Oxygen and ROS in Photosynthesis


Abstract
1. Introduction
2. ROS Properties and Basic Reactions
2.1. Singlet Oxygen, 1O2
2.1.1. Formation of 1O2
2.1.2. Physical Deactivation of 1O2
2.1.3. Chemical Reactions of 1O2






2.1.4. Lifetime and Diffusion Distance of 1O2
2.2. Superoxide Anion Radical, O2•−
2.2.1. Formation of O2•−
2.2.2. Reactions of O2•−


2.2.3. Lifetime and Diffusion Distance of O2•−
2.3. Hydrogen Peroxide, H2O2
2.3.1. Formation of H2O2.

2.3.2. Reactions of H2O2

2.3.3. Lifetime and Diffusion Distance of H2O2
2.4. Hydroxyl Radical, HO•
2.4.1. Formation of HO•
2.4.2. Reactions of HO•




2.4.3. Lifetime and Diffusion Distance of HO•
3. Production of ROS in Chloroplasts
3.1. ROS Production in Chloroplast Stroma
3.1.1. Formation of 1O2 in the Stroma
3.1.2. Formation of Reduced Forms of Oxygen, O2•−, H2O2, HO•, by Fd in the Stroma
3.1.3. Formation of Reduced Forms of Oxygen, O2•−, H2O2, HO•, by Flavins in the Stroma
3.1.4. Formation of H2O2 in the Stroma
3.2. Formation of ROS in Thylakoid Membranes
3.2.1. Formation of 1O2 in Thylakoids
3.2.2. Oxygen Reduction in PETC
3.2.3. Formation of Reduced Forms of Oxygen, O2•−, H2O2, HO•, in PSII
- HP form: 350–450 mV;
- IP form: 150–260 mV;
- LP form: −50–110 mV.
3.2.4. Formation of Reduced Forms of Oxygen, O2•−, H2O2, HO•, in PSI
3.2.5. Formation of Reduced Forms of Oxygen, O2•−, H2O2, HO•, in the PQ Pool and by Cyt b6f
4. Damage Caused by ROS in the Chloroplast
4.1. Damage to PSII
4.2. Damage to PSI
4.3. Oxidation of Membrane Lipids by ROS
4.4. Damage to Stromal Proteins
4.5. Damage to Chloroplast DNA
5. Detoxification of ROS in Plant Chloroplasts
5.1. Detoxification of O2•− and H2O2
5.2. Detoxification of 1O2
6. ROS Produced by Plant Chloroplasts Function as Signaling Molecules
6.1. Signaling by 1O2
6.2. Signaling by H2O2
6.3. Signaling by O2•−
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| β-CC | β-cyclocitral |
| 1Chl, 1Chl* and 3Chl | respectively, singlet state, singlet excited state and triplet excited state of chlorophyll |
| 1O2 | singlet oxygen (1∆gO2) |
| 2-Cys PRX | two-cysteine peroxiredoxin |
| A | acceptor |
| A1 | phylloquinone of PSI |
| APX | ascorbate peroxidase |
| AscH2 | ascorbate, ascorbic acid |
| Car and 3Car | respectively, singlet and triplet state of carotenoid |
| CAT | catalase |
| Chl and Chl* | respectively, chlorophyll and excited chlorophyll |
| Chl a | chlorophyll a |
| cyt | cytochrome |
| cyt b559 | cytochrome b559 |
| Cyt b6f | cytochrome b6/f complex |
| DBMIB | 2,5-dibromo-6-isopropyl-3-methyl-1,4-benzoquinone |
| DCMU | 3-(3,4-di-chlorophenyl)-1,1-dimethyl urea |
| DHA | dehydroascorbate; DMF, dimethylformamide |
| DNP-INT | 2-(2,4-dinitrophenoxy)-3-iodo-4-methyl-1-(1-methylethyl)-5-nitro-benzene |
| Em | midpoint redox potential |
| E0′ | standard redox potential |
| EPR | electron paramagnetic resonance |
| EX1 | Executer1 |
| Fd, Fdox and Fdred | ferredoxin, and oxidized and reduced ferredoxin, respectively |
| FL | flavin |
| FL•− | anion form of flavin semiquinone |
| FLH• | flavin semiquinone radical |
| FLHOOH | flavin hydroperoxide |
| FLU | a protein important in control of chlorophyll biosynthesis |
| FNR | ferredoxin-NADP+ reductase |
| FtsH | a protease involved in PSII repair; FX, FA, and FB, 4Fe-4S clusters of PSI |
| GSH | reduced glutathione |
| GSSG | glutathione disulfide |
| H2O2 | hydrogen peroxide |
| HO• | hydroxyl radical |
| HO2• | hydroperoxyl radical |
| HO2− | hydroperoxyl anion |
| HP, IP, LP and VLP | respectively, high, intermediate, low and very low potential forms of cytochrome b559 |
| hν | energy of a photon |
| ISC | intersystem crossing |
| kforward and kreverse• | forward and reverse rate constant, respectively |
| LHC | light harvesting complex; LHCI and LHCII, respectively, light harvesting complex of PSI and PSII |
| LOO• | lipid peroxyl radical; M, metal |
| MDA | monodehydroascorbate |
| MDAR | monodehydroascorbate reductase |
| MenB | 1,4-hydroxynaphthoyl-coenzyme A synthase |
| NHE | Normal Hydrogen Electrode |
| NPQ | non-photochemical quenching of excitation energy |
| O2 | molecular oxygen (3Σ+gO2); O3, ozone |
| O2•− | superoxide anion radical |
| OEC | oxygen-evolving complex |
| P680 | primary donor of PSII |
| P700 | primary donor of PSI |
| PAP | phosphoadenosine phosphate |
| PC | plastocyanin |
| PCD | programmed cell death |
| PETC | photosynthetic electron transport chain |
| Pheo | pheophytin |
| PheoD1 and PheoD2 | respectively, pheophytins bound to D1 and D2 proteins of PSII |
| PPFD | photosynthetic photon flux density |
| PQ | plastoquinone |
| PQ•− | plastosemiquinone anion radical |
| PQH2 | plastoquinol |
| PRX | peroxiredoxin; PSI and PSII, Photosystems I and II, respectively |
| PsbS | a chloroplast-localized protein required for NPQ |
| PTOX | plastid terminal oxidase |
| PX | peroxidase |
| Q | quinone |
| Q•− | semiquinone anion radical |
| R• | organic radical |
| RC | reaction center |
| ROO• | peroxyl radical |
| ROOH | organic peroxide |
| ROOOOR | linear tetraoxide |
| ROS | reactive oxygen species |
| RS | thiol |
| rubisco | ribulose-1,5-bisphosphate carboxylase oxygenase |
| S | sensitizer |
| SAL1 | an inositol polyphosphate phosphatase |
| SOD | superoxide dismutase |
| TRX | thioredoxin |
| TyrZ | the redox active tyrosine of PSII |
References
- Steiger, H.M.; Beck, E.; Beck, R. Oxygen concentration in isolated chloroplasts during photosynthesis. Plant Physiol. 1977, 60, 903–906. [Google Scholar] [CrossRef]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press: New York, NY, USA, 2015. [Google Scholar]
- Tyystjärvi, E. Phototoxicity. In Plant Cell Death Processes; Noodén, L.D., Ed.; Academic Press: San Diego, CA, USA, 2004; pp. 271–283. [Google Scholar]
- Triantaphylidès, C.; Krischke, M.; Hoeberichts, F.A.; Ksas, B.; Gresser, G.; Havaux, M.; Van Breusegem, F.; Mueller, M.J. Singlet oxygen is the major reactive oxygen species involved in photooxidative damage to plants. Plant Physiol. 2008, 148, 960–968. [Google Scholar] [CrossRef]
- Asada, K. Production and scavenging of reactive oxygen species in chloroplasts and their functions. Plant Physiol. 2006, 141, 391–396. [Google Scholar] [CrossRef]
- Miyake, C.; Yonekura, K.; Kobayashi, Y.; Yokota, A. Cyclic electron flow within PSII functions in intact chloroplasts from spinach leaves. Plant Cell Physiol. 2002, 43, 951–957. [Google Scholar] [CrossRef]
- Heber, U. Irrungen, Wirrungen? The Mehler reaction in relation to cyclic electron transport in C3 plants. Photosynth. Res. 2002, 73, 223–231. [Google Scholar] [CrossRef]
- Peltier, G.; Tolleter, D.; Billon, E.; Cournac, L. Auxiliary electron transport pathways in chloroplasts of microalgae. Photosynth. Res. 2010, 106, 19–31. [Google Scholar] [CrossRef]
- Noctor, G.; Reichheld, J.P.; Foyer, C.H. ROS-related redox regulation and signaling in plants. Semin. Cell Dev. Biol. 2018, 80, 3–12. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H. Reactive oxygen species, oxidative signaling and the regulation of photosynthesis. Environ. Exp. Bot. 2018, 154, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Noctor, G. Ascorbate and glutathione: The heart of the redox hub. Plant Physiol. 2011, 155, 2–18. [Google Scholar] [CrossRef] [PubMed]
- Asada, K. The water–water cycle in chloroplasts: Scavenging of active oxygens and dissipation of excess photons. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1999, 50, 601–639. [Google Scholar] [CrossRef] [PubMed]
- Myake, C. Alternative electron flows (water-water cycle and cyclic electron flow around PSI) in photosynthesis: Molecular mechanisms and physiological functions. Plant Cell Physiol. 2010, 51, 1951–1963. [Google Scholar] [CrossRef] [PubMed]
- Krinsky, N.I. Singlet oxygen in biological systems. Trends Biochem. Sci. 1977, 2, 35–38. [Google Scholar] [CrossRef]
- Krasnovsky, A.A., Jr. Singlet oxygen and primary mechanisms of photodynamic therapy and photodynamic diseases. In Photodynamic Therapy at the Cellular Level; Uzdensky, A.B., Ed.; Research Signpost: Kerala, India, 2007; pp. 17–62. [Google Scholar]
- Krasnovsky, A.A., Jr. Singlet molecular oxygen and primary mechanisms of photo-oxidative damage of chloroplasts. Studies based on detection of oxygen and pigment phosphorescence. Proc. R. Soc. Edinb. 1994, 102, 219–235. [Google Scholar] [CrossRef]
- Schweitzer, C.; Schmidt, R. Physical mechanisms of generation and deactivation of singlet oxygen. Chem. Rev. 2003, 5, 1685–1757. [Google Scholar] [CrossRef] [PubMed]
- You, Y. Chemical tools for the generation and detection of singlet oxygen. Org. Biomol. Chem. 2018, 16, 4044–4060. [Google Scholar] [CrossRef]
- Haber, F.; Weiss, J. The catalytic decomposition of hydrogen peroxide by iron salts. Proc. R. Soc. Lond. A 1934, 147, 332–351. [Google Scholar]
- Kellogg, E.W., III; Fridovich, I. Superoxide, hydrogen peroxide, and singlet oxygen in lipid peroxidation by a xanthine oxidase system. J. Biol. Chem. 1975, 250, 8812–8817. [Google Scholar]
- Weinstein, J.; Bielski, B.H.J. Kinetics of the interaction of HO2 and O2 radicals with hydrogen peroxide; The Haber–Weiss reaction. J. Am. Chem. Soc. 1979, 101, 58–62. [Google Scholar] [CrossRef]
- Melhuish, W.H.; Sutton, H.C. Study of the Haber–Weiss reaction using a sensitive method for detection of OH radicals. J. Chem. Soc. Chem. Commun. 1978, 22, 970–971. [Google Scholar] [CrossRef]
- Mattila, H.; Khorobrykh, S.; Havurinne, V.; Tyystjärvi, E. Reactive oxygen species: Reactions and detection from photosynthetic tissues. J. Photochem. Photobiol. B Biol. 2015, 152, 176–214. [Google Scholar] [CrossRef]
- MacManus-Spencer, L.A.; Edhlund, B.L.; McNeill, K. Singlet oxygen production in the reaction of superoxide with organic peroxides. J. Org. Chem. 2006, 71, 796–799. [Google Scholar] [CrossRef] [PubMed]
- Mayeda, E.A.; Bard, A.J. The production of singlet oxygen in electrogenerated radical ion electron transfer reactions. J. Am. Chem. Soc. 1973, 95, 6223–6226. [Google Scholar] [CrossRef]
- Takahama, U.; Nishimura, M. Formation of singlet molecular oxygen in illuminated chloroplasts. Effects on photoinactivation and lipid peroxidation. Plant Cell Physiol. 1975, 16, 737–748. [Google Scholar]
- Khan, A.U.; Kasha, M. Singlet molecular oxygen in the Haber-Weiss reaction. Proc. Natl. Acad. Sci. USA 1994, 91, 12362–12367. [Google Scholar] [CrossRef] [PubMed]
- Danen, W.C.; Arudi, R.L. Generation of singlet oxygen in the reaction of superoxide anion radical with diacyl peroxides. J. Am. Chem. Soc. 1978, 100, 3944–3945. [Google Scholar] [CrossRef]
- Russell, G.A. Deuterium-isotope effects in the autoxidation of aralkylhydrocarbons-mechanism of the interaction of peroxy radicals. J. Am. Chem. Soc. 1957, 79, 3871–3877. [Google Scholar] [CrossRef]
- Mendenhall, G.D.; Sheng, X.C.; Wilson, T. Yields of excited carbonyl species from alkoxyl and from alkylperoxyl radical dismutations. J. Am. Chem. Soc. 1991, 113, 8976–8977. [Google Scholar] [CrossRef]
- Niu, Q.J.; Mendenhall, G.D. Yields of singlet molecular oxygen from peroxyl radical termination. J. Am. Chem. Soc. 1992, 114, 165–172. [Google Scholar] [CrossRef]
- Miyamoto, S.; Martinez, G.R.; Medeiros, M.H.; Di Mascio, P. Singlet molecular oxygen generated by biological hydroperoxides. J. Photochem. Photobiol. B. 2014, 139, 24–33. [Google Scholar] [CrossRef]
- Kanofsky, J.R. Singlet oxygen production from the reactions of alkylperoxy radicals. Evidence from 1268-nm chemiluminescence. J. Org. Chem. 1986, 51, 3386–3388. [Google Scholar] [CrossRef]
- Khan, A.U. The discovery of the chemical evolution of singlet oxygen. Some current chemical, photochemical, and biological applications. Int. J. Quantum Chem. 1991, 39, 251–267. [Google Scholar] [CrossRef]
- Adam, W.; Kazakov, D.V.; Kazakov, V.P. Singlet-oxygen chemiluminescence in peroxide reactions. Chem. Rev. 2005, 105, 3371–3387. [Google Scholar] [CrossRef] [PubMed]
- Aubry, J.M.; Cazin, B. Chemical sources of singlet oxygen. 2. Quantitative generation of singlet oxygen from hydrogen peroxide disproportionation catalyzed by molybdate ions. Inorg. Chem. 1988, 27, 2013–2014. [Google Scholar] [CrossRef]
- Khan, A.U. Singlet molecular oxygen spectroscopy: Chemical and photosensitized. In Singlet O2; Frimer, A.A., Ed.; CRC Press: Boca Raton, FL, USA, 1985; pp. 39–79. [Google Scholar]
- Ogilby, P.R.; Foote, C.S. Chemistry of singlet oxygen. 42. Effect of solvent, solvent isotopic substitution and temperature on the lifetime of singlet molecular oxygen (1∆gO2). J. Am. Chem. Soc. 1983, 105, 3423–3430. [Google Scholar] [CrossRef]
- Arellano, J.B.; Yousef, Y.A.; Melø, T.B.; Mohamad, S.B.; Cogdell, R.J.; Naqvi, K.R. Formation and geminate quenching of singlet oxygen in purple bacterial reaction center. J. Photochem. Photobiol. B Biol. 2007, 87, 105–112. [Google Scholar] [CrossRef][Green Version]
- Bregnhøj, M.; Westberg, M.; Jensen, F.; Ogilby, P.R. Solvent-dependent singlet oxygen lifetimes: Temperature effects implicate tunneling and charge-transfer interactions. Phys. Chem. Chem. Phys. 2016, 18, 22946–22961. [Google Scholar] [CrossRef]
- Conn, P.F.; Schalch, W.; Truscott, T.G. The singlet oxygen and carotenoid interaction. J. Photochem. Photobiol. B Biol. 1991, 11, 41–47. [Google Scholar] [CrossRef]
- Koppenol, W.H. Reactions involving singlet oxygen and the superoxide anion. Nature 1976, 262, 420–421. [Google Scholar] [CrossRef]
- Triantaphylidès, C.; Havaux, M. Singlet oxygen in plants: Production, detoxification and signaling. Trends Plant. Sci. 2009, 14, 219–228. [Google Scholar] [CrossRef]
- Davies, M.J. Reactive species formed on proteins exposed to singlet oxygen. Photochem. Photobiol. Sci. 2004, 3, 17–25. [Google Scholar] [CrossRef]
- Jiang, G.; Chen, J.; Huang, J.S.; Che, C.M. Highly efficient oxidation of amines to imines by singlet oxygen and its application in Ugi-type reactions. Org. Lett. 2009, 11, 4568–4571. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Hoffman, M.Z. Oxidation of phenol by singlet oxygen photosensitized by the Tris(2,2′-bipyridine)ruthenium(II) ion. J. Phys. Chem. A 2000, 104, 5998–6002. [Google Scholar] [CrossRef]
- Kramarenko, G.G.; Hummel, S.G.; Martin, S.M.; Buettner, G.R. Ascorbate reacts with singlet oxygen to produce hydrogen peroxide. Photochem. Photobiol. 2006, 82, 1634–1637. [Google Scholar] [CrossRef]
- Khorobrykh, S.A.; Karonen, M.; Tyystjärvi, E. Experimental evidence suggesting that H2O2 is produced within the thylakoid membrane in a reaction between plastoquinol and singlet oxygen. FEBS Lett. 2015, 589, 779–786. [Google Scholar] [CrossRef] [PubMed]
- Bisby, R.H.; Morgan, C.G.; Hamblett, I.; Gormahn, A.A. Quenching of singlet oxygen by trolox C, ascorbate and amino acids: Effects of pH and temperature. J. Chem. Phys. A 1999, 103, 7454–7459. [Google Scholar] [CrossRef]
- Gruszka, J.; Pawlak, A.; Kruk, J. Tocochromanols, plastoquinol, and other biological prenyllipids as singlet oxygen quenchers–determination of singlet oxygen quenching rate constants and oxidation products. Free Radic. Biol. Med. 2008, 45, 920–928. [Google Scholar] [CrossRef] [PubMed]
- Krasnovsky, A.A., Jr. Singlet molecular oxygen in photobiochemical systems: IR phosphorescence studies. Membr. Cell Biol. 1998, 12, 665–690. [Google Scholar]
- Skovsen, E.; Snyder, J.W.; Lambert, J.D.; Ogilby, P.R. Lifetime and diffusion of singlet oxygen in a cell. J. Phys. Chem. B 2005, 109, 8570–8573. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Gibian, M.J. The chemistry of superoxide ion. Tetrahedron 1979, 35, 1471–1481. [Google Scholar] [CrossRef]
- Afanas’ev, I.B. Superoxide Ion: Chemistry and Biological Implications; CRC Press: Boca Raton, FL, USA, 1989; p. 296. [Google Scholar]
- Frimer, A.A. Superoxide chemistry in non-aqueous media. In Oxygen Radicals in Biology and Medicine. Basic Life Sciences; Simic, M.G., Taylor, K.A., Ward, J.F., von Sonntag, C., Eds.; Springer: Boston, MA, USA, 1988; Volume 49, pp. 29–38. [Google Scholar]
- Todres, Z.V. Ion-Radical Organic Chemistry: Principles and Applications, 2nd ed.; CRC Press: Boca Raton, FL, USA, 2008; p. 496. [Google Scholar]
- Koppenol, W.H. Solvation of the superoxide anion. In Oxy Radicals and Their Scavenger System; Gohen, G., Greenwald, R.A., Eds.; Elsevier Biomedical: New York, NY, USA, 1983; pp. 274–277. [Google Scholar]
- Armstrong, D.A.; Huie, R.E.; Koppenol, W.H.; Lymar, S.V.; Merenyi, G.; Neta, P.; Ruscic, B.; Stanbury, D.M.; Steenken, S.; Wardman, P. Standard electrode potentials involving radicals in aqueous solution: Inorganic radicals (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1139–1150. [Google Scholar] [CrossRef]
- Sawyer, D.T.; Roberts, J.L., Jr. Hydroxide ion: An effective one-electron reducing agent? Acc. Chem. Res. 1988, 21, 469–476. [Google Scholar] [CrossRef]
- Peover, M.E.; White, B.S. Electrolytic reduction of oxygen in aprotic solvents: The superoxide ion. Electrochim. Acta 1966, 11, 1061–1067. [Google Scholar] [CrossRef]
- Song, Y.; Buettner, G.R. Thermodynamic and kinetic considerations for the reaction of semiquinone radicals to form superoxide and hydrogen peroxide. Free Radic. Biol. Med. 2010, 49, 919–962. [Google Scholar] [CrossRef] [PubMed]
- Gotoh, N.; Niki, E. Rates of interactions of superoxide with vitamin E, vitamin C and related compounds as measured by chemiluminescence. Biochim. Biophys. Acta 1992, 1115, 201–207. [Google Scholar] [CrossRef]
- Cabelli, D.E.; Bielski, B.H.J. Kinetics and mechanism for the oxidation of ascorbic acid/ascorbate by HO2/O2− (hydroperoxyl/superoxide) radicals. A pulse radiolysis and stopped-flow photolysis study. J. Phys. Chem. 1983, 87, 1809–1812. [Google Scholar] [CrossRef]
- Afanas’ev, I.B.; Grabovetskii, V.V.; Kuprianova, N.S. Kinetics and mechanism of the reactions of superoxide ion in solution. Part 5. Kinetics and mechanism of the interaction of superoxide ion with vitamin E and ascorbic acid. J. Chem. Soc. Perkin Trans. 1987, 2, 281–285. [Google Scholar] [CrossRef]
- Wefers, H.; Sies, H. Oxidation of glutathione by the superoxide radical to the disulfide and the sulfonate yielding singlet oxygen. Eur. J. Biochem. 1983, 137, 29–36. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Metodiewa, D. The reaction of superoxide with reduced glutathione. Arch. Biochem. Biophys. 1994, 314, 284–290. [Google Scholar] [CrossRef]
- Winterbourn, C.C.; Metodiewa, D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999, 27, 322–328. [Google Scholar] [CrossRef]
- Bielski, B.H.J.; Cabelli, D.E.; Arudi, R.L.; Ross, A.B. Reactivity of HO2/O2− radicals in aqueous solution. J. Phys. Chem. Ref. Data 1985, 14, 1041–1100. [Google Scholar] [CrossRef]
- Frimer, A.A. The organic chemistry of superoxide anion radical. In The Chemistry of Functional Groups: Peroxides; Patai, S., Ed.; Wiley: Chichester, UK, 1983; pp. 429–461. [Google Scholar]
- Kobayashi, S.; Tezuka, T.; Ando, W. Nucleophilic and electron transfer oxidations of troponoid compounds by superoxide ion. J. Chem. Soc. Chem. Commun. 1979, 11, 508–510. [Google Scholar] [CrossRef]
- Shibata, K.; Saito, Y.; Urano, K.; Matsui, M. Reaction of Schiff bases with superoxide ion in acetonitrile. Bull. Chem. Soc. Jpn. 1986, 59, 3323–3325. [Google Scholar] [CrossRef]
- Pospíšil, P. Production of reactive oxygen species by photosystem II. Biochim. Biophys. Acta 2009, 1787, 1151–1160. [Google Scholar] [CrossRef] [PubMed]
- Korshunov, S.; Imlay, J.A. Detection and quantification of superoxide formed within the periplasm of Escherichia coli. J. Bacteriol. 2006, 188, 6326–6334. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. The biological chemistry of hydrogen peroxide. Methods Enzymol. 2013, 528, 3–25. [Google Scholar]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen Peroxide Synthesis: An Outlook beyond the Anthraquinone Process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Flaherty, D.W. Direct Synthesis of H2O2 from H2 and O2 on Pd Catalysts: Current Understanding, Outstanding Questions, and Research Needs. ACS Catal. 2018, 82, 1520–1527. [Google Scholar] [CrossRef]
- Melis, A.; Zhang, L.; Forestier, M.; Ghirardi, M.L.; Seibert, M. Sustained photobiological hydrogen gas production upon reversible inactivation of oxygen evolution in the green alga Chlamydomonas reinhardtii. Plant Physiol. 2000, 122, 127–136. [Google Scholar] [CrossRef]
- Kosourov, S.; Jokel, M.; Aro, E.-M.; Allahverdiyeva, Y. A new approach for sustained and efficient H2 photoproduction by Chlamydomonas reinhardtii. Energy Environ. Sci. 2018, 11, 1431–1436. [Google Scholar] [CrossRef]
- Claiborne, A.; Miller, H.; Parsonage, D.; Ross, R.P. Protein-sulfenic acid stabilization and function in enzyme catalysis and gene regulation. FASEB J. 1993, 7, 1483–1490. [Google Scholar] [CrossRef]
- Salmeen, A.; Andersen, J.N.; Myers, M.P.; Meng, T.C.; Hinks, J.A.; Tonks, N.K.; Barford, D. Redox regulation of protein tyrosine phosphatase 1B involves a sulphenyl-amide intermediate. Nature 2003, 423, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Stone, J.R. An assessment of proposed mechanisms for sensing hydrogen peroxide in mammalian systems. Arch. Biochem. Biophys. 2004, 422, 119–124. [Google Scholar] [CrossRef] [PubMed]
- Vasquez-Vivar, J.; Denicola, A.; Radi, R.; Augusto, O. Peroxynitrite-mediated decarboxylation of pyruvate to both carbon dioxide and carbon dioxide radical anion. Chem. Res. Toxicol. 1997, 10, 786–794. [Google Scholar] [CrossRef] [PubMed]
- Bakhmutova-Albert, E.V.; Yao, H.; Denevan, D.E.; Richardson, D.E. Kinetics and mechanism of peroxymonocarbonate formation. Inorg. Chem. 2010, 49, 11287–11296. [Google Scholar] [CrossRef] [PubMed]
- Mizuta, Y.; Masumizu, T.; Kohno, M.; Mori, A.; Packer, L. Kinetic analysis of the Fenton reaction by ESR-spin trapping. Biochem. Mol. Biol. Int. 1997, 43, 1107–1120. [Google Scholar] [CrossRef]
- Masarwa, M.; Cohen, H.; Meyerstein, D.; Hickman, D.L.; Bakac, A.; Espenson, J.H. Reactions of low valent transition metal complexes with hydrogen peroxide. Are they ‘‘Fenton-like” or not? 1. The case of Cu+aq and Cr2+aq. J. Am. Chem. Soc. 1988, 110, 4293–4297. [Google Scholar] [CrossRef]
- Moffett, J.W.; Zika, R.G. Reaction kinetics of hydrogen peroxide with copper and iron in seawater. Environ. Sci. Technol. 1987, 21, 804–810. [Google Scholar] [CrossRef]
- Salgado, P.; Melin, V.; Contreras, D.; Moreno, Y.; Mansilla, H.D. Fenton reaction driven by iron ligands. J. Chil. Chem. Soc. 2013, 58, 2096–2101. [Google Scholar] [CrossRef]
- Winterbourn, C.C. Toxicity of iron and hydrogen peroxide: The Fenton reaction. Toxicol. Lett. 1995, 82/83, 969–974. [Google Scholar] [CrossRef]
- Goldstein, S.; Meyerstein, D.; Czapski, G. The Fenton reagents. Free Radic. Biol. Med. 1993, 15, 435–445. [Google Scholar] [CrossRef]
- Davies, M.J.; Hawkins, C.L.; Pattison, D.I.; Rees, M.D. Mammalian heme peroxidases: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 1199–1234. [Google Scholar] [CrossRef] [PubMed]
- Miayke, C.; Michihata, F.; Asada, K. Scavenging of hydrogen peroxide in prokaryotic and eukaryotic algae: Acquisition of ascorbate peroxidase during the evolution of cyanobacteria. Plant Cell Physiol. 1991, 32, 33–43. [Google Scholar]
- Asada, K. The water–water cycle as alternative photon and electron sinks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 1419–1431. [Google Scholar] [CrossRef]
- Deyhimi, F.; Nami, F. Peroxidase-catalyzed electrochemical assay of hydrogen peroxide: A ping-pong mechanism. Int. J. Chem. Kinet. 2012, 44, 699–704. [Google Scholar] [CrossRef]
- Antunes, F.; Cadenas, E. Estimation of H2O2 gradients across biomembranes. FEBS Lett. 2000, 475, 121–126. [Google Scholar] [CrossRef]
- Bienert, G.P.; Møller, A.L.; Kristiansen, K.A.; Schulz, A.; Møller, I.M.; Schjoerring, J.K.; Jahn, T.P. Specific aquaporins facilitate the diffusion of hydrogen peroxide across membranes. J. Biol. Chem. 2007, 282, 1183–1192. [Google Scholar] [CrossRef]
- Bienert, G.P.; Schjoerring, J.K.; Jahn, T.P. Membrane transport of hydrogen peroxide. Biochim. Biophys. Acta 2006, 1758, 994–1003. [Google Scholar] [CrossRef]
- Weller, J.; Kizina, K.M.; Can, K.; Bao, G.; Müller, M. Response properties of the genetically encoded optical H2O2 sensor HyPer. Free Radic. Biol. Med. 2014, 76, 227–241. [Google Scholar] [CrossRef]
- Costa, A.; Drago, I.; Behera, S.; Zottini, M.; Pizzo, P.; Schroeder, J.I.; Pozzan, T.; Lo Schiavo, F. H2O2 in plant peroxisomes: An In Vivo analysis uncovers a Ca2+- dependent scavenging system. Plant J. 2010, 62, 760–772. [Google Scholar] [CrossRef]
- Buettner, G.R. The pecking order of free radicals and antioxidants: Lipid peroxidation, α-tocopherol, and ascorbate. Arch. Biochem. Biophys. 1993, 300, 535–543. [Google Scholar] [CrossRef]
- Buxton, G.V.; Greenstock, C.L.; Helman, W.P.; Ross, A.B. Critical review of rate constants for reactions of hydrated electrons, hydrogen atoms and hydroxyl radicals (•OH/•O−) in aqueous solution. J. Phys. Chem. Ref. Data. 1988, 17, 513–886. [Google Scholar] [CrossRef]
- Haag, W.R.; Yao, C.C.D. Rate constants for reaction of hydroxyl radicals with several drinking water contaminants. Environ. Sci. Technol. 1992, 26, 1005–1013. [Google Scholar] [CrossRef]
- Gligorovski, S.; Strekowski, R.; Barbati, S.; Vione, D. Environmental implications of hydroxyl radicals (•OH). Chem. Rev. 2015, 115, 13051–13092. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, H. On the Photolysis of simple anions and neutral molecules as sources of O−/OH, SOx− and Cl in Aqueous Solution. Phys. Chem. Chem. Phys. 2007, 9, 3935–3964. [Google Scholar] [CrossRef]
- Kwon, B.G.; Kwon, J.-H. Measurement of the hydroxyl radical formation from H2O2, NO3−, and Fe(III) using a continuous flow Injection analysis. J. Ind. Eng. Chem. 2010, 16, 193–199. [Google Scholar] [CrossRef]
- Peyton, G.R.; Glaze, W.H. Destruction of pollutants in water with ozone in combination with ultraviolet radiation. 3. Photolysis of aqueous ozone. Environ. Sci. Technol. 1988, 22, 761–767. [Google Scholar] [CrossRef]
- Tripathi, G.N.R. Electron-transfer component in hydroxyl radical reactions observed by time resolved resonance Raman spectroscopy. J. Am. Chem. Soc. 1998, 120, 4161–4166. [Google Scholar] [CrossRef]
- Fisher, S.C.; Schoonen, M.A.; Brownawell, B.J. Phenylalanine as a hydroxyl radical-specific probe in pyrite slurries. Geochem. Trans. 2012, 13, 3. [Google Scholar] [CrossRef]
- Leeuwenburgh, C.; Hansen, P.; Shaish, A.; Holloszy, J.O.; Heinecke, J.W. Markers of protein oxidation by hydroxyl radical and reactive nitrogen species in tissues of aging rats. Am. J. Physiol. 1998, 274, 453–461. [Google Scholar] [CrossRef]
- Koppenol, W.H.; Butler, J. Energetics of interconversion reactions of oxyradicals. Adv. Free Radic. Biol. Med. 1985, 1, 91–131. [Google Scholar] [CrossRef]
- Pryor, W.A. Oxy-radicals and related species: Their formation, lifetimes, and reactions. Annu. Rev. Physiol. 1986, 48, 657–667. [Google Scholar] [CrossRef] [PubMed]
- Codorniu-Hernández, E.; Kusalik, P.G. Mobility mechanism of hydroxyl radicals in aqueous solution via hydrogen transfer. J. Am. Chem. Soc. 2012, 134, 532–538. [Google Scholar] [CrossRef] [PubMed]
- Peterhansel, C.; Horst, I.; Niessen, M.; Blume, C.; Kebeish, R.; Kürkcüoglu, S.; Kreuzaler, F. Photorespiration. Arab. Book 2010, 8, e0130. [Google Scholar] [CrossRef]
- Noctor, G.; Veljovic-Jovanovic, S.; Driscoll, S.; Novitskaya, L.; Foyer, C.H. Drought and oxidative load in the leaves of C3 plants: A predominant role for photorespiration? Ann. Bot. 2002, 89, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Krieger-Liszkay, A. Singlet oxygen production in photosynthesis. J. Exp. Bot. 2005, 56, 337–346. [Google Scholar] [CrossRef] [PubMed]
- op den Camp, R.G.L.; Przybyla, D.; Ochsenbein, C.; Laloi, C.; Kim, C.; Danon, A.; Wagner, D.; Hideg, E.; Göbel, C.; Feussner, I.; et al. Rapid induction of distinct stress responses after the release of singlet oxygen in Arabidopsis. Plant Cell 2003, 15, 2320–2332. [Google Scholar] [CrossRef]
- Prasad, A.; Sedlářová, M.; Kale, R.S.; Pospíšil, P. Lipoxygenase in singlet oxygen generation as a response to wounding: In Vivo imaging in Arabidopsis thaliana. Sci. Rep. 2017, 7, 9831. [Google Scholar] [CrossRef]
- Carrillo, N.; Ceccarelli, E.A. Open questions in ferredoxin-NADP+ reductase catalytic mechanism. Eur. J. Biochem. 2003, 270, 1900–1915. [Google Scholar] [CrossRef]
- Zanetti, G.; Curti, B. Interactions between ferredoxin-NADP+ reductase and ferredoxin at different reduction levels of the two proteins. FEBS Lett. 1981, 129, 201–204. [Google Scholar] [CrossRef]
- Pagani, S.; Vecchio, G.; Iametti, S.; Bianchi, R.; Bonomi, F. On the role of the 2Fe-2S cluster in the formation of the structure of spinach ferredoxin. Biochim. Biophys. Acta 1986, 870, 538–544. [Google Scholar] [CrossRef]
- Cammack, R.; Rao, K.K.; Bargeron, C.P.; Hutson, K.G.; Andrew, P.W.; Rogers, L. Midpoint redox potentials of plant and algal ferredoxins. Biochem. J. 1977, 168, 205–209. [Google Scholar] [CrossRef]
- Hosein, B.; Palmer, G. The kinetics and mechanism of oxidation of reduced spinach ferredoxin by molecular oxygen and its reduced products. Biochim. Biophys. Acta 1983, 723, 383–390. [Google Scholar] [CrossRef]
- Allen, J.F. Oxygen reduction and optimum production of ATP in photosynthesis. Nature 1975, 256, 599–600. [Google Scholar] [CrossRef]
- Furbank, R.; Badger, M. Oxygen exchange associated with electron transport and photophosphorilation in spinach thylakoids. Biochim. Biophys. Acta 1983, 723, 400–409. [Google Scholar] [CrossRef]
- Asada, K.; Kiso, K.; Yoshikawa, K. Univalent reduction of molecular oxygen by spinach chloroplasts on illumination. J. Biol. Chem. 1974, 249, 2175–2181. [Google Scholar]
- Golbeck, J.H.; Radmer, R. Is the rate of oxygen uptake by reduced ferredoxin sufficient to account for photosystem I-mediated O2 reduction? In Advances in Photosynthesis Research; Sybesma, C., Ed.; Martinus Nijhoff/Dr W Junk Publishers: The Hague, The Netherlands, 1984; pp. 561–564. [Google Scholar]
- Kozuleva, M.A.; Ivanov, B.N. Evaluation of the participation of ferredoxin in oxygen reduction in the photosynthetic electron transport chain of isolated pea thylakoids. Photosynth. Res. 2010, 105, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F.; Hall, D.O. The relationship of oxygen uptake to electron transport in photosystem I of isolated chloroplasts: The role of superoxide and ascorbate. Biochem. Biophys. Res. Commun. 1974, 58, 579–585. [Google Scholar] [CrossRef]
- Khorobrykh, S.A.; Ivanov, B.N. Oxygen reduction in a plastoquinone pool of isolated pea thylakoids. Photosynth. Res. 2002, 71, 209–219. [Google Scholar] [CrossRef] [PubMed]
- Allen, J.F. A two-step mechanism for photosynthetic reduction of oxygen by ferredoxin. Biochem. Biophys. Res. Commun. 1975, 66, 36–43. [Google Scholar] [CrossRef]
- Gibson, Q.H.; Hastings, J.W. The oxidation of reduced flavin mononucleotide by molecular oxygen. Biochem. J. 1962, 83, 368–377. [Google Scholar] [CrossRef]
- Kemal, C.; Chan, T.W.; Bruice, T.C. Reaction of 3O2 with dihydroflavins. 1. N3,5-Dimethyl-1,5-dihydrolumiflavin and 1,5-dihydroisoalloxazines. J. Am. Chem. Soc. 1977, 99, 7272–7286. [Google Scholar] [CrossRef] [PubMed]
- Kemal, C.; Bruice, T.C. The chemistry of an N5-methyl-1,5-dihydroflavin and its aminium cation radical. J. Am. Chem. Soc. 1976, 98, 3955–3964. [Google Scholar] [CrossRef] [PubMed]
- Massey, V. Activation of molecular oxygen by flavins and flavoproteins. J. Biol. Chem. 1994, 269, 22459–22462. [Google Scholar] [PubMed]
- Massey, V. The reactivity of oxygen with flavoproteins. Int. Congr. Ser. 2002, 1233, 3–11. [Google Scholar] [CrossRef]
- Massey, V. The chemical and biological versatility of riboflavin. Biochem. Soc. Trans. 2000, 28, 283–296. [Google Scholar] [CrossRef] [PubMed]
- Mayhew, S.G. The effects of pH and semiquinone formation on the oxidation-reduction potentials of flavin mononucleotide. A reappraisal. Eur. J. Biochem. 1999, 265, 698–702. [Google Scholar] [CrossRef]
- Hille, R.; Nishino, T. Flavoprotein structure and mechanism. 4. Xanthine oxidase and xanthine dehydrogenase. FASEB J. 1995, 9, 995–1003. [Google Scholar] [CrossRef]
- Wohlfahrt, G.; Trivić, S.; Zeremski, J.; Peričin, D.; Leskovac, V. The chemical mechanism of action of glucose oxidase from Aspergillus niger. Mol. Cell Biochem. 2004, 260, 69–83. [Google Scholar] [CrossRef]
- Romero, E.; Gómez Castellanos, J.R.; Gadda, G.; Fraaije, M.W.; Mattevi, A. Same substrate, many reactions: Oxygen activation in flavoenzymes. Chem. Rev. 2018, 118, 1742–1769. [Google Scholar] [CrossRef]
- Miyake, C.; Schreiber, U.; Hormann, H.; Sano, S.; Asada, K. The FAD-enzyme monodehydroascorbate radical reductase mediates photoproduction of superoxide radicals in spinach thylakoid membranes. Plant Cell Physiol. 1998, 39, 821–829. [Google Scholar] [CrossRef]
- Sano, S.; Miyake, C.; Mikami, B.; Asada, K. Molecular characterization of monodehydroascorbate radical reductase from cucumber highly expressed in Eschericia coli. J. Biol. Chem. 1995, 270, 21354–21361. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Tagawa, S.; Sano, S.; Asada, K. A direct demonstration of the catalytic action of monodehydroascorbate reductase by pulse radiolysis. J. Biol. Chem. 1995, 270, 27551–27554. [Google Scholar] [CrossRef] [PubMed]
- Goetze, D.C.; Carpentier, R. Ferredoxin-NADP+ reductase is the site of oxygen reduction in pseudocyclic electron transport. Can. J. Bot. 1994, 72, 256–260. [Google Scholar] [CrossRef]
- Hossain, M.A.; Asada, K. Monodehydroascorbate reductase from cucumber is a flavin adenine dinucleotide enzyme. J. Biol. Chem. 1985, 260, 12920–12926. [Google Scholar]
- Ptushenko, V.V.; Cherepanov, D.A.; Krishtalik, L.I.; Semenov, A.Y. Semi-continuum electrostatic calculations of redox potentials in photosystem I. Photosynth. Res. 2008, 97, 55–74. [Google Scholar] [CrossRef]
- Osmond, C.B.; Grace, S.C. Perspectives on photoinhibition and photorespiration in the field: Quintessential inefficiencies of the light and dark reactions of photosynthesis? J. Exp. Bot. 1995, 48, 1351–1362. [Google Scholar] [CrossRef]
- Badger, M.; Sharkey, T.D.; Von Caemmerer, S. The relationship between steady-state gas-exchange of bean leaves and the levels of carbon-reducing-cycle intermediates. Planta 1984, 160, 305–313. [Google Scholar] [CrossRef]
- Helman, Y.; Tchernov, D.; Reinhold, L.; Shibata, M.; Ogawa, T.; Schwarz, R.; Ohad, I.; Kakplan, A. Genes encoding A-type flavoproteins are essetial for photoreduction of O2 in cyanobacteria. Curr. Biol. 2003, 13, 230–235. [Google Scholar] [CrossRef]
- Zhang, P.; Allahverdiyeva, Y.; Eisenhut, M.; Aro, E.-M. Flavodiiron proteins in oxygenic photosynthetic organisms: Photoprotection of Photosystem II by Flv2 and Flv4 in Synechocystis sp. PCC 6803. PLoS ONE 2009, 4, e5331. [Google Scholar] [CrossRef] [PubMed]
- Santana-Sanchez, A.; Solymosi, D.; Mustila, M.; Bersanini, L.; Aro, E.-M.; Allahverdiyeva, Y. Flavodiiron proteins 1–to-4 function in versatile combinations in O2 photoreduction in cyanobacteria. eLife 2019, 8, e45766. [Google Scholar] [CrossRef]
- Shimakawa, G.; Miyake, C. Oxidation of P700 ensures robust photosynthesis. Front. Plant. Sci. 2018, 9, 1617. [Google Scholar] [CrossRef]
- Asada, K. Radical production and scavenging in the chloroplasts. In Photosynthesis and the Environment; Baker, N.R., Ed.; Kluwer Academic Publisher: Dordrecht, The Netherlands, 1996; pp. 128–150. [Google Scholar]
- Yruela, I. Transition metals in plant photosynthesis. Metallomics 2013, 5, 1090–1109. [Google Scholar] [CrossRef] [PubMed]
- Waldo, G.S.; Wright, E.; Whang, Z.H.; Briat, J.F.; Theil, E.C.; Sayers, D.E. Formation of the ferritin iron mineral occurs in plastids. Plant Physiol. 1995, 109, 797–802. [Google Scholar] [CrossRef] [PubMed]
- Theil, E.C. Iron, ferritin, and nutrition. Annu. Rev. Nutr. 2004, 24, 327–343. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.E.; Morehouse, L.A.; Aust, S.D. Ferritin and superoxide-dependent lipid peroxidation. J. Biol. Chem. 1985, 260, 3275–3280. [Google Scholar] [PubMed]
- Šnyrychová, I.; Pospíšil, P.; Nauša, J. Reaction pathways involved in the production ofhydroxyl radicals in thylakoid membrane: EPR spin-trapping study. Photochem. Photobiol. Sci. 2006, 5, 472–476. [Google Scholar] [CrossRef]
- Jakob, B.; Heber, U. Photoproduction and detoxification of hydroxyl radicals in chloroplasts and leaves and relation to photoinactivation of photosystems I and II. Plant Cell Physiol. 1996, 37, 629–635. [Google Scholar] [CrossRef]
- Santabarbara, S.; Agostini, G.; Casazza, A.P.; Syme, C.D.; Heathcote, P.; Böhles, F.; Evans, M.C.; Jennings, R.C.; Carbonera, D. Chlorophyll triplet states associated with photosystem I and photosystem II in thylakoids of the green alga Chlamydomonas reinhardtii. Biochim. Biophys. Acta 2007, 1767, 88–105. [Google Scholar] [CrossRef]
- Pospíšil, P. Molecular mechanisms of production and scavenging of reactive oxygen species by photosystem II. Biochim. Biophys. Acta 2012, 1817, 218–231. [Google Scholar] [CrossRef]
- Hoff, A.J.; Rademaker, H.; van Grondelle, R.; Duysens, L.N.M. On the magnetic field dependence of the yield of the triplet state in reaction centers of photosynthetic bacteria. Biochim. Biophys. Acta 1977, 460, 547–554. [Google Scholar] [CrossRef]
- Sétif, P.; Brettel, K. Photosystem I photochemistry under highly reducing conditions: Study of the P700 triplet state formation from the secondary radical pair (P700+-A−1). Biochim. Biophys. Acta 1990, 1020, 232–238. [Google Scholar] [CrossRef]
- Antal, T.K.; Kovalenko, I.B.; Rubin, A.B.; Tyystjärvi, E. Photosynthesis-related quantities for education and modeling. Photosynth. Res. 2013, 117, 1–30. [Google Scholar] [CrossRef] [PubMed]
- Rinalducci, S.; Pedersen, J.Z.; Zolla, L. Formation of radicals from singlet oxygen produced during photoinhibition of isolated light-harvesting proteins of photosystem II. Biochim. Biophys. Acta 2004, 1608, 63–73. [Google Scholar] [CrossRef] [PubMed]
- Zolla, L.; Rinalducci, S. Involvement of active oxygen species in degradation of light-harvesting proteins under light stresses. Biochemistry 2002, 41, 14391–14402. [Google Scholar] [CrossRef] [PubMed]
- Santabarbara, S.; Cazzalini, I.; Rivadossi, A.; Garlaschi, F.M.; Zucchelli, G.; Jennings, R.C. Photoinhibition In Vivo and In Vitro involves weakly coupled chlorophyll-protein complexes. Photochem. Photobiol. 2002, 6, 613–618. [Google Scholar] [CrossRef]
- Santabarbara, S.; Neverov, K.; Garlaschi, F.M.; Zucchelli, G.; Jennings, R.C. Involvement of uncoupled antenna chlorophylls in photoinhibition in thylakoids. FEBS Lett. 2001, 491, 109–113. [Google Scholar] [CrossRef]
- Schmidt, K.; Fufezan, C.; Krieger-Liszkay, A.; Satoh, H.; Paulsen, H. Recombinant watersoluble chlorophyll protein from Brassica oleracea var. botrys binds various chlorophyll derivatives. Biochemistry 2003, 42, 7427–7433. [Google Scholar] [CrossRef]
- Cardona, T.; Sedoud, A.; Cox, N.; Rutherford, A.W. Charge separation in Photosystem II: A comparative and evolutionary overview. Biochim. Biophys. Acta 2012, 1817, 26–43. [Google Scholar] [CrossRef]
- Durrant, J.R.; Giorgi, L.B.; Barber, J.; Klug, D.R.; Porter, G. Characterization of triplet-states in isolated photosystem II reaction centres—Oxygen quenching as a mechanism for photodamage. Biochim. Biophys. Acta 1990, 1017, 167–175. [Google Scholar]
- Macpherson, A.N.; Telfer, A.; Truscott, T.G.; Barber, J. Direct detection of singlet oxygen from isolated photosystem II reaction centers. Biochim. Biophys. Acta 1993, 1143, 301–309. [Google Scholar] [CrossRef]
- Krieger-Liszkay, A.; Fufezan, C.; Trebst, A. Singlet oxygen production in photosystem II and related protection mechanism. Photosynth. Res. 2008, 98, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Rappaport, F.; Lavergne, J. Thermoluminescence: Theory. Photosynth. Res. 2009, 101, 205–216. [Google Scholar] [CrossRef] [PubMed]
- Pospíšil, P. Production of reactive oxygen species by photosystem II as a response to light and temperature stress. Front. Plant Sci. 2016, 7, 1950. [Google Scholar] [CrossRef] [PubMed]
- Diner, B.A.; Schlodder, E.; Nixon, P.J.; Coleman, W.J.; Rappaport, F.; Lavergne, J.; Vermaas, W.F.J.; Chisholm, D.A. Site-directed mutations at D1-His198 and D2-His197 of photosystem II in Synechocystis PCC 6803: Sites of primary charge separation and cation and triplet stabilization. Biochemistry 2001, 40, 9265–9281. [Google Scholar] [CrossRef]
- Noguchi, T.; Tomo, T.; Inoue, Y. Triplet formation on a monomeric chlorophyll in the photosystem II reaction center as studied by time-resolved infrared spectroscopy. Biochemistry 2001, 40, 2176–2185. [Google Scholar] [CrossRef]
- Loll, B.; Kern, J.; Saenger, W.; Zouni, A.; Biesiadka, J. Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II. Nature 2005, 438, 1040–1044. [Google Scholar] [CrossRef]
- Telfer, A.; Barber, J. Role of carotenoid bound to the photosystem II reaction centre. In Photosynthesis: From Light to Biosphere; Mathis, P., Ed.; Kluwer: Dordrecht, The Netherlands, 1995; Volume 5, pp. 15–20. [Google Scholar]
- Hideg, E.; Spetea, C.; Vass, I. Singlet oxygen production in thylakoid membranes during photoinhibition as detected by EPR spectroscopy. Photosynth. Res. 1994, 39, 191–199. [Google Scholar] [CrossRef]
- Hideg, E.; Spetea, C.; Vass, I. Singlet oxygen and free radical production during acceptor- and donor-side-induced photoinhibition. Studies with spin trapping EPR spectroscopy. Biochim. Biophys. Acta 1994, 1186, 143–152. [Google Scholar] [CrossRef]
- Hideg, E.; Vass, I. Singlet oxygen is not produced by photosystem I under photoinhibitory conditions. Photochem. Photobiol. 1995, 62, 949–952. [Google Scholar] [CrossRef]
- Fischer, B.B.; Krieger-Liszkay, A.; Eggen, R.L. Photosensitizers neutral red (type I) and rose bengal (type II) cause light-dependent toxicity in Chlamydomonas reinhardtii and induce the Gpxh gene via increased singlet oxygen formation. Environ. Sci. Technol. 2004, 38, 6307–6313. [Google Scholar] [CrossRef]
- Hakala-Yatkin, M.; Tyystjärvi, E. Inhibition of Photosystem II by the singlet oxygen sensor compounds TEMP and TEMPD. Biochim. Biophys. Acta 2011, 1807, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Hideg, E.; Déak, Z.; Hakala-Yatkin, M.; Karonen, M.; Rutherford, A.W.; Tyystjärvi, E.; Vass, I.; Krieger-Liszkay, A. Pure forms of the singlet oxygen sensors TEMP and TEMPD do not inhibit Photosystem II. Biochim. Biophys. Acta 2011, 1807, 1658–1661. [Google Scholar] [CrossRef] [PubMed]
- Karonen, M.; Mattila, H.; Huang, P.; Mamedov, F.; Styring, S.; Tyystjärvi, E. A tandem mass spectrometric method for singlet oxygen measurement. Photochem. Photobiol. 2014, 90, 965–971. [Google Scholar] [CrossRef] [PubMed]
- Fischer, B.B.; Eggen, R.I.; Trebst, A.; Krieger-Liszkay, A. The glutathione peroxidase homologous gene Gpxh in Chlamydomonas reinhardtii is upregulated by singlet oxygen produced in photosystem II. Planta 2006, 223, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Hideg, E.; Kálai, T.; Hideg, K.; Vass, I. Photoinhibition of photosynthesis in vivo results in singlet oxygen production detection via nitroxide-induced fluorescence quenching in broad bean leaves. Biochemistry 1998, 37, 11405–11411. [Google Scholar] [CrossRef]
- Hideg, E.; Ogawa, K.; Kálai, T.; Hideg, K. Singlet oxygen imaging in Arabidopsis thaliana leaves under photoinhibition by excess photosynthetically active radiation. Physiol. Plant. 2001, 112, 10–14. [Google Scholar] [CrossRef]
- Hideg, E.; Kós, P.B.; Vass, I. Photosystem II damage induced by chemically generated singlet oxygen in tobacco leaves. Physiol. Plant. 2007, 131, 33–40. [Google Scholar] [CrossRef]
- Hideg, E. A comparative study of fluorescent singlet oxygen probes in plant leaves. Cent. Eur. J. Biol. 2008, 330, 273–284. [Google Scholar] [CrossRef]
- Telfer, A.; Bishop, S.M.; Phillips, D.; Barber, J. Isolated photosynthetic reaction center of photosystem II as a sensitizer for the formation of singlet oxygen. Detection and quantum yield determination using a chemical trapping technique. J. Biol. Chem. 1994, 269, 13244–13253. [Google Scholar]
- Rehman, A.U.; Czer, K.; Sass, L.; Vass, I. Characterization of singlet oxygen production and its involvement in photodamage of Photosystem II in the cyanobacterium Synechocystis PCC 6803 by histidine-mediated chemical trapping. Biochim. Biophys. Acta 2013, 1827, 689–698. [Google Scholar] [CrossRef]
- Hakkila, K.; Antal, T.; Rehman, A.U.; Kurkela, J.; Wada, H.; Vass, I.; Tyystjärvi, E.; Tyystjärvi, T. Oxidative stress and photoinhibition can be separated in the cyanobacterium Synechocystis sp. PCC 6803. Biochim. Biophys. Acta 2014, 1837, 217–225. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Davis, G.A.; Kanazawa, A.; Schöttler, M.A.; Kohzuma, K.; Froelich, J.E.; Rutherford, A.W.; Satoh-Cruz, M.; Minhas, D.; Tietz, S.; Dhingra, A.; et al. Limitations to photosynthesis by proton motive force-induced photosystem II photodamage. eLife 2016, 5, e16921. [Google Scholar] [CrossRef] [PubMed]
- Prasad, A.; Sdelářová, M.; Pospíšil, P. Singlet oxygen imaging using fluorescent probe Singlet Oxygen Sensor Green in photosynthetic organisms. Sci. Rep. 2018, 8, 13685. [Google Scholar] [CrossRef] [PubMed]
- Telfer, A.; Dhami, S.; Bishop, S.M.; Phillips, D.; Barber, J. β-Carotene quenches singlet oxygen formed by isolated photosystem II reaction centers. Biochemistry 1994, 33, 14469–14474. [Google Scholar] [CrossRef]
- Caffarri, S.; Tibiletti, T.; Jennings, R.C.; Santabarbara, S. A comparison between plant photosystem I and photosystem II architecture and functioning. Curr. Protein Pept. Sci. 2014, 15, 296–331. [Google Scholar] [CrossRef]
- Barber, J.; Chapman, D.J.; Telfer, A. Characterisation of a PS II reaction centre isolated from the chloroplasts of Pisum sativum. FEBS Lett. 1987, 220, 67–73. [Google Scholar] [CrossRef]
- Yin, L.; Fristedt, R.; Herdean, A.; Solymosi, K.; Bertrand, M.; Andersson, M.X.; Mamedov, F.; Vener, A.V.; Schoefs, B.; Spetea, C. Photosystem II function and dynamics in three widely used Arabidopsis thaliana accessions. PLoS ONE 2012, 7, e46206. [Google Scholar] [CrossRef]
- Khorobrykh, S.A.; Khorobrykh, A.A.; Yanykin, D.V.; Ivanov, B.N.; Klimov, V.V.; Mano, J. Photoproduction of catalase-insensitive peroxides on the donor side of manganese-depleted photosystem II: Evidence with a specific fluorescent probe. Biochemistry 2011, 50, 10658–10665. [Google Scholar] [CrossRef]
- Yadav, D.K.; Pospíšil, P. Evidence on the formation of singlet oxygen in the donor side photoinhibition of photosystem II: EPR spin-trapping study. PLoS ONE 2012, 7, e45883. [Google Scholar] [CrossRef]
- Pathak, V.; Prasad, A.; Pospíšil, P. Formation of singlet oxygen by decomposition of protein hydroperoxide in photosystem II. PLoS ONE 2017, 12, e0181732. [Google Scholar] [CrossRef]
- Shuvalov, V.A.; Nuijs, A.M.; van Gorkom, H.J.; Smit, H.W.J.; Duysens, L.N.M. Picosecond absorbance changes upon selective excitation of the primary electron donor P-700 in photosystem I. Biochim. Biophys. Acta 1986, 850, 319–323. [Google Scholar] [CrossRef]
- Sétif, P.; Hervo, G.; Mathis, P. Flash-induced absorption changes in Photosystem I, Radical pair or triplet state formation? Biochim. Biophys. Acta 1981, 638, 257–267. [Google Scholar] [CrossRef]
- Rutherford, A.W.; Osyczka, A.; Rappaport, F. Back-reactions, short-circuits, leaks and other energy wasteful reactions in biological electron transfer: Redox tuning to survive life in O(2). FEBS Lett. 2012, 586, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Cazzaniga, S.; Li, Z.; Niyogi, K.K.; Bassi, R.; Dall’Osto, L. The Arabidopsis szl1 mutant reveals a critical role of β-carotene in photosystem I photoprotection. Plant Physiol. 2012, 159, 1745–1758. [Google Scholar] [CrossRef]
- Jung, J.; Kim, H.-S. The chromophores as endogenous sensitizers involved in the photogeneration of singlet oxygen in spinach thylakoids. Photochem. Photobiol. 1990, 52, 1003–1009. [Google Scholar] [CrossRef]
- Mehler, A.H. Studies on reactivity of illuminated chloroplasts. Mechanism of the reduction of oxygen and other Hill reagents. Arch. Biochem. Biophys. 1951, 33, 65–77. [Google Scholar] [CrossRef]
- Glidewell, S.M.; Raven, J.A. Measurement of simultaneous oxygen evolution and uptake in Hydrodictyon africanum. J. Exp. Bot. 1975, 26, 479–488. [Google Scholar] [CrossRef]
- Patterson, C.O.P.; Myers, J. Photosynthetic production of hydrogen peroxide by Anacystis nidulans. Plant Physiol. 1973, 51, 104–109. [Google Scholar] [CrossRef]
- Egneus, H.; Heber, U.; Matthiesen, U.; Kirk, M. Reduction of oxygen by the electron transport chain of chloroplasts during assimilation of carbon dioxide. Biochim. Biophys. Acta 1975, 408, 252–268. [Google Scholar] [CrossRef]
- Radmer, R.; Kok, B. Photoreduction of O2 primes and replaces CO2 assimilation. Plant Physiol. 1976, 58, 336–340. [Google Scholar] [CrossRef]
- Elstner, E.F. Oxygen activation and oxygen toxicity. Annu. Rev. Plant Physiol. 1982, 33, 73–96. [Google Scholar] [CrossRef]
- Badger, M.R. Photosynthetic oxygen exchange. Annu. Rev. Plant Physiol. 1985, 36, 27–53. [Google Scholar] [CrossRef]
- Robinson, J.M. Does O2 photoreduction occur within chloroplasts In Vivo? Physiol. Plant. 1988, 72, 666–680. [Google Scholar] [CrossRef]
- Asada, K. Production and action of active oxygen species in photosynthesis tissues. In Causes of Photooxidative Stress and Amelioration of Defense Systems in Plants; Foyer, C.H., Mullineaux, P.M., Eds.; CRC Press: Boca Raton, FL, USA, 1994; pp. 77–104. [Google Scholar]
- Ananyev, G.M.; Renger, G.; Wacker, U.; Klimov, V. The photoproduction of superoxide radicals and the superoxide-dismutase activity of photosystem II. The possible involvement of cytochrome b559. Photosynth. Res. 1994, 41, 327–338. [Google Scholar] [CrossRef] [PubMed]
- Nugent, J.H.A. Photoreducible high spin iron electron paramagnetic resonance signals in dark-adapted Photosystem II: Are they oxidised non-haem iron formed from interaction of oxygen with PSII electron acceptors? Biochim. Biophys. Acta 2001, 1504, 288–298. [Google Scholar] [CrossRef]
- Ishikita, H.; Biesiadka, J.; Loll, B.; Saenger, W.; Knapp, E.W. Cationic state of accessory chlorophyll and electron transfer through pheophytin to plastoquinone in photosystem II. Angew. Chem. Int. Ed. 2006, 45, 1964–1965. [Google Scholar] [CrossRef]
- Rutherford, A.W.; Mullet, J.E.; Crofts, A.R. Measurement of the midpoint potential of the pheophytin acceptor of photosystem II. FEBS Lett. 1981, 123, 235–237. [Google Scholar] [CrossRef]
- Klimov, V.V.; Allakhverdiev, S.I.; Demeter, S.; Krasnovskii, A.A. Photoreduction of pheophytin in the photosystem 2 of chloroplasts with respect to the redox potential of the medium. Dokl. Akad. Nauk SSSR 1979, 249, 227–230. [Google Scholar]
- Mamedov, M.D.; Kurashov, V.N.; Cherepanov, D.A.; Semenov, A.Y. Photosysem II: Where does the light-induced voltage come from? Front. Biosci. 2010, 15, 1007–1017. [Google Scholar] [CrossRef]
- Kato, Y.; Sugiura, M.; Oda, A.; Watanabe, T. Spectroelectrochemical determination of the redox potential of pheophytin a, the primary electron acceptor in photosystem II. Proc. Natl. Acad. Sci. USA 2009, 106, 17365–17370. [Google Scholar] [CrossRef]
- Crofts, A.R.; Wraight, C.A. The electrochemical domain of photosynthesis. Biochim. Biophys. Acta 1983, 726, 149–185. [Google Scholar] [CrossRef]
- Allakhverdiev, S.I.; Tsuchiya, T.; Watabe, K.; Kojima, A.; Los, D.A.; Tomo, T.; Klimov, V.V.; Mimuro, M. Redox potentials of primary electron acceptor quinone molecule (QA)− and conserved energetics of photosystem II in cyanobacteria with chlorophyll a and chlorophyll d. Proc. Natl. Acad. Sci. USA 2011, 108, 8054–8058. [Google Scholar] [CrossRef] [PubMed]
- Tyystjärvi, E.; Vass, I. Light emission as a probe of charge separation and recombination in the photosynthetic apparatus: Relation of prompt fluorescence to delayed light emission and thermoluminescence. In Chlorophyll a Fluorescence: A Signature of Photosynthesis; Papageorgiou, G.C., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 2004; pp. 363–388. [Google Scholar]
- Brinkert, K.; De Causmaecker, S.; Krieger-Liszkay, A.; Fantuzzi, A.; Rutherford, A.W. Bicarbonate-induced redox tuning in Photosystem II for regulation and protection. Proc. Natl. Acad. Sci. USA 2016, 113, 12144–12149. [Google Scholar] [CrossRef]
- Renger, G.; Eckert, H.J.; Bergmann, A.; Bernarding, J.; Liu, B.; Napiwotzki, A.; Reifarth, F.; Eichler, H.J. Fluorescence and spectroscopic studies of exciton trapping and electron transfer in photosystem II of higher plants. Aust. J. Plant Physiol. 1995, 22, 167–181. [Google Scholar] [CrossRef]
- de Wijn, R.; van Gorkom, H.J. Kinetics of electron transfer from QA to QB in photosystem II. Biochemistry 2001, 40, 11912–11922. [Google Scholar] [CrossRef]
- Rich, P.R.; Moss, D.A. The reaction of quinones in higher plant photosynthesis. In The Light Reaction; Barber, J., Ed.; Elsevier: Amsterdam, The Netherlands, 1987; pp. 421–445. [Google Scholar]
- Crofts, A.R.; Robinson, H.H.; Snozzi, M. Reactions of quinols at catalytic sites: A diffusionrole in H-transfer. In Advances in Photosynthesis Research; Sybesma, C., Ed.; M. Nijhoff/Dr. W. Junk Publisher: The Hague, The Netherlands, 1984; pp. 1461–1468. [Google Scholar]
- Kato, M.; Zhang, J.Z.; Paul, N.; Reisner, E. Protein film photoelectrochemistry of the water oxidation enzyme photosystem II. Chem. Soc. Rev. 2014, 43, 6485–6497. [Google Scholar] [CrossRef]
- Zhu, Z.; Gunner, M.R. Energetics of quinone-dependent electron and proton transfers in Rhodobacter sphaeroides photosynthetic reaction centers. Biochemistry 2005, 44, 82–96. [Google Scholar] [CrossRef]
- Klimov, V.V.; Ananyev, G.M.; Zastrizhnaya, O.M.; Wydrzynski, T.; Renger, G. Photoproduction of hydrogen peroxide in Photosystem II membrane fragments: A comparison of four signals. Photosynth. Res. 1993, 38, 409–416. [Google Scholar] [CrossRef]
- Arato’, A.; Bondarava, N.; Krieger-Liszkay, A. Production of reactive oxygen species in chloride- and calcium-depleted photosystem II and their involvement in photoinhibition. Biochim. Biophys. Acta 2004, 1608, 171–180. [Google Scholar] [CrossRef]
- Cleland, R.E.; Grace, S.C. Voltammetric detection of superoxide production by photosystem II. FEBS Lett. 1999, 457, 348–352. [Google Scholar] [CrossRef]
- Zulfugarov, I.S.; Tovuu, A.; Eu, Y.J.; Dogsom, B.; Poudyal, R.S.; Nath, K.; Hall, M.; Banerjee, M.; Yoon, U.C.; Moon, Y.H.; et al. Production of superoxide from Photosystem II in a rice (Oryza sativa L.) mutant lacking PsbS. BMC Plant Biol. 2014, 14, 242. [Google Scholar] [CrossRef] [PubMed]
- Vass, I.; Gatzen, G.; Holzwarth, A.R. Picosecond time-resolved fluorescence studies on photoinhibition and double reduction of QA in Photosystem II. Biochim. Biophys. Acta 1993, 1183, 388–396. [Google Scholar] [CrossRef]
- Müh, F.; Zouni, A. Cytochrome b 559 in photosystem II. In Cytochrome Complexes: Evolution, Structures, Energy Transduction, and Signaling; Cramer, W.A., Kallas, T., Eds.; Springer: Dordrecht, The Netherlands, 2015; pp. 147–175. [Google Scholar]
- Shuvalov, V.A. Composition and function of cytochrome b559 in reaction centers of Photosystem II of green plants. J. Bioenerg. Biomembr. 1994, 26, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, T. Modification of oxygen evolving center by Tris-washing. Photosynth. Res. 1986, 10, 473–481. [Google Scholar] [CrossRef] [PubMed]
- Horton, P.; Croze, E. The relationship between activity of chloroplast Photosystem II and the midpoint oxidation-reduction potential of cytochrome b-559. Biochim. Biophys. Acta 1977, 462, 86–101. [Google Scholar] [CrossRef]
- Berthold, D.A.; Babcock, G.T.; Yocum, C.F. A highly resolved, oxygen-evolving photosystem II preparation from spinach thylakoid membranes: EPR and electron-transport properties. FEBS Lett. 1981, 134, 231–234. [Google Scholar] [CrossRef]
- Babcock, G.T.; Widger, W.R.; Cramer, W.A.; Oertling, W.A.; Metz, J.G. Axial ligands of chloroplast cytochrome b-559: Identification and requirement for a heme-cross-linked polypeptide structure. Biochemistry 1985, 24, 3638–3645. [Google Scholar] [CrossRef]
- Pospíšil, P.; Šnyrychová, I.; Kruk, J.; Strzałka, K.; Nauš, J. Evidence that cytochrome b559 is involved in superoxide production in Photosystem II: Effect of synthetic short-chain plastoquinones in a cytochrome b559 tobacco mutant. Biochem. J. 2006, 397, 321–327. [Google Scholar] [CrossRef]
- Whitmarsh, J.; Cramer, W.A. A pathway for the reduction of cytochrome b-559 by Photosystem II in chloroplasts. Biochim. Biophys. Acta 1978, 501, 83–93. [Google Scholar] [CrossRef]
- Gounaris, K.; Chapman, D.J.; Barber, J. Reconstitution of plastoquinone in the D1/D2/cytochrome b-559 Photosystem II reaction centre complex. FEBS Lett. 1988, 240, 143–147. [Google Scholar] [CrossRef]
- Kruk, J.; Strzałka, K. Dark reoxidation of the plastoquinone-pool is mediated by the low-potential form of cytochrome b-559 in spinach thylakoids. Photosynth. Res. 1999, 62, 273–279. [Google Scholar] [CrossRef]
- Kruk, J.; Strzałka, K. Redox changes of cytochrome b559 in the presence of plastoquinones. J. Biol. Chem. 2001, 276, 86–91. [Google Scholar] [CrossRef]
- Yadav, D.K.; Prasad, A.; Kruk, J.; Pospíšil, P. Evidence for the involvement of loosely bound plastosemiquinones in superoxide anion radical production in photosystem II. PLoS ONE 2014, 9, e115466. [Google Scholar] [CrossRef]
- Guskov, A.; Kern, J.; Gabdulkhakov, A.; Broser, M.; Zouni, A.; Saenger, W. Cyanobacterial Photosystem II at 2.9 Å resolution: Role of quinones, lipids, channels and chloride. Nat. Struct. Mol. Biol. 2009, 16, 334–342. [Google Scholar] [CrossRef]
- Khorobrykh, S.; Tyystjärvi, E. Plastoquinol generates and scavenges reactive oxygen species in organic solvent: Potential relevance for thylakoids. Biochim. Biophys. Acta 2018, 1859, 1119–1131. [Google Scholar] [CrossRef]
- Tiwari, A.; Pospíšil, P. Superoxide oxidase and reductase activity of cytochrome b559 in photosystem II. Biochim. Biophys. Acta 2009, 1787, 985–994. [Google Scholar] [CrossRef]
- Fine, P.L.; Frasch, W.D. The oxygen-evolving complex requires chloride to prevent hydrogen-peroxide formation. Biochemistry 1992, 31, 12204–12210. [Google Scholar] [CrossRef]
- Antal, T.K.; Sarvikas, P.; Tyystjärvi, E. Two-Electron Reactions S2QB → S0QB and S3QB → S1QB are involved in deactivation of higher S states of the oxygen-evolving complex of photosystem II. Biophys. J. 2009, 96, 1–9. [Google Scholar] [CrossRef][Green Version]
- Pospíšil, P.; Šnyrychová, I.; Nauš, J. Dark production of reactive oxygen species in photosystem II membrane particles at elevated temperature: EPR spin-trapping study. Biochim. Biophys. Acta 2007, 1767, 854–859. [Google Scholar] [CrossRef]
- Yamashita, A.; Nijo, N.; Pospísil, P.; Morita, N.; Takenaka, D.; Aminaka, R.; Yamamoto, Y.; Yamamoto, Y. Quality control of photosystem II: Reactive oxygen species are responsible for the damage to photosystem II under moderate heat stress. J. Biol. Chem. 2008, 283, 28380–28391. [Google Scholar] [CrossRef]
- Navari-Izzo, F.; Pinzino, C.; Quartacci, M.F.; Sgherri, C.L. Superoxide and hydroxyl radical generation, and superoxide dismutase in PSII membrane fragments from wheat. Free Radic. Res. 1999, 33, 3–9. [Google Scholar] [CrossRef]
- Zhang, S.; Weng, J.; Pan, J.; Tu, T.; Yao, S.; Xu, C. Study on the photo-generation of superoxide radicals in Photosystem II with EPR spin trapping techniques. Photosynth. Res. 2003, 75, 41–48. [Google Scholar] [CrossRef]
- Pospísil, P.; Arató, A.; Krieger-Liszkay, A.; Rutherford, A.W. Hydroxyl radical generation by photosystem II. Biochemistry 2004, 43, 6783–6792. [Google Scholar] [CrossRef]
- Khorobrykh, S.A.; Khorobrykh, A.A.; Klimov, V.V.; Ivanov, B.N. Photoconsumption of oxygen in photosystem II preparations under impairment of the water-oxidizing complex. Biochemistry 2002, 67, 683–688. [Google Scholar]
- Yanykin, D.V.; Khorobrykh, A.A.; Khorobrykh, S.A.; Klimov, V.V. Photoconsumption of molecular oxygen on both donor and acceptor sides of photosystem II in Mn-depleted subchloroplast membrane fragments. Biochim. Biophys. Acta 2010, 1797, 516–523. [Google Scholar] [CrossRef]
- Asada, K.; Kiso, K. The photooxidation of epinephrine by spinach chloroplasts and its inhibition by superoxide dismutase: Evidence for the formation of superoxide radicals in chloroplasts. Agric. Biol. Chem. 1973, 37, 453–454. [Google Scholar] [CrossRef]
- Khorobrykh, S.; Mubarakshina, M.; Ivanov, B. Photosystem I is not solely responsible for oxygen reduction in isolated thylakoids. Biochim. Biophys. Acta 2004, 1657, 164–167. [Google Scholar] [CrossRef]
- Fork, D.C.; Heber, U.W. Studies on electron-transport reactions of photosynthesis in plastome mutants of Oenothera. Plant Physiol. 1968, 43, 606–612. [Google Scholar] [CrossRef]
- Kruk, J.; Jemiola-Rzeminska, M.; Burda, K.; Schmid, G.; Strzalka, K. Scavenging of superoxide generated in photosystem I by plastoquinol and other prenyllipids in thylakoid membranes. Biochemistry 2003, 42, 8501–8505. [Google Scholar] [CrossRef]
- Nelson, N.; Yocum, C.F. Structure and function of photosystems I and II. Annu. Rev. Plant Biol. 2006, 57, 521–565. [Google Scholar] [CrossRef]
- Brettel, K. Electron transfer and arrangement of the redox cofactors in photosystem I. Biochim. Biophys. Acta 1997, 1318, 322–373. [Google Scholar] [CrossRef]
- Brettel, K.; Leibl, W. Electron transfer in photosystem I. Biochim. Biophys. Acta 2001, 1507, 100–114. [Google Scholar] [CrossRef]
- Kirchhoff, H.; Schöttler, M.A.; Maurer, J.; Weis, E. Plastocyanin redox kinetics in spinach chloroplasts: Evidence for disequilibrium in the high potential chain. Biochim. Biophys. Acta 2004, 1659, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, A.; Suzawa, T.; Kato, Y.; Watanabe, T. Species dependence of the redox potential of the primary electron donor p700 in photosystem I of oxygenic photosynthetic organisms revealed by spectroelectrochemistry. Plant Cell Physiol. 2011, 52, 815–823. [Google Scholar] [CrossRef]
- Takahashi, M.; Asada, K. Superoxide production in aprotic interior of chloroplast thylakoids. Arch. Biochem. Biophys. 1988, 267, 714–722. [Google Scholar] [CrossRef]
- Kozuleva, M.A.; Ivanov, B.N. The mechanisms of oxygen reduction in the terminal reducing segment of the chloroplast photosynthetic electron transport chain. Plant Cell Physiol. 2016, 57, 1397–1404. [Google Scholar] [CrossRef]
- Kozuleva, M.; Klenina, I.; Proskuryakov, I.; Kirilyuk, I.; Ivanov, B. Production of superoxide in chloroplast thylakoid membranes: ESR study with cyclic hydroxylamines of different lipophilicity. FEBS Lett. 2011, 585, 1067–1071. [Google Scholar] [CrossRef]
- Kozuleva, M.A.; Petrova, A.A.; Mamedov, M.D.; Semenov, A.Y.; Ivanov, B.N. O2 reduction by photosystem I involves phylloquinone under steady-state illumination. FEBS Lett. 2014, 588, 4364–4368. [Google Scholar] [CrossRef]
- Sauer, K.; Mathis, P.; Acker, S.; van Best, J.A. Electron acceptors associated with P-700 in Triton solubilized photosystem I particles from spinach chloroplasts. Biochim. Biophys. Acta 1978, 503, 120–134. [Google Scholar] [CrossRef]
- Hiyama, T.; Ke, B. A further study of P430: A possible primary electron acceptor of photosystem I. Arch. Biochem. Biophys. 1971, 147, 99–108. [Google Scholar] [CrossRef]
- Asada, K.; Nakano, Y. Affinity for oxygen in photoreduction of molecular oxygen and scavenging of hydrogen peroxide in chloroplasts. Photochem. Photobiol. 1978, 28, 917–920. [Google Scholar] [CrossRef]
- Takahashi, M.; Asada, K. Dependence of oxygen affinity for Mehler reaction on photochemical activity of chloroplast thylakoids. Plant Cell Physiol. 1982, 23, 1457–1461. [Google Scholar]
- Semenov, A.Y.; Mamedov, M.D.; Chamorovsky, S.K. Photoelectric studies of the transmembrane charge transfer reactions in photosystem I pigment-protein complexes. FEBS Lett. 2003, 553, 223–228. [Google Scholar] [CrossRef][Green Version]
- Semenov, A.Y.; Vassiliev, I.R.; van der Est, A.; Mamedov, M.D.; Zybailov, B.; Shen, G.; Stehlik, D.; Diner, B.A.; Chitnis, P.R.; Golbeck, J.H. Recruitment of a foreign quinone into the A1 site of photosystem I. Altered kinetics of electron transfer in phylloquinone biosynthetic pathway mutants studied by time-resolved optical, EPR, and electrometric techniques. J. Biol. Chem. 2000, 275, 23429–23438. [Google Scholar] [CrossRef]
- Ivanov, B.N.; Ignatova, L.K.; Ovchinnikova, V.I.; Khorobrykh, S.A. Photoreduction of acceptor generated in an ascorbate peroxidase reaction in pea thylakoids. Biochemistry 1997, 62, 1082–1088. [Google Scholar]
- Miyake, C.; Asada, K. Ferredoxin-dependent photoreduction of the monodehydroascorbate radical in spinach thylakoids. Plant Cell Physiol. 1994, 35, 539–549. [Google Scholar] [CrossRef]
- Mubarakshina, M.; Khorobrykh, S.; Ivanov, B. Oxygen reduction in chloroplast thylakoids results in production of hydrogen peroxide inside the membrane. Biochim. Biophys. Acta 2006, 1757, 1496–1503. [Google Scholar] [CrossRef]
- Takag, D.; Takumi, S.; Hashiguchi, M.; Sejima, T.; Miyake, C. Superoxide and singlet oxygen produced within the thylakoid membranes both cause photosystem I photoinhibition. Plant Physiol. 2016, 171, 1626–1634. [Google Scholar] [CrossRef]
- Curci, R.; Edwards, J.O. Activation of hydrogen peroxide by organic compounds. In Catalytic Oxidations with Hydrogen Peroxide as Oxidant. Catalysis by Metal Complexes; Strukul, G., Ed.; Springer: Dordrecht, The Netherlands, 1992; Volume 9, pp. 45–95. [Google Scholar]
- Kruk, J.; Strzałka, K. Identification of plastoquinone-C in spinach and maple leaves by reverse-phase high-performance liquid chromatography. Phytochemistry 1998, 49, 2267–2271. [Google Scholar] [CrossRef]
- Kruk, J.; Karpinski, S. An HPLC-based method of estimation of the total redox state of plastoquinone in chloroplasts, the size of the photochemically active plastoquinone-pool and its redox state in thylakoids of Arabidopsis. Biochim. Biophys. Acta 2006, 1757, 1669–1675. [Google Scholar] [CrossRef]
- Kruk, J.; Karpinski, S. Redox state analysis of the plastoquinone-pool in Arabidopsis thaliana reveals unexpected changes under different light conditions. In Photosynthesis: Fundamental Aspects to Global Perspectives; van der Est, A., Bruce, D., Eds.; International Society of Photosynthesis Research: Montreal, QC, Canada, 2005; pp. 568–570. [Google Scholar]
- Lichtenthaler, H.K. Localization and functional concentrations of lipoquinones in chloroplasts. In Progress in Photosynthesis Research; Metzner, H., Ed.; Laupp: Tübingen, Germany, 1969; Volume 1, pp. 304–314. [Google Scholar]
- Lichtenthaler, H.K.; Prenzel, U.; Douce, R.; Joyard, J. Localization of prenylquinones in the envelope of spinach chloroplasts. Biochim. Biophys. Acta 1981, 641, 99–105. [Google Scholar] [CrossRef]
- Lichtenthaler, H.K. Biosynthesis, accumulation and emission of carotenoids, α-tocopherol, plastoquinone, and isoprene in leaves under high photosynthetic irradiance. Photosynth. Res. 2007, 92, 163–179. [Google Scholar] [CrossRef] [PubMed]
- Block, M.A.; Douce, R.; Joyard, J.; Rolland, N. Chloroplast envelope membranes: A dynamic interface between plastids and the cytosol. Photosynth. Res. 2007, 92, 225–244. [Google Scholar] [CrossRef] [PubMed]
- Austin, J.R., 2nd; Frost, E.; Vidi, P.A.; Kessler, F.; Staehelin, L.A. Plastoglobules are lipoprotein subcompartments of the chloroplast that are permanently coupled to thylakoid membranes and contain biosynthetic enzymes. Plant Cell 2006, 18, 1693–1703. [Google Scholar] [CrossRef] [PubMed]
- McCauley, S.W.; Melis, A. Quantitation of plastoquinone photoreduction in spinach chloroplasts. Photosynth. Res. 1986, 8, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Graan, T.; Ort, D.R. Quantitation of the rapid electron donors to P700, the functional plastoquinone pool, and the ratio of the photosystems in spinach chloroplasts. J. Biol. Chem. 1984, 259, 14003–14010. [Google Scholar]
- Joliot, P.; Lavergne, J.; Beal, D. Plastoquinone compartmentation in chloroplasts. I. Evidence for domains with different rates of photo-reduction. Biochim. Biophys. Acta 1992, 1101, 1–12. [Google Scholar] [CrossRef]
- Chapman, D.J.; Barber, J. Analysis of plastoquinone-9 levels in appressed and non-appressed thylakoid membrane regions. Biochim. Biophys. Acta 1986, 850, 170–172. [Google Scholar] [CrossRef]
- Rich, P.R.; Bendall, D.S. The kinetics and thermodynamics of the reduction of cytochrome c by substituted p-benzoquinols in solution. Biochim. Biophys. Acta 1980, 592, 506–518. [Google Scholar] [CrossRef]
- Elstner, E.F.; Frommeyer, D. Production of hydrogen peroxide by Photosystem II of spinach chloroplast lamellae. FEBS Lett. 1978, 86, 143–147. [Google Scholar] [CrossRef]
- Hauska, G.; Hurt, E.; Gabellini, N.; Lockau, W. Comparative aspects of quinol-cytochrome c/plastocyanin oxidoreductases. Biochim. Biophys. Acta 1983, 726, 97–133. [Google Scholar] [CrossRef]
- Prince, R.C.; Dutton, P.L.; Bruce, J.M. Electrochemistry of ubiquinones: Menaquinones and plastoquinones in aprotic solvents. FEBS Lett. 1983, 160, 273–276. [Google Scholar] [CrossRef]
- Wardman, P. Bioreductive activation of quinones: Redox properties and thiol reactivity. Free Radic. Res. Commun. 1990, 8, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Lawlor, D.W. Photosynthesis: Metabolism, Control and Physiology, 1st ed.; Longman Scientific & Technica: Essex, UK, 1987; p. 262. [Google Scholar]
- Kruk, J.; Trebst, A. Plastoquinol as a singlet oxygen scavenger in photosystem II. Biochim. Biophys. Acta 2008, 1777, 154–162. [Google Scholar] [CrossRef] [PubMed]
- Yadav, D.K.; Kruk, J.; Sinha, R.K.; Pospíšil, P. Singlet oxygen scavenging activity of plastoquinol in photosystem II of higher plants: Electron paramagnetic resonance spin-trapping study. Biochim. Biophys. Acta 2010, 1797, 1807–1811. [Google Scholar] [CrossRef]
- Nowicka, B.; Kruk, J. Plastoquinol is more active than α-tocopherol in singlet oxygen scavenging during high light stress of Chlamydomonas reinhardtii. Biochim. Biophys. Acta 2012, 1817, 389–394. [Google Scholar] [CrossRef]
- Vetoshkina, D.V.; Ivanov, B.N.; Khorobrykh, S.A.; Proskuryakov, I.I.; Borisova-Mubarakshina, M.M. Involvement of the chloroplast plastoquinone pool in the Mehler reaction. Physiol. Plant. 2017, 161, 45–55. [Google Scholar] [CrossRef]
- Cournac, L.; Josse, E.-M.; Joet, T.; Rumeu, D.; Redding, K.; Kuntz, M.; Peltier, G. Flexibility in photosynthetic electron transport: A newly identified chloroplasts oxidase involved in chlororespiration. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 1447–1454. [Google Scholar] [CrossRef]
- Lennon, A.M.; Prommeenate, P.; Nixon, P.J. Location, expression and orientation of the putative chlororespiratory enzymes, Ndh and IMMUTANS, in higher-plant plastids. Planta 2003, 218, 254–260. [Google Scholar] [CrossRef]
- McDonald, A.E.; Ivanov, A.G.; Bode, R.; Maxwell, D.P.; Rodermel, S.R.; Huner, N.P. Flexibility in photosynthetic electron transport: The physiological role of plastoquinol terminal oxidase (PTOX). Biochim. Biophys. Acta 2011, 1807, 954–967. [Google Scholar] [CrossRef]
- Nixon, P.J.; Rich, P.R. Chlororespiratory pathways and their physiological significance. In The Structure and Function of Plastids. Advances in Photosynthesis and Respiration; Wise, R.R., Hoober, J.K., Eds.; Springer: Dordrecht, The Netherlands, 2006; Volume 23, pp. 237–251. [Google Scholar]
- Trouillard, M.; Shahbazi, M.; Moyet, L.; Rappaport, F.; Joliot, P.; Kuntz, M.; Finazzi, G. Kinetic properties and physiological role of the plastoquinone terminal oxidase (PTOX) in a vascular plant. Biochim. Biophys. Acta 2012, 1817, 2140–2148. [Google Scholar] [CrossRef] [PubMed]
- Krieger-Liszkay, A.; Feilke, K. The dual role of the plastid terminal oxidase PTOX: Between a protective and a pro-oxidant function. Front. Plant Sci. 2016, 6, 1147. [Google Scholar] [CrossRef] [PubMed]
- Yu, Q.; Feilke, K.; Krieger-Liszkay, A.; Beyer, P. Functional and molecular characterization of plastid terminal oxidase from rice (Oryza sativa). Biochim. Biophys. Acta 2014, 1837, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Baniulis, D.; Hasan, S.S.; Stofleth, J.T.; Cramer, W.A. Mechanism of enhanced superoxide production in the cytochrome b6f complex of oxygenic photosynthesis. Biochemistry 2013, 52, 8975–8983. [Google Scholar] [CrossRef] [PubMed]
- Sang, M.; Xie, J.; Qin, X.C.; Wang, W.D.; Chen, X.B.; Wang, K.B.; Zhang, J.P.; Li, L.B.; Kuang, T.Y. High-light induced superoxide radical formation in cytochrome b₆f complex from Bryopsis corticulans as detected by EPR spectroscopy. J. Photochem. Photobiol. B 2011, 102, 177–181. [Google Scholar] [CrossRef] [PubMed]
- Mishra, N.P.; Francke, C.; van Gorkom, H.J.; Ghanotakis, D.F. Destructive role of singlet oxygen during aerobic illumination of the Photosystem II core complex. Biochim. Biophys. Acta 1994, 1186, 81–90. [Google Scholar] [CrossRef]
- Tyystjärvi, E. Photoinhibition of Photosystem II. Int. Rev. Cell Mol. Biol. 2013, 300, 243–303. [Google Scholar]
- Vass, I. Role of charge recombination processes in photodamage and photoprotection of the photosystem II complex. Physiol. Plant. 2011, 142, 6–16. [Google Scholar] [CrossRef]
- Hakala, M.; Tuominen, I.; Keränen, M.; Tyystjärvi, T.; Tyystjärvi, E. Evidence for the role of the oxygen-evolving manganese complex in photoinhibition of Photosystem II. Biochim. Biophys. Acta 2005, 1706, 68–80. [Google Scholar] [CrossRef]
- Fan, D.Y.; Ye, Z.P.; Wang, S.C.; Chow, W. Multiple roles of oxygen in the photoinactivation and dynamic repair of Photosystem II in spinach leaves. Photosynth. Res. 2016, 127, 307–319. [Google Scholar] [CrossRef]
- Jahns, P.; Depka, B.; Trebst, A. Xanthophyll cycle mutants from Chlamydomonas reinhardtii indicate a role for zeaxanthin in the D1 protein turnover. Plant Physiol. Biochem. 2000, 38, 371–376. [Google Scholar] [CrossRef]
- Hakala-Yatkin, M.; Sarvikas, P.; Paturi, P.; Mäntysaari, M.; Mattila, H.; Tyystjärvi, T.; Nedbal, L.; Tyystjärvi, E. Magnetic field protects plants against high light by slowing down production of singlet oxygen. Physiol. Plant. 2011, 142, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Inoue, S.; Ejima, K.; Iwai, E.; Hayashi, H.; Appel, J.; Tyystjärvi, E.; Murata, N.; Nishiyama, Y. Protection by α-tocopherol of the repair of photosystem II during photoinhibition in Synechocystis sp. PCC 6803. Biochim. Biophys. Acta 2011, 1807, 236–241. [Google Scholar] [CrossRef] [PubMed]
- Fufezan, C.; Gross, C.M.; Sjödin, M.; Rutherford, A.W.; Krieger-Liszkay, A.; Kirilovsky, D. Influence of the redox potential of the primary quinone electron acceptor on photoinhibition of photosystem II. J. Biol. Chem. 2007, 282, 12492–12502. [Google Scholar] [CrossRef]
- Sarvikas, P.; Hakala, M.; Pätsikkä, E.; Tyystjärvi, T.; Tyystjärvi, E. Action spectrum of photoinhibition in leaves of wild type and npq1-2 and npq4-1 mutants of Arabidopsis thaliana. Plant Cell Physiol. 2006, 47, 391–400. [Google Scholar] [CrossRef]
- Ohnishi, N.; Allakhverdiev, S.I.; Takahashi, S.; Higashi, S.; Watanabe, M.; Nishiyama, Y.; Murata, N. Two-step mechanism of photodamage to photosystem II: Step I occurs at the oxygen-evolving complex and step 2 occurs at the photochemical reaction center. Biochemistry 2005, 44, 8494–8499. [Google Scholar] [CrossRef] [PubMed]
- Treves, H.; Raanan, H.; Kedem, I.; Murik, O.; Keren, N.; Zer, H.; Berkowicz, S.M.; Giordano, M.; Norici, A.; Shotland, Y.; et al. The mechanisms whereby the green alga Chlorella ohadii, isolated from desert soil crust, exhibits unparalleled photodamage resistance. New Phytol. 2016, 210, 1229–1243. [Google Scholar] [CrossRef]
- Mishra, N.P.; Ghanotakis, D.F. Exposure of Photosystem II complex to chemically generated singlet oxygen results in D1 fragments similar to the ones observed during aerobic photoinhibition. Biochim. Biophys. Acta 1994, 1187, 296–300. [Google Scholar] [CrossRef]
- Okada, K.; Ikeuchi, M.; Yamamoto, N.; Ono, T.; Miyao, M. Selective and specific cleavage of the D1 and D2 proteins of Photosystem II by exposure to singlet oxygen: Factors responsible for the susceptibility to cleavage of the proteins. Biochim. Biophys. Acta 1996, 1274, 73–79. [Google Scholar] [CrossRef]
- Miyao, M.; Ikeuchi, M.; Yamamoto, N.; Ono, T. Specific degradation of the D1 protein of photosystem II by treatment with hydrogen peroxide in darkness: Implications for the mechanism of degradation of the D1 protein under illumination. Biochemistry 1995, 34, 10019–10026. [Google Scholar] [CrossRef]
- Miyao, M. Involvement of active oxygen species in degradation of the D1 protein under strong illumination in isolated subcomplexes of photosystem II. Biochemistry 1994, 33, 9722–9730. [Google Scholar] [CrossRef] [PubMed]
- Nixon, P.J.; Michoux, F.; Yu, J.; Boehm, M.; Komenda, J. Recent advances in understanding the assembly and repair of photosystem II. Ann. Bot. 2010, 106, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kale, R.; Hebert, A.E.; Frankel, L.K.; Sallans, L.; Bricker, T.M.; Pospíšil, P. Amino acid oxidation of the D1 and D2 proteins by oxygen radicals during photoinhibition of Photosystem II. Proc. Natl. Acad. Sci. USA 2017, 114, 2988–2993. [Google Scholar] [CrossRef] [PubMed]
- Terashima, I.; Funayama, S.; Sonoike, K. The site of photoinhibition in leaves of Cucumis sativus L. at low temperatures is photosystem I, not photosystem II. Planta 1994, 193, 300–306. [Google Scholar] [CrossRef]
- Sonoike, K. Degradation of psaB gene product, the reaction center subunit of photosystem I, is caused during photoinhibition of photosystem I: Possible involvement of active oxygen species. Plant Sci. 1996, 115, 157–164. [Google Scholar] [CrossRef]
- Tiwari, A.; Mamedov, F.; Grieco, M.; Suorsa, M.; Jajoo, A.; Styring, S.; Tikkanen, M.; Aro, E.-M. Photodamage of iron-sulphur clusters in photosystem I induces non-photochemical energy dissipation. Nat. Plants 2016, 2, 16035. [Google Scholar] [CrossRef] [PubMed]
- Sonoike, K. Photoinhibition of photosystem I. Physiol. Plant. 2011, 142, 56–64. [Google Scholar] [CrossRef]
- Sejima, T.; Takagi, D.; Fukayama, H.; Makino, A.; Miyake, C. Repetitive short-pulse light mainly inactivates photosystem I in sunflower leaves. Plant Cell Physiol. 2014, 55, 1184–1193. [Google Scholar] [CrossRef]
- Tikkanen, M.; Grebe, S. Switching off photoprotection of photosystem I–a novel tool for gradual PSI photoinhibition. Physiol. Plant. 2018, 162, 156–161. [Google Scholar] [CrossRef]
- Blée, E. Phytooxylipins and plant defense reactions. Prog. Lipid Res. 1998, 37, 33–72. [Google Scholar] [CrossRef]
- Farmer, E.E.; Davoine, C. Reactive electrofile species. Curr. Opin. Plant Biol. 2007, 10, 380–386. [Google Scholar] [CrossRef]
- Mêne-Saffraneé, L.; Davoine, C.; Stolz, S.; Majcherczyk, P.; Farmer, E.E. Genetic removal of tri-unsaturated fatty acids suppresses developmental and molecular phenotypes of an Arabidopsis tocopherol-deficient mutant. Whole-body mapping of malondialdehyde pools in a complex eukaryote. J. Biol. Chem. 2007, 49, 35749–35756. [Google Scholar] [CrossRef] [PubMed]
- Mano, J.; Khorobrykh, S.; Matsui, K.; Iijima, Y.; Sakurai, N.; Suzuki, H.; Shibata, D. Acrolein is formed from trienoic fatty acids in chloroplast: A targeted metabolimics approach. Plant Biotechnol. 2014, 31, 535–543. [Google Scholar] [CrossRef]
- Farmer, E.E.; Mueller, M.J. ROS-mediated lipid peroxidation and RES-activated signaling. Annu. Rev. Plant. Biol. 2013, 64, 429–450. [Google Scholar] [CrossRef] [PubMed]
- Mêne-Saffraneé, L.; Dubugnon, L.; Chételat, A.; Stolz, S.; Gouhier-Darimont, C.; Farmer, E.E. Nonenzymatic oxidation of trienoic fatty acids contributes to reactive oxygen species management in Arabidopsis. J. Biol. Chem. 2009, 284, 1702–1708. [Google Scholar] [CrossRef]
- Kojima, K.; Oshita, M.; Nanjo, Y.; Kasai, K.; Tozawa, Y.; Hayashi, H.; Nishiyama, Y. Oxidation of elongation factor G inhibits the synthesis of the D1 protein of photosystem II. Mol. Microbiol. 2007, 65, 936–947. [Google Scholar] [CrossRef]
- Yutthanasirikul, R.; Nagano, T.; Jimbo, H.; Hihara, Y.; Kanamori, T.; Ueda, T.; Haruyama, T.; Konno, H.; Yoshida, K.; Hisabori, T.; et al. Oxidation of a cysteine residue in elongation factor EF-Tu reversibly inhibits translation in the cyanobacterium Synechocystis sp. PCC 6803. J. Biol. Chem. 2016, 291, 5860–5870. [Google Scholar] [CrossRef]
- Jimbo, H.; Yutthanasirikul, R.; Nagano, T.; Hisabori, T.; Hiharo, Y.; Nishiyama, Y. Oxidation of translation factor EF-Tu inhibits the repair of photosystem II. Plant Physiol. 2018, 176, 2691–2699. [Google Scholar] [CrossRef]
- Nishiyama, Y.; Allakhverdiev, S.I.; Yamamamoto, H.; Hayashi, H.; Murata, N. Singlet oxygen inhibits the repair of photosystem II by suppressing the translation elongation of the D1 protein in Synechocystis sp. PCC 6803. Biochemistry 2004, 43, 11321–11330. [Google Scholar] [CrossRef]
- Kaiser, W. The effect of hydrogen peroxide on CO2 fixation of isolated intact chloroplasts. Biochim. Biophys. Acta 1976, 440, 476–482. [Google Scholar] [CrossRef]
- Muthuramalingam, M.; Matros, A.; Scheibe, R.; Mock, H.-P.; Dietz, K.-J. The hydrogen peroxide-sensitive proteome of the chloroplast in vitro and in vivo. Front. Plant Sci. 2013, 4, 54. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.; Oldenburg, D.J.; Bendich, A.J. Molecular integrity of chloroplast DNA and mitochondrial DNA in mesophyll and bundle sheath cells of maize. Planta 2015, 241, 1221–1230. [Google Scholar] [CrossRef] [PubMed]
- Balasaraswathi, K.; Jayaveni, S.; Sridevi, J.; Sujatha, D.; Aaron, K.P.; Rose, C. Cr-induced cellular injury and necrosis in Glycine max L.: Biochemical mechanism of oxidative damage in chloroplast. Plant Physiol. Biochem. 2017, 118, 653–666. [Google Scholar] [CrossRef] [PubMed]
- Noctor, G.; Foyer, C.H. Intracellular redox compartmentation and ROS-related communication in regulation and signaling. Plant Physiol. 2016, 171, 1581–1592. [Google Scholar] [CrossRef] [PubMed]
- Foyer, C.H.; Ruban, A.V.; Noctor, G. Viewing oxidative stress through the lens of oxidative signalling rather than damage. Biochem. J. 2017, 474, 877–883. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, K.; Kanematsu, S.; Takabe, K.; Asada, K. Attachment of CuZn-superoxide dismutase to thylakoid membranes at the site of superoxide generation (PSI) in spinach chloroplasts: Detection by immuno-gold labeling after rapid freezing and substitution method. Plant Cell Physiol. 1995, 36, 565–573. [Google Scholar]
- Gray, B.; Carmichael, A.J. Kinetics of superoxide scavenging by dismutase enzymes and manganese mimics determined by electron spin resonance. Biochem. J. 1992, 281, 795–802. [Google Scholar] [CrossRef]
- Barnese, K.; Gralla, E.B.; Valentine, J.S.; Cabelli, D.E. Biologically relevant mechanism for catalytic superoxide removal by simple manganese compounds. Proc. Natl. Acad. Sci. USA 2012, 109, 6892–6897. [Google Scholar] [CrossRef]
- Giacomelli, L.; Masi, A.; Ripoll, D.R.; Lee, M.J.; van Wijk, K.J. Arabidopsis thaliana deficient in two chloroplast ascorbate peroxidases shows accelerated light-induced necrosis when levels of cellular ascorbate are low. Plant Mol. Biol. 2007, 65, 627–644. [Google Scholar] [CrossRef]
- Kangasjärvi, S.; Lepistö, A.; Hännikäinen, K.; Piippo, M.; Luomala, E.M.; Aro, E.M.; Rintamäki, E. Diverse roles for chloroplast stromal and thylakoid-bound ascorbate peroxidases in plant stress responses. Biochem. J. 2008, 412, 275–285. [Google Scholar] [CrossRef]
- Maruta, T.; Tanouchi, A.; Tamoi, M.; Yabuta, Y.; Yoshimura, K.; Ishikawa, T.; Shigeoka, S. Arabidopsis chloroplastic ascorbate peroxidase isoenzymes play a dual role in photoprotection and gene regulation under photooxidative stress. Plant Cell Physiol. 2010, 51, 190–200. [Google Scholar] [CrossRef] [PubMed]
- Shigeoka, S.; Ishikawa, T.; Tamoi, M.; Miyagawa, Y.; Takeda, T.; Yabuta, Y.; Yoshimura, K. Regulation and function of ascorbate peroxidase isoenzymes. J. Exp. Bot. 2002, 53, 1305–1319. [Google Scholar] [CrossRef]
- Pandey, P.; Singh, J.; Achary, V.M.M.; Reddy, M.K. Redox homeostasis via gene families of ascorbate-glutathione pathway. Front. Environ. Sci. 2015, 3, 25. [Google Scholar] [CrossRef]
- Awad, J.; Stotz, H.U.; Fekete, A.; Krischke, M.; Engert, C.; Havaux, M.; Berger, S.; Mueller, M.J. 2-Cysteine peroxiredoxins and thylakoid ascorbate peroxidase create a water-water cycle that is essential to protect the photosynthetic apparatus under high light stress conditions. Plant Physiol. 2015, 167, 1592–1603. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Ruiz, J.M.; Spínola, M.C.; Kirchsteiger, K.; Moreno, J.; Sahrawy, M.; Cejudo, F.J. Rice NTRC is a high-efficiency redox system for chloroplast protection against oxidative damage. Plant Cell 2006, 18, 2356–2368. [Google Scholar] [CrossRef]
- Bernal-Bayard, P.; Ojeda, V.; Hervás, M.; Cejudo, F.J.; Navarro, J.A.; Velázquez-Campoy, A.; Pérez-Ruiz, J.M. Molecular recognition in the interaction of chloroplast 2-Cys peroxiredoxin with NADPH-thioredoxin reductase C (NTRC) and thioredoxin x. FEBS Lett. 2014, 588, 4342–4347. [Google Scholar] [CrossRef]
- Pulido, P.; Spínola, M.C.; Kirchsteiger, K.; Guinea, M.; Pascual, M.B.; Sahrawy, M.; Sandalio, L.M.; Dietz, K.J.; González, M.; Cejudo, F.J. Functional analysis of the pathways for 2-Cys peroxiredoxin reduction in Arabidopsis thaliana chloroplasts. J. Exp. Bot. 2010, 61, 4043–4054. [Google Scholar] [CrossRef]
- Dietz, K.J. Peroxiredoxins in plants and cyanobacteria. Antioxid. Redox Signal. 2011, 15, 1129–1159. [Google Scholar] [CrossRef]
- Spínola, M.C.; Pérez-Ruiz, J.M.; Pulido, P.; Kirchsteiger, K.; Guinea, M.; González, M.; Cejudo, F.J. NTRC new ways of using NADPH in the chloroplast. Physiol. Plant. 2008, 133, 516–524. [Google Scholar] [CrossRef]
- Crawford, N.A.; Droux, M.; Kosower, N.S.; Buchanan, B.B. Evidence for function of the ferredoxin/thioredoxin system in the reductive activation of target enzymes of isolated intact chloroplasts. Arch. Biochem. Biophys. 1989, 271, 223–239. [Google Scholar] [CrossRef]
- Herbette, S.; Lenne, C.; Leblanc, N.; Julien, J.L.; Drevet, J.R.; Roeckel-Drevet, P. Two GPX-like proteins from Lycopersicon esculentum and Helianthus annuus are antioxidant enzymes with phospholipid hydroperoxide glutathione peroxidase and thioredoxin peroxidase activities. Eur. J. Biochem. 2002, 269, 2414–2420. [Google Scholar] [CrossRef] [PubMed]
- Jung, B.G.; Lee, K.O.; Lee, S.S.; Chi, Y.H.; Jang, H.H.; Kang, S.S.; Lee, K.; Lim, D.; Yoon, S.C.; Yun, D.J.; et al. A Chinese cabbage cDNA with high sequence identity to phospholipid hydroperoxide glutathione peroxidases encodes a novel isoform of thioredoxin-dependent peroxidase. J. Biol. Chem. 2002, 277, 12572–12578. [Google Scholar] [CrossRef] [PubMed]
- Chang, C.C.C.; Ślesak, I.; Jordá, L.; Sotnikov, A.; Melzer, M.; Miszalski, Z.; Mullineaux, P.M.; Parker, J.E.; Karpińska, B.; Karpiński, S. Arabidopsis chloroplastic glutathione peroxidases play a role in cross talk between photooxidative stress and immune responses. Plant Physiol. 2009, 150, 670–683. [Google Scholar] [CrossRef]
- Ruban, A.V. Nonphotochemical chlorophyll fluorescence quenching: Mechanism and effectiveness in protecting plants from photodamage. Plant Phys. 2016, 170, 1903–1916. [Google Scholar] [CrossRef] [PubMed]
- Malnoë, A. Photoinhibition or photoprotection of photosynthesis? Update on the (newly termed) sustained quenching component qH. Environ. Exp. Bot. 2018, 154, 123–133. [Google Scholar] [CrossRef]
- Liu, Z.; Yan, H.; Wang, K.; Kuang, T.; Zhang, J.; Gui, L.; An, X.; Chang, W. Crystal structure of spinach major light-harvesting complex at 2.72 Å resolution. Nature 2004, 428, 287–292. [Google Scholar] [CrossRef]
- Peterman, E.J.; Dukker, F.M.; van Grondelle, R.; van Amerongen, H. Chlorophyll a and carotenoid triplet states in light-harvesting complex II of higher plants. Biophys. J. 1995, 69, 2670–2678. [Google Scholar] [CrossRef]
- Barzda, V.; Peterman, E.J.; van Grondelle, R.; van Amerongen, H. The influence of aggregation on triplet formation in light-harvesting chlorophyll a/b pigment-protein complex II of green plants. Biochemistry 1998, 37, 546–551. [Google Scholar] [CrossRef]
- Mozzo, M.; Dall’Osto, L.; Hienerwadel, R.; Bassi, R.; Croce, R. Photoprotection in the antenna complexes of photosystem II: Role of individual xanthophylls in chlorophyll triplet quenching. J. Biol. Chem. 2008, 283, 6184–6192. [Google Scholar] [CrossRef]
- Croce, R.; Remelli, R.; Varotto, C.; Breton, J.; Bassi, R. The neoxanthin binding site of the major light harvesting complex (LHCII) from higher plants. FEBS Lett. 1999, 456, 1–6. [Google Scholar] [CrossRef]
- Caffarri, S.; Croce, R.; Breton, J.; Bassi, R. The major antenna complex of photosystem II has a xanthophyll binding site not involved in light harvesting. J. Biol. Chem. 2001, 276, 35924–35933. [Google Scholar] [CrossRef] [PubMed]
- Dall’Osto, L.; Holt, N.E.; Kaligotla, S.; Fuciman, M.; Cazzaniga, S.; Carbonera, D.; Frank, H.A.; Alric, J.; Bassi, R. Zeaxanthin protects plant photosynthesis by modulating chlorophyll triplet yield in specific light-harvesting antenna subunits. J. Biol. Chem. 2012, 287, 41820–41834. [Google Scholar] [CrossRef] [PubMed]
- Niyogi, K.K.; Grossman, A.R.; Björkman, O. Arabidopsis mutants define a central role for the xanthophyll cycle in the regulation of photosynthetic energy conversion. Plant Cell 1998, 10, 1121–1134. [Google Scholar] [CrossRef] [PubMed]
- Umena, Y.; Kawakami, K.; Shen, J.R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef]
- Santabarbara, S.; Agostini, A.; Casazza, A.P.; Zucchelli, G.; Carbonera, D. Carotenoid triplet states in photosystem II: Coupling with low-energy states of the core complex. Biochim. Biophys. Acta 2015, 1847, 262–275. [Google Scholar] [CrossRef]
- Fischer, B.B.; Hideg, É.; Krieger-Liszkay, A. Production, detection, and signaling of singlet oxygen in photosynthetic organisms. Antioxid. Redox Signal. 2013, 18, 2145–2162. [Google Scholar] [CrossRef]
- Di Mascio, P.; Devasagayam, T.P.; Kaiser, S.; Sies, H. Carotenoids, tocopherols and thiols as biological singlet molecular oxygen quenchers. Biochem. Soc. Trans. 1990, 18, 1054–1056. [Google Scholar] [CrossRef]
- Ramel, F.; Birtic, S.; Cuiné, S.; Triantaphylidès, C.; Ravanat, J.L.; Havaux, M. Chemical quenching of singlet oxygen by carotenoids in plants. Plant Physiol. 2012, 158, 1267–1278. [Google Scholar] [CrossRef]
- Ramel, F.; Birtic, S.; Ginies, C.; Soubigou-Taconnat, L.; Triantaphylidès, C.; Havaux, M. Carotenoid oxidation products are stress signals that mediate gene responses to singlet oxygen in plants. Proc. Natl. Acad. Sci. USA 2012, 109, 5535–5540. [Google Scholar] [CrossRef]
- Johnson, M.P.; Havaux, M.; Triantaphylidès, C.; Ksas, B.; Pascal, A.A.; Robert, B.; Davison, P.A.; Ruban, A.V.; Horton, P. Elevated zeaxanthin bound to oligomeric LHCII enhances the resistance of Arabidopsis to photooxidative stress by a lipid-protective, antioxidant mechanism. J. Biol. Chem. 2007, 282, 22605–22618. [Google Scholar] [CrossRef]
- Foyer, C.H.; Trebst, A.; Noctor, G. Signaling and integration of defense functions of tocopherol, ascorbate and glutathione. In Photoprotection, Photoinhibition, Gene Regulation, and Environment. Advances in Photosynthesis and Respiration; Demmig-Adams, B., Adams, W.W., Mattoo, A.K., Eds.; Springer: Dordrecht, The Netherlands, 2008; Volume 21, pp. 241–268. [Google Scholar]
- Neely, W.C.; Martin, J.M.; Barker, S.A. Products and relative reaction rates of the oxidation of tocopherols with singlet molecular oxygen. Photochem. Photobiol. 1988, 48, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Krieger-Liszkay, A.; Trebst, A. Tocopherol is the scavenger of singlet oxygen produced by the triplet states of chlorophyll in the PSII reaction centre. J. Exp. Bot. 2006, 57, 1677–1684. [Google Scholar] [CrossRef] [PubMed]
- Nagai, S.; Ohara, K.; Mukai, K. Kinetic study of the quenching reaction of singlet oxygen by flavonoids in ethanol solution. J. Phys. Chem. B 2005, 109, 4234–4240. [Google Scholar] [CrossRef] [PubMed]
- Agati, G.; Matteini, P.; Goti, A.; Tattini, M. Chloroplast-located flavonoids can scavenge singlet oxygen. New Phytol. 2007, 174, 77–89. [Google Scholar] [CrossRef] [PubMed]
- Ferretti, U.; Ciura, J.; Ksas, B.; Rác, M.; Sedlářová, M.; Kruk, J.; Havaux, M.; Pospíšil, P. Chemical quenching of singlet oxygen by plastoquinols and their oxidation products in Arabidopsis. Plant J. 2018, 95, 848–861. [Google Scholar] [CrossRef]
- Affek, H.P.; Yakir, D. Protection by isoprene against singlet oxygen in leaves. Plant Physiol. 2002, 129, 269–277. [Google Scholar] [CrossRef]
- Velikova, V.; Edreva, A.; Loreto, F. Endogenous isoprene protects Phragmites australis leaves against singlet oxygen. Physiol. Plant. 2004, 122, 219–225. [Google Scholar] [CrossRef]
- Mignolet-Spruyt, L.; Xu, E.; Idänheimo, N.; Hoeberichts, F.; Mühlenbock, P.; Brosché, M.; Van Breusegem, F.; Kangasjärvi, J. Spreading the news: Subcellular and organellar reactive oxygen species production and signalling. J. Exp. Bot. 2016, 67, 3831–3844. [Google Scholar] [CrossRef]
- Crawford, T.; Lehotai, N.; Strand, Å. The role of retrograde signals during plant stress responses. J. Exp. Bot. 2018, 69, 2783–2795. [Google Scholar] [CrossRef]
- Dogra, V.; Rochaix, J.D.; Kim, C. Singlet oxygen-triggered chloroplast-to-nucleus retrograde signalling pathways: An emerging perspective. Plant Cell Environ. 2018, 41, 1727–1738. [Google Scholar] [CrossRef]
- Mullineaux, P.M.; Exposito-Rodriguez, M.; Laissue, P.P.; Smirnoff, N. ROS-dependent signalling pathways in plants and algae exposed to high light: Comparisons with other eukaryotes. Free Radic. Biol. Med. 2018, 122, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Borisova-Mubarakshina, M.M.; Vetoshkina, D.V.; Ivanov, B.N. Antioxidant and signaling functions of the plastoquinone pool in higher plants. Physiol. Plant. 2019, 166, 181–198. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.J.; Wesemann, C.; Wegener, M.; Seidel, T. Toward an integrated understanding of retrograde control of photosynthesis. Antioxid. Redox Signal. 2019, 30, 1186–1205. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Apel, K. Dose-dependent effects of 1O2 in chloroplasts are determined by its timing and localization of production. J. Exp. Bot. 2019, 70, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Černý, M.; Habánová, H.; Berka, M.; Luklová, M.; Brzobohatý, B. Hydrogen peroxide: Its role in plant biology and crosstalk with signalling networks. Int. J. Mol. Sci. 2018, 19, 2812. [Google Scholar] [CrossRef] [PubMed]
- Lv, F.; Zhou, J.; Zeng, L.; Xing, D. β-cyclocitral upregulates salicylic acid signalling to enhance excess light acclimation in Arabidopsis. J. Exp. Bot. 2015, 66, 4719–4732. [Google Scholar] [CrossRef]
- D’Alessandro, S.; Mizokami, Y.; Légeret, B.; Havaux, M. The apocarotenoid β-cyclocitric acid elicits drought tolerance in plants. iScience 2019, 19, 461–473. [Google Scholar] [CrossRef]
- Havaux, M. Small molecules: From structural diversity to signaling and regulatory roles. Carotenoid oxidation products as stress signals in plants. Plant J. 2013, 79, 597–606. [Google Scholar] [CrossRef]
- Shao, N.; Duan, G.Y.; Bock, R. A mediator of singlet oxygen responses in Chlamydomonas reinhardtii and Arabidopsis identified by a luciferase-based genetic screen in algal cells. Plant Cell 2013, 25, 4209–4226. [Google Scholar] [CrossRef]
- Shumbe, L.; D’alessandro, S.; Shao, N.; Chevalier, A.; Ksas, B.; Block, R.; Havaux, M. METHYLENE BLUE SENSITIVITY 1 (MBS1) is required for acclimation of Arabidopsis to singlet oxygen and acts downstream of β-cyclocitral. Plant Cell Environ. 2017, 40, 216–226. [Google Scholar] [CrossRef]
- Shumbe, L.; Bott, R.; Havaux, M. Dihydroactinidiolide, a high light-induced β-carotene derivative that can regulate gene expression and photoacclimation in Arabidopsis. Mol. Plant 2014, 7, 1248–1251. [Google Scholar] [CrossRef] [PubMed]
- Wagner, D.; Przybyla, D.; Op den Camp, R.; Kim, C.; Landgraf, F.; Lee, K.P.; Würsch, M.; Laloi, C.; Nater, M.; Hideg, E.; et al. The genetic basis of singlet oxygen-induced stress responses of Arabidopsis thaliana. Science 2004, 306, 1183–1185. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.P.; Kim, C.; Landgraf, F.; Apel, K. EXECUTER1- and EXECUTER2-dependent transfer of stress-related signals from the plastid to the nucleus of Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2007, 104, 10270–10275. [Google Scholar] [CrossRef] [PubMed]
- Dogra, V.; Li, M.; Singh, S.; Li, M.; Kim, C. Oxidative post-translational modification of EXECUTER1 is required for singlet oxygen sensing in plastids. Nat. Commun. 2019, 10, 2834. [Google Scholar] [CrossRef] [PubMed]
- Dogra, V.; Duan, J.; Lee, K.P.; Lv, S.; Liu, R.; Kim, C. FtsH2-dependent proteolysis of EXECUTER1 is essential in mediating singlet oxygen-triggered retrograde signaling in Arabidopsis thaliana. Front. Plant Sci. 2017, 8, 1145. [Google Scholar] [CrossRef] [PubMed]
- Danon, A.; Coll, N.S.; Apel, K. Cryptochrome-1-dependent execution of programmed cell death induced by singlet oxygen in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2006, 103, 17036–17041. [Google Scholar] [CrossRef]
- Carmody, M.; Crisp, P.A.; d’Alessandro, S.; Ganguly, D.; Gordon, M.; Havaux, M.; Albrecht-Borth, V.; Pogson, B.J. Uncoupling high light responses from singlet oxygen retrograde signaling and spatial-temporal systemic acquired acclimation. Plant Physiol. 2016, 171, 1734–1749. [Google Scholar] [CrossRef]
- Ramel, F.; Ksas, B.; Havaux, M. Jasmonate: A decision maker between cell death and acclimation in the response of plants to singlet oxygen. Plant Signal. Behav. 2013, 8, e26655. [Google Scholar] [CrossRef]
- Shumbe, L.; Chevalier, A.; Legeret, B.; Taconnat, L.; Monnet, F.; Havaux, M. Singlet oxygen-induced cell death in Arabidopsis under high-light stress is controlled by OXI1 kinase. Plant Physiol. 2016, 170, 1757–1771. [Google Scholar] [CrossRef]
- D’Alessandro, S.; Havaux, M. Sensing β-carotene oxidation in photosystem II to master plant stress tolerance. New Phytol. 2019, 223, 1776–1783. [Google Scholar] [CrossRef]
- Armond, P.A.; Arntzen, C.J. Localization and characterization of photosystem II in grana and stroma lamellae. Plant Physiol. 1977, 59, 398–404. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Kim, C.; Xu, X.; Piskurewicz, U.; Dogra, V.; Singh, S.; Mahler, H.; Apel, K. Singlet oxygen- and EXECUTER1-mediated signaling is initiated in grana margins and depends on the protease FtsH2. Proc. Natl. Acad. Sci. USA 2016, 113, E3792–E3800. [Google Scholar] [CrossRef] [PubMed]
- Järvi, S.; Suorsa, M.; Aro, E.M. Photosystem II repair in plant chloroplasts–Regulation, assisting proteins and shared components with photosystem II biogenesis. Biochim. Biophys. Acta 2015, 1847, 900–909. [Google Scholar] [CrossRef] [PubMed]
- Feierabend, J.; Dahne, S. Fate of the porphyrin cofactors during the light-dependent turnover of catalase and of the photosystem II reaction center protein D1 in mature rye leaves. Planta 1996, 198, 413–422. [Google Scholar] [CrossRef]
- Hernandez-Prieto, M.A.; Tibiletti, T.; Abasova, L.; Kirilovsky, D.; Vass, I.; Funk, C. The small CAB-like proteins of the cyanobacterium Synechocystis sp. PCC 6803: Their involvement in chlorophyll biogenesis for Photosystem II. Biochim. Biophys. Acta 2011, 1807, 1143–1151. [Google Scholar] [CrossRef]
- Zaltsman, A.; Feder, A.; Adam, Z. Developmental and light effects on the accumulation of FtsH protease in Arabidopsis chloroplasts: Implications for thylakoid formation and photosystem II maintenance. Plant J. 2005, 42, 609–617. [Google Scholar] [CrossRef]
- Stael, S.; Kmiecik, P.; Willems, P.; Van Der Kelen, K.; Coll, N.S.; Teige, M.; Van Breusegem, F. Plant innate immunity–sunny side up? Trends Plant Sci. 2015, 20, 3–11. [Google Scholar] [CrossRef]
- Mur, L.A.; Aubry, S.; Mondhe, M.; Kingston-Smith, A.; Gallagher, J.; Timms-Taravella, E.; James, C.; Papp, I.; Hörtensteiner, S.; Thomas, H.; et al. Accumulation of chlorophyll catabolites photosensitizes the hypersensitive response elicited by Pseudomonas syringae in Arabidopsis. New Phytol. 2010, 188, 161–174. [Google Scholar] [CrossRef]
- Tarahi Tabrizi, S.; Sawicki, A.; Zhou, S.; Luo, M.; Willows, R.D. GUN4-Protoporphyrin IX is a singlet oxygen generator with consequences for plastid retrograde signaling. J. Biol. Chem. 2016, 291, 8978–8984. [Google Scholar] [CrossRef]
- Kato, Y.; Hyodo, K.; Sakamoto, W. The photosystem II repair cycle requires FtsH turnover through the EngA GTPase. Plant Physiol. 2018, 178, 596–611. [Google Scholar] [CrossRef]
- de Torres Zabala, M.; Littlejohn, G.; Jayaraman, S.; Studholme, D.; Bailey, T.; Lawson, T.; Tillich, M.; Licht, D.; Bölter, B.; Delfino, L.; et al. Chloroplasts play a central role in plant defence and are targeted by pathogen effectors. Nat. Plants 2015, 1, 15074. [Google Scholar] [CrossRef] [PubMed]
- Järvi, S.; Isojärvi, J.; Kangasjärvi, S.; Salojärvi, J.; Mamedov, F.; Suorsa, M.; Aro, E.M. Photosystem II repair and plant immunity: Lessons learned from Arabidopsis mutant lacking the THYLAKOID LUMEN PROTEIN 18.3. Front. Plant Sci. 2016, 7, 405. [Google Scholar] [CrossRef] [PubMed]
- Exposito-Rodriguez, M.; Laissue, P.P.; Yvon-Durocher, G.; Smirnoff, N.; Mullineaux, P.M. Photosynthesis dependent H2O2 transfer from chloroplasts to nuclei provides a high-light signalling mechanism. Nat. Commun. 2017, 8, 49. [Google Scholar] [CrossRef] [PubMed]
- Mubarakshina-Borisova, M.M.; Kozuleva, M.A.; Rudenko, N.N.; Naydov, I.A.; Klenina, I.B.; Ivanov, B.N. Photosynthetic electron flow to oxygen and diffusion of hydrogen peroxide through the chloroplast envelope via aquaporins. Biochim. Biophys. Acta 2012, 1817, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Sadhukhan, A.; Kobayashi, Y.; Nakano, Y.; Iuchi, S.; Kobayashi, M.; Sahoo, L.; Koyama, H. Genomewide association study reveals that the aquaporin NIP1;1 contributes to variation in hydrogen peroxide sensitivity in Arabidopsis thaliana. Mol. Plant 2017, 10, 1082–1094. [Google Scholar] [CrossRef]
- Brunkard, J.O.; Runkel, A.M.; Zambryski, P.C. Chloroplasts extend stromules independently and in response to internal redox signals. Proc. Natl. Acad. Sci. USA 2015, 112, 10044–10049. [Google Scholar] [CrossRef]
- Caplan, J.L.; Kumar, A.S.; Park, E.; Padmanabhan, M.S.; Hoban, K.; Modla, S.; Czymmek, K.; Dinesh-Kumar, S.P. Chloroplast stromules function during innate immunity. Dev. Cell 2015, 34, 45–57. [Google Scholar] [CrossRef]
- Ivanov, B.N.; Borisova-Mubarakshina, M.M.; Kozuleva, M.A. Formation mechanisms of superoxide radical and hydrogen peroxide in chloroplasts, and factors determining the signalling by hydrogen peroxide. Funct. Plant Biol. 2017, 45, 102–110. [Google Scholar] [CrossRef]
- Willems, P.; Mhamdi, A.; Stael, S.; Storme, V.; Kerchev, P.; Noctor, G.; Gevaert, K.; Van Breusegem, F. The ROS wheel: Refining ROS transcriptional footprints. Plant Physiol. 2016, 171, 1720–1733. [Google Scholar] [CrossRef]
- Sewelam, N.; Jaspert, N.; Van Der Kelen, K.; Tognetti, N.B.; Schmitz, J.; Frerigmann, H.; Stahl, E.; Zeier, J.; Van Breusegem, F.; Maurino, V.G. Spatial H2O2 signaling specificity: H2O2 from chloroplasts and peroxisomes modulates the plant transcriptome differentially. Mol. Plant 2014, 7, 1191–1210. [Google Scholar] [CrossRef]
- Wang, W.H.; He, E.M.; Chen, J.; Guo, Y.; Chen, J.; Liu, X.; Zheng, H.L. The reduced state of the plastoquinone pool is required for chloroplast-mediated stomatal closure in response to calcium stimulation. Plant J. 2016, 86, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Borisova-Mubarakshina, M.M.; Ivanov, B.N.; Vetoshkina, D.V.; Lubimov, V.Y.; Fedorchuk, T.P.; Naydov, I.A.; Kozuleva, M.A.; Rudenko, N.N.; Dall’Osto, L.; Cazzaniga, S.; et al. Long-term acclimatory response to exess excitation energy: Evidence for a role of hydrogen peroxide in the regulation of photosystem II antenna size. J. Exp. Bot. 2015, 66, 7151–7164. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.X.; Mabbitt, P.D.; Phua, S.Y.; Mueller, J.W.; Nisar, N.; Gigolashvili, T.; Stroeher, E.; Grassl, J.; Arlt, W.; Estavillo, G.M.; et al. Sensing and signaling of oxidative stress in chloroplasts by inactivation of the SAL1 phosphoadenosine phosphatase. Proc. Natl. Acad. Sci. USA 2016, 113, E4567–E4576. [Google Scholar] [CrossRef] [PubMed]
- Estavillo, G.M.; Crisp, P.A.; Pornsiriwong, W.; Wirtz, M.; Collinge, D.; Carrie, C.; Giraud, E.; Whelan, J.; David, P.; Javot, H.; et al. Evidence for a SAL1-PAP chloroplast retrograde pathway that functions in drought and high light signaling in Arabidopsis. Plant Cell 2011, 23, 3992–4012. [Google Scholar] [CrossRef] [PubMed]
- Gigolashvili, T.; Geier, M.; Ashykhmina, N.; Frerigmann, H.; Wulfert, S.; Krueger, S.; Mugford, S.G.; Kopriva, S.; Haferkamp, I.; Flügge, U.-I. The arabidopsis thylakoid ADP/ATP carrier TAAC has an additional role in supplying plastidic phosphoadenosine 5′-phosposulfate to the cytosol. Plant Cell 2012, 24, 4187–4204. [Google Scholar] [CrossRef] [PubMed]
- Miao, Y.; Lv, D.; Wang, P.; Wang, X.C.; Chen, J.; Miao, C.; Song, C.P. An Arabidopsis glutathione peroxidase functions as both a redox transducer and a scavenger in abscisic acid and drought stress responses. Plant Cell 2006, 18, 2749–2766. [Google Scholar] [CrossRef]
- Miller, G.; Mittler, R. Could heat shock transcription factors function as hydrogen peroxide sensors in plants? Ann. Bot. 2006, 98, 279–288. [Google Scholar] [CrossRef]
- Bheri, M.; Pandey, G.K. Protein phosphatases meet reactive oxygen species in plant signaling networks. Environ. Exp. Bot. 2019, 161, 26–40. [Google Scholar] [CrossRef]
- Laloi, C.; Stachowiak, M.; Pers-Kamczyc, E.; Warzych, E.; Murgia, I.; Apel, K. Cross-talk between singlet oxygen- and hydrogen peroxide-dependent signaling of stress responses in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2007, 104, 672–677. [Google Scholar] [CrossRef]
- Šimková, K.; Moreau, F.; Pawlak, P.; Vriet, C.; Baruah, A.; Alexandre, C.; Hennig, L.; Apel, K.; Laloi, C. Integration of stress-related and reactive oxygen species-mediated signals by Topoisomerase VI in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2012, 109, 16360–16365. [Google Scholar] [CrossRef]
- Rentel, M.C.; Lecourieux, D.; Ouaked, F.; Usher, S.L.; Petersen, L.; Okamoto, H.; Knight, H.; Peck, S.C.; Grierson, C.S.; Hirt, H.; et al. OXI1 kinase is necessary for oxidative burst-mediated signalling in Arabidopsis. Nature 2004, 427, 858–861. [Google Scholar] [CrossRef] [PubMed]
- Gadjev, I.; Vanderauwera, S.; Gechev, T.S.; Laloi, C.; Minkov, I.N.; Shulaev, V.; Apel, K.; Inzé, D.; Mittler, R.; Van Breusegem, F. Transcriptomic footprints disclose specificity of reactive oxygen species signaling in Arabidopsis. Plant Physiol. 2006, 141, 436–445. [Google Scholar] [CrossRef]
- Scarpeci, T.E.; Zanor, M.I.; Carrillo, N.; Mueller-Roeber, B.; Valle, E.M. Generation of superoxide anion in chloroplasts of Arabidopsis thaliana during active photosynthesis: A focus on rapidly induced genes. Plant Mol. Biol. 2008, 66, 361–378. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.J.; Choi, H.J.; Moon, M.E.; Chi, Y.T.; Ji, K.Y.; Choi, D. Superoxide serves as a putative signal molecule for plant cell division: Overexpression of CaRLK1 promotes the plant cell cycle via accumulation of O2− and decrease in H2O2. Physiol. Plant. 2017, 159, 228–243. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Tran, T.; Padilla Marcia, C.S.; Braun, D.M.; Goggin, F.L. Superoxide-responsive gene expression in Arabidopsis thaliana and Zea mays. Plant Physiol. Biochem. 2017, 117, 51–60. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
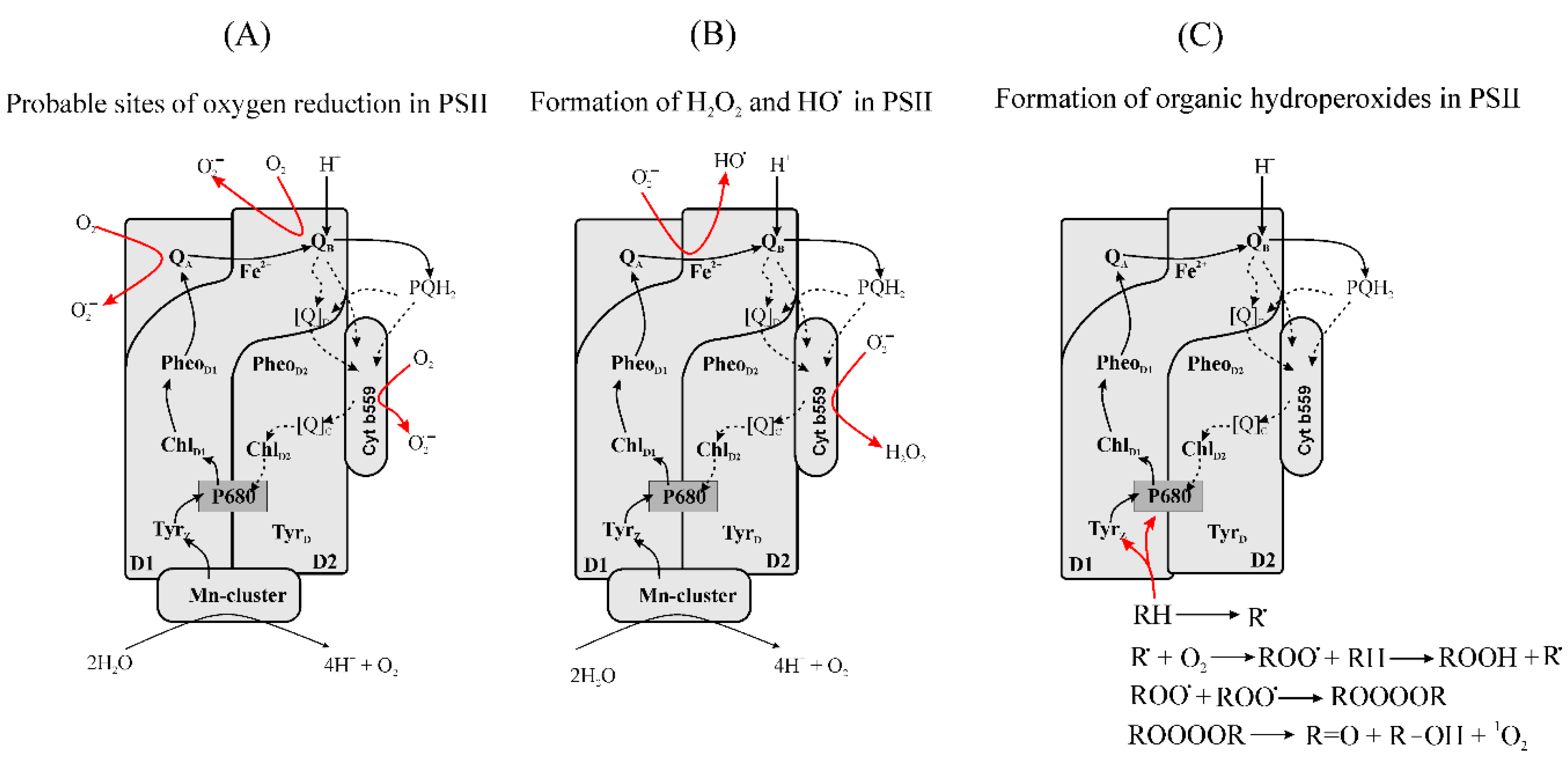{kind=link}
{kind=link}
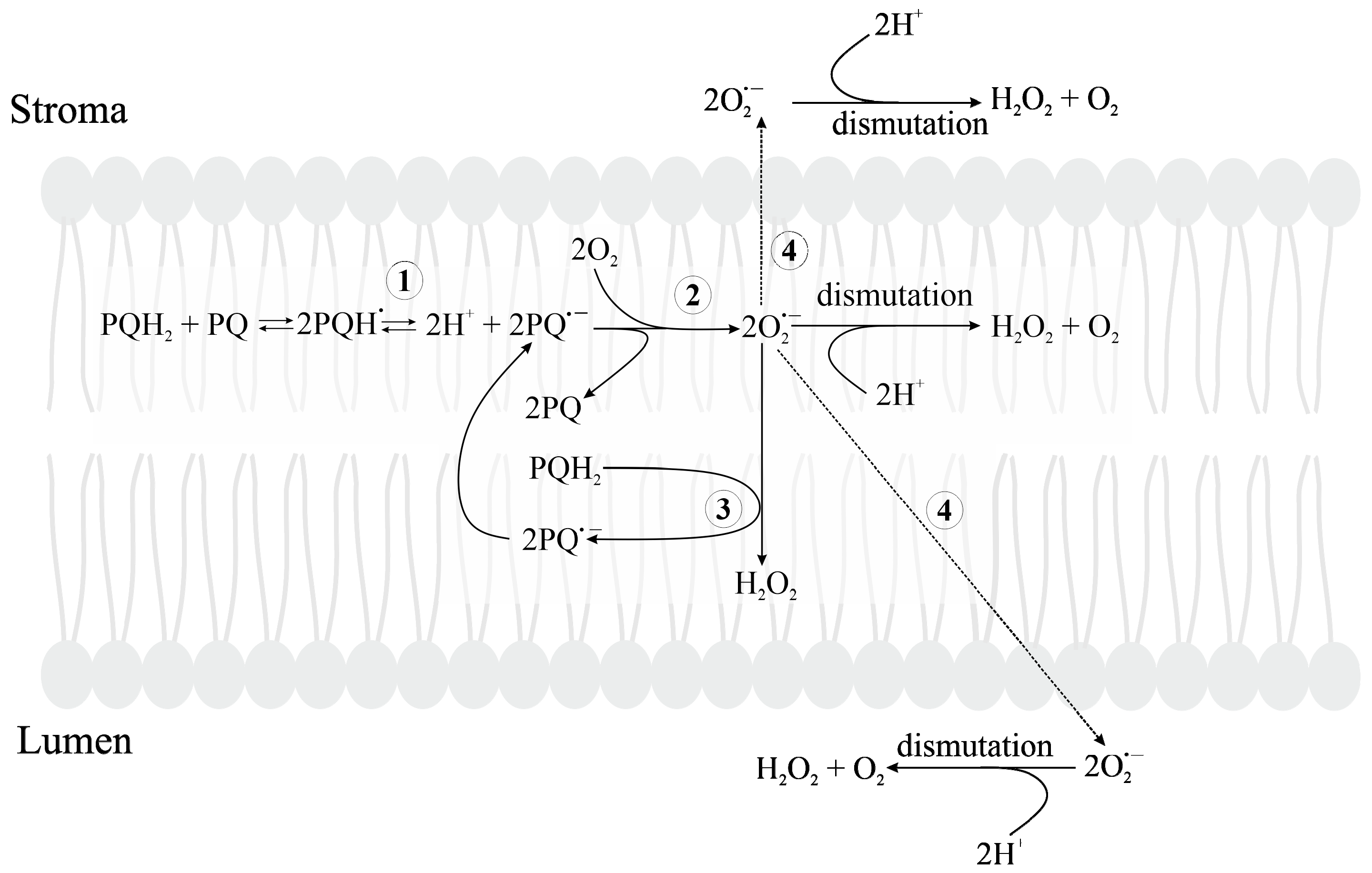{kind=link}
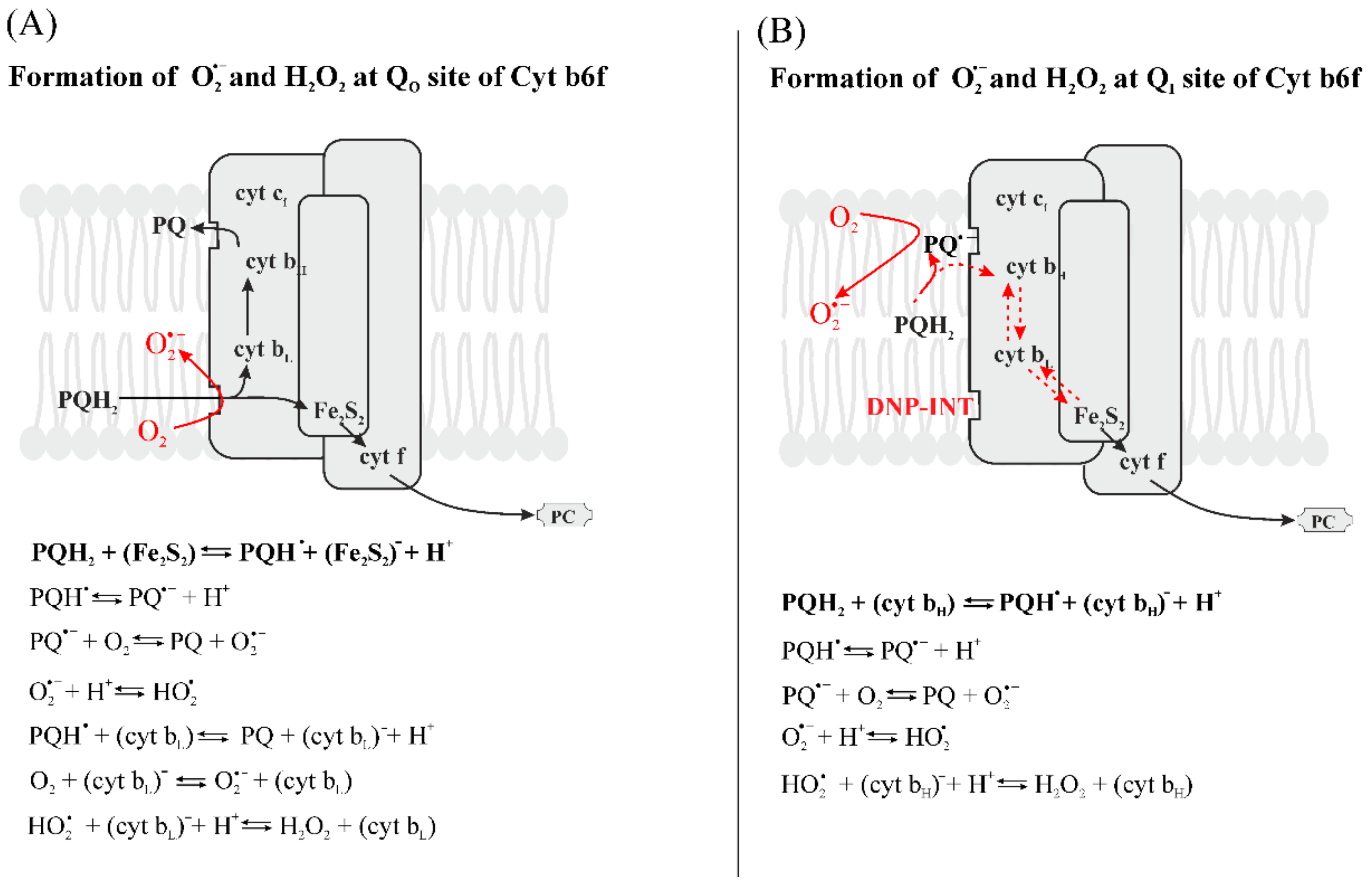{kind=link}
| Radicals | Non-Radicals |
|---|---|
| Reactive oxygen species | |
| Superoxide anion radical, O2•− | Singlet oxygen, 1O2 |
| Hydroperoxyl radical, HO2• | Hydrogen peroxide, H2O2 |
| Hydroxyl radical, HO• | Ozone, O3 |
| ROS derivatives and reactive nitrogen species | |
| Peroxyl radical, ROO• | Organic peroxides, ROOH |
| Alkoxyl radical, RO• | Peroxynitrite ion, ONOO− |
| Nitric Oxide, NO• | Alkyl peroxynitrite, ROONO |
| Nitrogen dioxide, NO2• | |
| Redox Active Cofactors | Midpoint Redox Potential vs. Normal Hydrogen Electrode (NHE), mV | Lifetime, s | Remarks |
|---|---|---|---|
| Pheo/Pheo− | ≈−610 [220,221] | (2–5) × 10−10 [222] | Reoxidation of Pheo− via forward electron transfer to QA |
| −588 [219] | |||
| −505 [223] | (4–30) × 10−8 [222] | Reoxidation of Pheo− via recombination of [P680+Pheo−] | |
| QA/QA− | −80–−200 [224,225] | (0.1–0.2) × 10−3 [224,226] | Reoxidation of QA− via forward electron transfer to QB |
| Shift from −145 to −70 [227] | (0.3–0.5) × 10−3 [224,226,228] | Removal of HCO3− bound to acceptor side of PSII. | |
| (2–4.6) × 10−3 [226,228,229] | Reoxidation of QA− via forward electron transfer to QB− and protonation. | ||
| (0.2–2) × 10−1 [222] | Reoxidation of QA− by PQ that binds to an empty QB site. | ||
| 1–2 [226] | Reoxidation of QA− via charge recombination with oxidized TyrZ. | ||
| Reoxidation of QA− via charge recombination with S2 | |||
| QA−/QA2− | −500 [230] | Double reduction achieved either by chemical treatment or by strong illumination in anaerobic conditions. No doubly reduced QA accumulates during aerobic light treatment. | |
| QB/QB− | −45–−60 [220,231,232] | (0.3–0.5) × 10−3 [224,226,228] | Reduction of QB− via electron transfer from QA− |
| >0.4 [222] | Reoxidation of QB− via charge recombination with oxidized TyrZ. | ||
| 30 [222] | Reoxidation of QB− via charge recombination with S2 | ||
| QB/QBH | 100 [220] | (0.3–0.5) × 10−3 [224,226,228] | Assuming the same time as for QB− |
| >0.4 [222] | |||
| 30 [222] | |||
| QB−/QB2− | −200–−464 [233] | ||
| QBH/QBH− | 290–373 [233] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khorobrykh, S.; Havurinne, V.; Mattila, H.; Tyystjärvi, E. Oxygen and ROS in Photosynthesis. Plants 2020, 9, 91. https://doi.org/10.3390/plants9010091
Khorobrykh S, Havurinne V, Mattila H, Tyystjärvi E. Oxygen and ROS in Photosynthesis. Plants. 2020; 9(1):91. https://doi.org/10.3390/plants9010091
Chicago/Turabian StyleKhorobrykh, Sergey, Vesa Havurinne, Heta Mattila, and Esa Tyystjärvi. 2020. "Oxygen and ROS in Photosynthesis" Plants 9, no. 1: 91. https://doi.org/10.3390/plants9010091
APA StyleKhorobrykh, S., Havurinne, V., Mattila, H., & Tyystjärvi, E. (2020). Oxygen and ROS in Photosynthesis. Plants, 9(1), 91. https://doi.org/10.3390/plants9010091