- and macro-evolutionary time scales in palms. Mol. Ecol. Resour. 2019, 19, 221–234. [Google Scholar] [CrossRef]
- Schmid, S.; Genevest, R.; Gobet, E.; Suchan, T.; Sperisen, C.; Tinner, W.; Alvarez, N. HyRAD-X, a versatile method combining exome capture and RAD sequencing to extract genomic information from ancient DNA. Methods Ecol. Evol. 2017, 8, 1374–1388. [Google Scholar] [CrossRef]
- Schulte, L.; Bernhardt, N.; Stoof-Leichsenring, K.R.; Zimmermann, H.H.; Pestryakova, L.A.; Epp, L.S.; Herzschuh, U. Hybridization capture of larch (Larix Mill) chloroplast genomes from sedimentary ancient DNA reveals past changes of Siberian forests. bioRxiv 2020. [Google Scholar] [CrossRef]
- Parducci, L.; Alsos, I.G.; Unneberg, P.; Pedersen, M.W.; Han, L.; Lammers, Y.; Salonen, J.S.; Valiranta, M.M.; Slotte, T.; Wohlfarth, B. Shotgun environmental DNA, pollen, and macrofossil analysis of Lateglacial lake sediments from southern Sweden. Front. Ecol. Evol. 2019, 7. [Google Scholar] [CrossRef]
- Johnson, J.S.; Krutovsky, K.V.; Rajora, O.P.; Gaddis, K.D.; Cairns, D.M. Advancing biogeography through population genomics. In Population Genomics: Concepts, Approaches and Applications; Rajora, O.P., Ed.; Springer: Cham, Switzerland, 2019; pp. 539–585. ISBN 9783030045890. [Google Scholar]
- Lammers, Y. Sedimentary ancient DNA: Exploring Methods of Ancient DNA Analysis for Different Taxonomic Groups. Ph.D. Thesis, UiT—The Arctic University of Norway, Tromsø, Norway, 2020. [Google Scholar]
- Slon, V.; Hopfe, C.; Weiß, C.L.; Mafessoni, F.; de la Rasilla, M.; Lalueza-Fox, C.; Rosas, A.; Soressi, M.; Knul, M.V.; Miller, R.; et al. Neandertal and Denisovan DNA from Pleistocene sediments. Science 2017, 356, 605–608. [Google Scholar] [CrossRef]
- Berger, B.A.; Han, J.; Sessa, E.B.; Gardner, A.G.; Shepherd, K.A.; Ricigliano, V.A.; Jabaily, R.S.; Howarth, D.G. The unexpected depths of genome-skimming data: A case study examining Goodeniaceae floral symmetry genes. Appl. Plant. Sci. 2017, 5, 1700042. [Google Scholar] [CrossRef] [PubMed]
- FastX Package. Available online: http://hannonlab.cshl.edu/fastx_toolkit/index.html (accessed on 27 March 2020).
- Organelle Assembler. Available online: http://metabarcoding.org/asm (accessed on 27 March 2020).
- ORG.Annot Pipeline. Available online: https://metabarcoding.org/annot (accessed on 27 March 2020).
- OrthoSkim Pipeline. Available online: https://github.com/cpouchon/OrthoSkim (accessed on 27 March 2020).
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed]
- Buchfink, B.; Xie, C.; Huson, D.H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods 2015, 12, 59–60. [Google Scholar] [CrossRef] [PubMed]
- Slater, G.S.C.; Birney, E. Automated generation of heuristics for biological sequence comparison. BMC Bioinform. 2005, 6, 31. [Google Scholar] [CrossRef]
- BOLD Systems. Available online: https://www.boldsystems.org (accessed on 27 March 2020).
- Zhang, Z.; Schwartz, S.; Wagner, L.; Miller, W. A greedy algorithm for aligning DNA sequences. J. Comput. Biol. 2000, 7, 203–214. [Google Scholar] [CrossRef]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef]
- Kim, D.; Song, L.; Breitwieser, F.P.; Salzberg, S.L. Centrifuge: Rapid and sensitive classification of metagenomic sequences. Genome Res. 2016, 26, 1721–1729. [Google Scholar] [CrossRef]
- R_Core_Team. R: A Language and Environment for Statistical Computing; Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- European Nucleotide Archive (ENA-EBI). Available online: https://www.ebi.ac.uk/ena (accessed on 27 March 2020).
- PhyloAlps. Available online: https://data.phyloalps.org/browse/ (accessed on 27 March 2020).








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All | PhyloAlps + Carp | Phylo Norway | All (+ 20 fam) | PhyloAlps + Carp (+ 20 fam) | PhyloNorway (+ 20 fam) | |
|---|---|---|---|---|---|---|
| Specimens | 6655 | 4604 | 2051 | 5893 | 4057 | 1836 |
| Libraries | 6817 | 4726 | 2091 | 6018 | 4147 | 1871 |
| Families | 161 | 158 | 112 | 43 | 43 | 43 |
| Genera | 1037 | 922 | 576 | 804 | 705 | 461 |
| Taxa | 5575 | 4437 | 1899 | 4957 | 3914 | 1689 |
| Complete genome cpDNA | 4439 | 3303 | 1136 | 3944 | 2922 | 1022 |
| Average sequencing depth cpDNA | 278 | 318 | 187 | 265 | 300 | 188 |
| Complete nrDNA cluster | 5748 | 4021 | 1727 | 5092 | 3543 | 1549 |
| Average sequencing depth nrDNA | 603 | 674 | 444 | 579 | 638 | 450 |
| Average library insert size | 316 | 346 | 249 | 318 | 350 | 249 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alsos, I.G.; Lavergne, S.; Merkel, M.K.F.; Boleda, M.; Lammers, Y.; Alberti, A.; Pouchon, C.; Denoeud, F.; Pitelkova, I.; Pușcaș, M.; et al. The Treasure Vault Can be Opened: Large-Scale Genome Skimming Works Well Using Herbarium and Silica Gel Dried Material. Plants 2020, 9, 432. https://doi.org/10.3390/plants9040432
Alsos IG, Lavergne S, Merkel MKF, Boleda M, Lammers Y, Alberti A, Pouchon C, Denoeud F, Pitelkova I, Pușcaș M, et al. The Treasure Vault Can be Opened: Large-Scale Genome Skimming Works Well Using Herbarium and Silica Gel Dried Material. Plants. 2020; 9(4):432. https://doi.org/10.3390/plants9040432
Chicago/Turabian StyleAlsos, Inger Greve, Sebastien Lavergne, Marie Kristine Føreid Merkel, Marti Boleda, Youri Lammers, Adriana Alberti, Charles Pouchon, France Denoeud, Iva Pitelkova, Mihai Pușcaș, and et al. 2020. "The Treasure Vault Can be Opened: Large-Scale Genome Skimming Works Well Using Herbarium and Silica Gel Dried Material" Plants 9, no. 4: 432. https://doi.org/10.3390/plants9040432
APA StyleAlsos, I. G., Lavergne, S., Merkel, M. K. F., Boleda, M., Lammers, Y., Alberti, A., Pouchon, C., Denoeud, F., Pitelkova, I., Pușcaș, M., Roquet, C., Hurdu, B.-I., Thuiller, W., Zimmermann, N. E., Hollingsworth, P. M., & Coissac, E. (2020). The Treasure Vault Can be Opened: Large-Scale Genome Skimming Works Well Using Herbarium and Silica Gel Dried Material. Plants, 9(4), 432. https://doi.org/10.3390/plants9040432