Abstract
Seed, resulting from reproductive development, is the main nutrient source for human beings, and reproduction has been intensively studied through genetic, molecular, and epigenetic approaches. However, how different epigenetic pathways crosstalk and integrate to regulate seed development remains unknown. Here, we review the recent progress of epigenetic changes that affect chromatin structure, such as DNA methylation, polycomb group proteins, histone modifications, and small RNA pathways in regulating plant reproduction. In gametogenesis of flowering plants, epigenetics is dynamic between the companion cell and gametes. Cytosine DNA methylation occurs in CG, CHG, CHH contexts (H = A, C, or T) of genes and transposable elements, and undergoes dynamic changes during reproduction. Cytosine methylation in the CHH context increases significantly during embryogenesis, reaches the highest levels in mature embryos, and decreases as the seed germinates. Polycomb group proteins are important transcriptional regulators during seed development. Histone modifications and small RNA pathways add another layer of complexity in regulating seed development. In summary, multiple epigenetic pathways are pivotal in regulating seed development. It remains to be elucidated how these epigenetic pathways interplay to affect dynamic chromatin structure and control reproduction.
1. Introduction
Seed represents the major nutrient source for humans and domesticated animals and produces many important industrial polymers such as oils and starches. Seed is the product of a double fertilization event in angiosperms. Unlike mammals, plants feature a haploid gametophytic growth involving postmeiotic cell divisions before fertilization. In the flowering plants, a haploid megaspore after meiosis goes through three mitotic divisions to form an eight-nucleus seven-cell female gametophyte containing the egg, central cell, two synergids, and three antipodal cells. Two haploid nuclei fuse to make the diploid central cell nucleus in the female gametophyte [1,2]. In the male gametophyte, a haploid microspore after meiosis goes through a mitotic division to form one vegetative and one generative cell. The generative cell then divides mitotically into two haploid sperm cells. After a pollen grain settles on top of the pistil, the vegetative cell grows into a pollen tube and transports two sperms to ovules at the base of pistils. Double fertilization event occurs in the female gametophyte of the ovule. During fertilization, one sperm fertilizes the egg cell to generate a diploid embryo. The second sperm fertilizes the diploid central cell to generate a triploid endosperm, which supports embryo and seedling growth [1,2,3,4]. Embryogenesis in vascular plants generates a mature and functional embryo that can eventually grow into a whole plant after seed germination. During this process, primary plant tissues are differentiated and the basic body plan is formed [5]. The embryo goes through a series of stages morphologically characterized as globular, heart, torpedo, bent-cotyledon, and walking-stick stages in many plant species such as Arabidopsis [5,6]. After completing rapid cell divisions and differentiation, the young embryo shifts into a seed-filling and seed-maturation stage at which the seed volume expands and storage reserve is synthesized [7]. Seed maturation is an important process for seed quality and yield during late development.
DNA methylation usually refers to modification of cytosine to become 5-methylcytosine (5mC). Cytosine methylation regulates transposable element (TE) silencing, genomic imprinting, and gene transcription in mammals and plants. DNA methylation at CG dinucleotides is evolutionarily conserved in plants, mammals, and some fungi [8,9,10,11]. De novo DNA methyltransferase 3 (Dnmt3) initiates and DNA methyltransferase 1 (Dnmt1) maintains cytosine methylation in mammals [12,13]. Flowering plants have DNA methylation at CG, CHG, and CHH contexts (H = A, C, or T). In Arabidopsis, CG and CHG methylation is mainly maintained by DNA METHYLTRANSFERASE1 (MET1), an ortholog of Dnmt1 in mammals, and CHROMOMETHYLASE3 (CMT3), respectively [14,15,16,17,18,19]; CHH methylation is maintained by the de novo DNA methyltransferases DOMAINS REARRANGED METHYLTRANSFERASE1 and 2 (DRM1 and DRM2) and CMT2 [18,19,20,21,22]. DRM2 is also responsible for de novo methylation in all sequence contexts through the RNA-directed DNA methylation (RdDM) pathway mediated by RNA polymerase IV (Pol IV) that produces small interfering RNAs (siRNAs) [21,23].
Polycomb group (PcG) proteins were originally identified in Drosophila and are involved in silencing homeotic genes by maintaining the repressed chromatin state [24,25]. PcG proteins recognize and interact with polycomb response elements (PREs) that are scattered throughout the genome to silence expression of genes containing PREs and nearby genes in Drosophila [26]. Most PcG proteins form two major polycomb repressive complexes (PRC)—PRC1 and PRC2 [27]. PRC2 consists of four key components: enhancer of zeste (EZ), suppressor of zeste 12 (Suz12), extra sex combs (Esc), and the histone-binding protein p55 in Drosophila. Homologous components of PcG proteins have been identified and function in plant development, which will be reviewed later in this paper. The Arabidopsis homologs of PcG proteins in seed development were identified by genetic screening, and they include FERTILIZATION INDEPENDENT ENDOSPERM (FIE), FERTILIZATION INDEPENDENT SEED2 (FIS2), MEDEA (MEA), and MULTICOPY SUPPRESSOR OF IRA1 (MSI1) [28,29,30,31,32]. Mutations in these PcG genes result in endosperm proliferation in the absence of fertilization.
Histone modifications refer to post-translational covalent changes on histone tails that are essential for dynamic chromatin structure and many cellular processes [33,34,35,36]. To date, more than 100 different histone modifications have been identified, from intensively studied lysine methylation and acetylation, and serine and threonine phosphorylation to recently discovered modifications, such as crotonylation [37,38]. With advancement of recent technologies, such as chromatin immunoprecipitation with tiled microarray (ChIP chip) and next-generation sequencing, global patterns of histone modifications have been mapped in many organisms [39,40,41,42,43]. These research studies have shown that histone acetylation and methylation in genes are important for gene transcription. Specifically, histone modifications such as acetylation and methylation in gene-coding regions are involved in plant reproduction [39].
Imprinting refers to differential expression of alleles on the basis of their parent of origin, contributing to reproductive success in animals and plants [4,44,45,46]. Gene imprinting primarily happens in the plant endosperm [47,48], whereas it can occur in the embryo and other tissues throughout post-embryonic stages in mammals [49]. Imprinted genes affect endosperm growth and seed size in Arabidopsis and crop plants [50,51,52,53,54,55]. In addition, a few imprinted genes have been found to occur in the embryo in both Arabidopsis [56] and maize [47]. Mutations in some imprinted genes from one parental allele can cause seed abortion, whereas the mutation on the other parental allele has mostly no effect in plants [57]. On the other hand, mutations in many recently found imprinted genes have no obvious phenotypes, or functions of these genes in seed development remain to be discovered [58].
The RdDM pathway has been shown to be involved in imprinting in Arabidopsis [59]. Non-coding small RNAs (sRNAs) have important functions in growth and development in both mammals and plants [60]. sRNAs are generated from double-stranded RNAs through the activities of RNA DEPENDENT RNA POLYMERASE (RDR), DICER-LIKE (DCL), and ARGONAUTE (AGO) proteins [61,62]. The two main classes of sRNAs are siRNAs and microRNAs (miRNAs) that both negatively regulate gene expression by binding to complementary sequences of their target genes [61,63]. The 24 nt siRNAs can trigger DNA methylation through the RdDM pathway [60] and affect seed development [64]. Briefly, Pol IV transcribes non-coding single-stranded RNAs (ssRNAs) that will be synthesized to double-stranded RNAs (dsRNAs) by RDR2. Then, DCL3 cuts the dsRNAs [65] into 24 nt siRNAs, which are methylated at their 3′ end by HUA ENCHANCER1 (HEN1) and are loaded onto ARGONAUTE4 (AGO4) [66]. Pol V and KOW DOMAIN-CONTAINING TRANSCRIPTION FACTOR1 (KTF1) recruit AGO4 through the C-terminal domain of Pol V’s largest subunit, NUCLEAR RNA POLYMERASE E1 (NRPE1) [67,68,69]. In Arabidopsis, CLASSY (CLSY) 1–4, SUCROSE NONFERMENTING2 (SNF2)-related, and putative chromatin remodeling proteins are necessary for global DNA methylation [41]. The CLSY proteins regulate 24 nt siRNA generation and are important for Pol IV chromatin association [41]. Lastly, the siRNAs bound to AGO4 are thought to complementarily pair with the Pol V transcripts and recruit DRM2 to methylate at homologous genomic sites.
Recent evidence has shown that a non-canonical RDR6-dependent RdDM pathway exists in plants [70,71,72,73,74,75]. When a TE is transcribed by Pol II, the TE transcripts are converted into dsRNAs by RDR6. The dsRNAs are then processed into 21–22 nt siRNAs by DCL2 and DCL4. The 21–22 nt siRNAs are then loaded onto AGO1 and can subsequently target Pol V-scaffolding transcripts, resulting in de novo DNA methylation.
DNA methylation was first shown to be crucial for seed development by regulating proper expression of genes important for cell identity and hormone gradient establishment [76]. Since then, substantial progress has been made in epigenetic control of gametogenesis, embryogenesis, and seed maturation through epigenetic and epigenomic approaches. With rapid advancement of this field in recent years, it is challenging to summarize all results, along with sometimes seemingly contradictory results. The authors hope that this review summarizes the major progress during the last decade and stimulates discussion about future perspectives in epigenetic regulation of seed development.
2. Differential DNA Methylation in the Egg and Central Cell of the Female Gametophyte
The female gametophyte is formed from meiosis of a diploid megaspore mother cell followed by three mitotic divisions. The mature female gametophyte contains seven cells and eight nuclei including an egg cell and diploid central cell, which give rise to the embryo and endosperm after fertilization by the sperm cells, respectively. Until recently, little was known about DNA methylation profiles in a specific cell type in the female gametophyte due to difficulty in isolating abundant amount of genomic DNA from the specific cell type. By studying whole genome methylation in the central cell of Arabidopsis and rice, as well as the egg cell in rice, Park et al. found that DNA demethylation is initiated in the central cell in both Arabidopsis and rice, and demethylation requires DEMETER (DME), a DNA glycosylase in Arabidopsis [77]. In the rice female gametophyte, DNA methylation in the central cell is approximately 39.4% CG, 18.2% CHG, and 6.3% CHH, lower than 41.9% CG, 26.2% CHG, and 7.4% CHH in the egg cell, which is likely due to demethylation in the central cell [77]. In the central cell of Arabidopsis, global CG methylation levels (26.6%) are slightly lower than those in the sperm (31.3%), but higher than those in the endosperm (20.9%); CHG methylation levels (12.6%) are similar to those in the sperm (12.5%), and slightly higher than those in the endosperm (8.9%); CHH methylation levels (3.7%) are significantly higher than those in the sperm (1.6%–2.1%) and endosperm (2.8%) (Figure 1). Without considering variations in different experiments, CG, CHG, and CHH methylation in reproductive tissues and cells except the endosperm (inflorescence, vegetative, and sperm cells; central cells; and embryos) is higher than that in somatic vegetative tissues (shoots, rosette leaves, and seedlings, except the sample of “Rosette leaf 2”) in Arabidopsis (Figure 1; no egg cell methylome data available). Due to DME activity in the central cell, maternal hypomethylation of some TEs can cause small RNA expression in the endosperm, which might be moved to the embryo in order to methylate the TEs sharing homology or nearby genes via RdDM, thus fortifying the silencing of TEs in the embryo that passes its genetic information to the next generation [78].
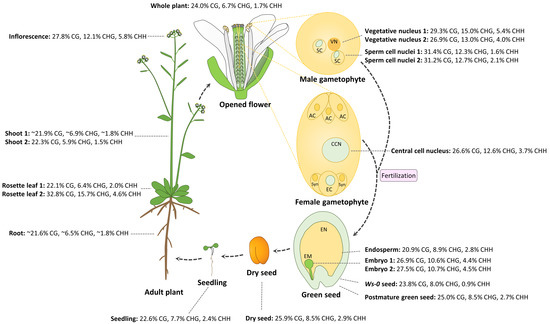
Figure 1.
Dynamic DNA methylation patterns in the life cycle of Arabidopsis. DNA methylation percentage in each of CG, CHG, and CHH contexts represent the number of methylated cytosine sites relative to all sequenced cytosine sites in the same context. Abbreviations are as follows: vegetative nucleus (VN), sperm cell (SC), antipodal cell (AC), central cell nuclei (CCN), egg cell (EC), synergid cell (Syn), embryo (EM), endosperm (EN). Materials and references for methylation data are listed as follows: all materials were from Col-0 wild type except green seed from Ws-0. Data were from the following: whole plant [79]; shoot 1 and root [80]; shoot 2 [81]; rosette leaf 1 [82]; rosette leaf 2 [83]; inflorescence, sperm cell nuclei 1, vegetative nucleus 1, and embryo 2 [84]; sperm cell nuclei 2 and vegetative nucleus 2 [78]; central cell nucleus (24 h after stage 12 flower) and seedling (2 week old) [77]; endosperm and embryo 1 at mid-torpedo to early seed maturation stage (7–9 days after pollination (DAP)) [85]; Ws-0 green seed at globular stage, postmature green seed (Col-0), and dry seed (Col-0) [86].
DNA methylation and demethylation can regulate expression of imprinted genes in Arabidopsis. MEA is imprinted and critical for seed development [57]. Expression of the maternal MEA allele is antagonistically controlled by MET1 and DME in the Arabidopsis central cell [53,87,88]. MET1 represses maternal MEA expression by adding the methyl group to cytosine residues in the maternal MEA promoter, whereas DME activates maternal MEA expression through DNA demethylation in the MEA promoter region (Figure 2). The antagonism between MET1 and DME also regulates other imprinted genes in the late endosperm development, such as FLOWERING WAGENINGEN (FWA), homeodomain leucine zipper transcription factor, and FIS2 [54,89]. It has been reported that a 200 bp MEA cis-regulatory region termed the imprinting control region (ICR) is required and sufficient for MEA imprinted expression by analyzing expression of the transgene pMEA:GUS, encoding β-glucuronidase (GUS) with the MEA promoter [90]. The authors suggested that maternal expression of MEA is indirectly controlled by antagonism between MET1 and DME, and proposed a model that maternal MEA expression depends on a yet unidentified positive transcription activator that is a direct target of MET1 and DME antagonism [90]. In short, it is critical to establish different methylation profiles in the egg and central cell during female gametogenesis, which are fertilized by sperm cells and develop into embryo and endosperm during embryogenesis, respectively.
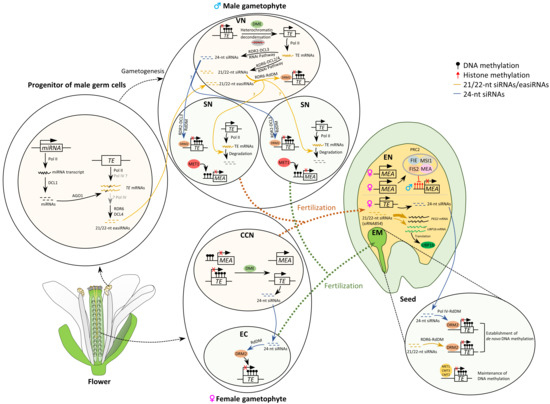
Figure 2.
A simplified model of DNA methylation and siRNA reprogramming in gametes and during early embryogenesis in Arabidopsis. During gametogenesis, global DNA demethylation occurs in companion cells—vegetative and central cells [78,84,85,91]. DEMETER (DME) removes DNA methylation from imprinted genes and transposable elements (TEs) through the base excision repair pathway [94]. Expressed TEs due to demethylation and heterochromatin decondensation produce 21–24 nt small RNAs in companion cells [93]. In the vegetative cell, a portion of 21–22 nt small interfering RNAs (siRNAs) is generated through the ARGONAUTE1 (AGO1)-AGO2-DICER-LIKE4 (DCL4) RNAi pathway [93], and 21–22 nt epigenetically activated small RNA (easiRNA) biogenesis occurs during meiosis or early during gametogenesis [74,75]. In a progenitor of male germ cells, microRNA (miRNA) targets TEs and triggers polymerase IV (Pol IV)-mediated 21–22 nt easiRNA production, and the process requires Pol IV, RNA DEPENDENT RNA POLYMERASE6 (RDR6), and DCL4 [74,75]. In the vegetative cell, both 21–22 nt siRNAs generated from the RNAi pathway and 21–22 nt easiRNAs can trigger TE methylation through the RDR6-RNA-directed DNA methylation (RdDM) pathway. These easiRNAs/siRNAs might move from the vegetative cell to sperm cells to cause TE mRNA degradation. On the other hand, 24 nt siRNAs generated by the canonical RDR2-DCL3 RNAi pathway due to TE demethylation by DME in the vegetative cell might also move to the sperm cell to trigger methylation in TE by the canonical RDR2-DCL3 RdDM pathway. In the sperm cells, gene methylation is maintained by DNA METHYLTRANSFERASE1 (MET1), and TE methylation at CG, CHG, and CHH sites is maintained by MET1, CHROMOMETHYLASE3 (CMT3), and DOMAINS REARRANGED METHYLTRANSFERASE2 (DRM2)/CMT2, respectively (MET1/CMT3/CMT2 were not shown). The 24 nt siRNAs generated from expressed TEs in the central cell can move to the egg cell, where the siRNAs help maintain hypermethylation and silencing of imprinted genes and TEs through the canonical Pol IV-RdDM pathway. After fertilization, the hypomethylated alleles of imprinted genes in the central cell are passed onto the endosperm, which causes differential DNA methylation between parental alleles. PcG PRC2 complex including maternal MEA inhibits paternal MEA expression. In the endosperm, the 21–22 nt siRNAs, such as siRNA854, target PATERNALLY EXPRESSED GENE2 (PEG2) mRNA to suppress its translation, and target OLIGOURIDYLATE BINDING PROTEIN1b (UBP1b) to regulate UBP1b protein levels by translation inhibition [95]. In the embryo, the 24 nt siRNAs can establish and silence TEs through the canonical Pol IV-RdDM pathway; the 21–22-nt siRNAs retained before fertilization help establish DNA methylation in TEs through the non-canonical RDR6-RdDM pathway. TE methylation at CG, CHG, and CHH sites is maintained by MET1, CMT3, and CMT2, respectively. Abbreviations are as follows: vegetative nucleus (VN), sperm nucleus (SN), central cell nucleus (CCN), egg cell (EC), embryo (EM), endosperm (EN).
3. Dynamic DNA Methylation in the Vegetative and Sperm Cells of Male Gametophyte
Compared with the seven-cell eight-nucleus female gametophyte in Arabidopsis, there are only three cells and two cell types in the male gametophyte. Recent studies reveal that vegetative and sperm cells have different methylation profiles, although they are only two divisions apart from the same precursor cell, microspore. The sperm nuclei have a slightly higher CG and slightly lower CHG methylation than the vegetative nucleus, but CHH DNA methylation in sperm nuclei (1.6%–2.1%) is only about half of that in the vegetative nucleus (4.0%–5.4%) (Figure 1) [78,84].
One major function of DNA methylation is to silence TEs. Surprisingly, some TEs in pollen vegetative nucleus are actually reactivated [91]. This is likely because DME is expressed in the vegetative cell but silenced in the sperm cell of the male gametophyte in addition to downregulation of DECREASE IN DNA METHYLATION1 (DDM1), a gene required for DNA methylation [92]. Examining Athila, a retrotransposon, showed that activation of the TEs in the vegetative cell results in accumulation of 21 nt siRNAs that are likely to be transported to the sperm nuclei (Figure 2) [91]. Recently, it has been further revealed that siRNAs generated from mRNA transcripts in the vegetative cell can move to sperm cells and silence TE reporters, revealing a potential mechanism in which reactivation of TEs in companion cells can regulate the TE activity in their associated gametes (Figure 2) [93]. Further evidence was gathered by sequencing the methylome of the sperm cell, vegetative cell, and their precursor [84]. The results showed that CG and CHG DNA methylation was stably retained whereas CHH methylation was lost in retrotransposons in the sperm cells but was later restored in the embryo after fertilization. It was hypothesized that loss of methylation in TEs in sperm cells would regain their methylation later in part by the maternal 24 nt siRNAs produced by the RdDM pathway (Figure 2) [84].
DNA demethylation in the male and female companion cells by an active DME-dependent mechanism occurs before these cells are differentiated. Ibarra and colleagues examined methylation in pollen from DME/dme plants and found that CG sites were hypomethylated in wild-type vegetative cells compared to somatic tissue, however, hypomethylation was abolished in the dme vegetative cell nuclei, suggesting that DME is required for demethylation in the vegetative cell (Figure 2) [78]. Loci that showed decreased CHH methylation in the dme sperm had an increased CG methylation in the dme vegetative cell. Because loss of DME affects methylation status of the vegetative cell, which in turns affects sperm cell methylation, the authors proposed that DME expression in the vegetative cell is necessary for full methylation of a certain number of sperm TEs (small, AT-rich, and nucleosome-depleted euchromatic), suggesting that DNA demethylation in the companion cell produces a potential signal in the form of sRNAs that help prevent TE activation in the gametes [78].
It has been recently shown that the paternal TE-derived 21–22 nt sRNAs termed epigenetically activated small RNAs (easiRNAs) increased in the crossed seed in response to paternally increased ploidy, and that mutations from the paternal Pol IV suppressed easiRNA production and generated viable triploid seeds by inducing methylation in TEs through RdDM [75]. It has been further shown that the 21–22 nt easiRNA accumulation through Pol IV resulted from a highly conserved miRNA in plants, miR845, and targeting tRNAMet primer-binding site (PBS) in long terminal repeats (LTR) [74]. These studies suggest that miRNAs produced in pollen affect siRNA accumulation, which can serve as a quantitative signal to balance parental dosage and affect seed development after fertilization.
4. Epigenetic Changes Regulate Early Embryogenesis in Plants
During early embryogenesis, body axes and major tissue layers are established. Although many factors involved in basic body plan establishment have been identified [96], underlying epigenetic mechanisms that potentially regulate cell differentiation in early embryogenesis have not been elucidated in many species. Patterns of DNA methylation during embryogenesis have been studied in many species, such as Arabidopsis (Figure 1), maize, and soybean [78,86,97]. DNA methylation can vary significantly at different developmental stages, especially during reproduction (Figure 1). DNA methylation in CG, CHG, and CHH contexts in the endosperm is lower than that in the embryo for each context, with an especially significant hypomethylation in CHH methylation (Figure 1) [78]. These dynamic patterns of DNA methylation appear to be important for successful reproduction, as abnormal DNA methylation affects normal planes and numbers of cell division and auxin hormone gradients in early embryogenesis in Arabidopsis [76].
Recently, multiple studies have clearly demonstrated that dynamic DNA changes occur during seed development [86,98,99,100,101]. Lin et al. profiled DNA methylome of Arabidopsis and soybean seeds at globular and cotyledon stages and in different seed regions, sub-regions, and tissues, and found that DNA methylation in the CHH context increases significantly during early seed development [86]. Analyzing the transcriptome and seed development of the quadruple drm1 drm2 cmt2 cmt3 (ddcc) mutant that lacks CHH and CHG methylation, the authors thought that CHH and CHG methylation might not play an important role in seed development [86]. The above result is not a surprise because no obvious morphological phenotype has been reported about the ddcc mutant [102]. However, we cannot conclude that methylation is not involved in seed development because CHH and CHG methylation is necessary for silencing of more than 100 TEs during early seed development [86]. Moreover, double mutants for the maize CMT3 and DDM1 orthologs are embryonic lethal [103]. More importantly, DNA methyltransferase MET1 regulates early seed development including pattern formation of early cell division [76]. Furthermore, a recent study demonstrated that Pol IV is important for seed development in Brassica rapa [104]. Mutations in Pol IV-mediated small RNA pathway caused defects in seed development in Brassica rapa, suggesting Pol IV-mediated RdDM is involved in seed development. When the maternal parent is the mutant of NUCLEAR RNA POLYMERASE D1 (nrpd1), a high percentage of seed abortion occurs. The self-pollinated nrpd1 has reduced seeds per silique and small seeds with irregular shape [104]. The authors further showed that RdDM is critical in maternal sporophytic tissues, not in the female gametophyte or zygote [104]. Thus, these results reveal that DNA methylation is crucial for regulating transcriptional activation of TEs [105], gene expression, and early seed development, although some species may not show any morphological phenotypes with loss of methylation.
5. Epigenetics Affects Late Embryogenesis, Seed Maturation, and Germination in Plants
Dynamic epigenetic changes occur during late embryogenesis and seed maturation and play a critical role in seed dormancy [86,98,99,100,101]. By using Arabidopsis as a model, DNA methylation dynamics during late seed development and germination have been revealed [86,98,99,100]. Although CG methylation does not show a significant change, CHH methylation increases significantly during embryogenesis and reaches the highest levels for most methylated sites in mature embryos [98,99]. Through whole-genome bisulfite sequencing, it has also been shown that methylation goes through dynamic changes during the process of seed maturation in soybean [86,101]. CHH methylation levels in soybean cotyledons changed from 6% at the early stage S2 to 10% and 11% at the late stages S6 and S8, respectively. Furthermore, genes with negative correlation between their promoter methylation and gene expression during soybean seed maturation were identified: 36 genes that have differentially methylated regions (DMRs) in the CG sites, 66 genes in the CHG sites, and 2136 genes in the CHH sites [101]. Profiling methylomes revealed that CHH methylation increases significantly in both soybean and Arabidopsis during seed development, but drops precipitously within the germinating seedling [86,98,99,100]. By comparing the methylation levels of TEs in germinating seeds and seedlings of wild type and ros1 dml2 dml3 (rdd) triple demethylase mutant, no significant difference was detected, suggesting that active demethylation might not play a major role in this process [99]. It is likely that this global demethylation is due to methylation dilution caused by rapid cell divisions. Relatively stable CG and CHG methylation is likely maintained by active MET1 and CMT3, whereas dynamic CHH methylation is not fully established or maintained through RdDM and CMT2 pathways during seed germination [86,98,99,100].
Epigenetic and genetic pathways can crosstalk during seed development. LEAFY COTYLEDON1 (LEC1) encodes a Heme Activator Protein (HAP3) subunit of the CCAAT binding factor and is required for cotyledon identity and embryo maturation [106,107]. LEC1 is only expressed in the embryo and endosperm. A recent study has shown that LEC1, as a master regulator of seed development, not only directly controls a distinct set of genes during seed development [7], but also helps to reverse a silent chromatin at the FLOWER LOCUS C (FLC) locus to an active chromatin state and activates its expression de novo during early embryogenesis from the gametes. Importantly, this active chromatin is transmitted from the embryonic stage to postembryonic stages [108], an example of epigenetic memory, thus suggesting that epigenetic status during embryogenesis can be transmitted to an adult life. Epigenetic reprogramming of the vernalized state has shown that EARLY FLOWERING6 (EFL6) has histone H3 lysine 27 trimethylation (H3K27me3) demethylase activity [109]. Mutations in EFL6 decrease its H3K27me3 demethylase activity in the reproductive tissues of adult plants, leaving H3K27me3 methylation at the FLC locus at higher levels and its expression at lower levels, and thus the epigenetic state of H3K27me3 at FLC in the reproductive tissues is maintained or passed onto the embryo of next generation [109]. These results suggest that an epigenetic reprogramming exists during plant reproduction.
6. PcG Proteins Regulate Seed Development
PcG proteins are critical for seed development. PcG proteins were identified and shown to function in seed development by genetic screening in Arabidopsis, and they include FIE, FIS2, MEA, and MSI1 [28,29,30,31]. Mutations in these PcG genes can bypass fertilization and initiate seed development in the absence of fertilization. The fis-class mutants (fie, fis2, mea, and msi1) show maternal gametophytic defects—seeds abort when the mutant allele is maternally inherited regardless of the paternal allele, and the mutants have a similar phenotype of an excessive endosperm proliferation resulting in early seed developmental arrest. However, obtaining the homozygous fie mutant plant from the cross of the maternal fie heterozygote with the paternal cdka;1 fie double heterozygote indicates that FIE is not essential for embryogenesis (CDKA: a cyclin-dependent kinase, a homologue of cdc2; the cdka;1 mutant generates bicellular pollen grains, consisting of a vegetative and a single sperm-like cell), but FIE functioning in the PRC2 complex plays an essential role in catalyzing H3K27me3 in the genome [110].
FIS PRC2 complexes can target genes encoding MADS-box proteins in plants [111]. Expression of PHERES1 (PHE1), a type I MADS-box gene, is regulated by the FIS2 PRC2 complex including MEA, FIE, and FIS2. Köhler et al. demonstrated that PHE1 was not expressed before fertilization, and was transiently expressed in the embryo and the endosperm. The FIS-class proteins are necessary for silencing the maternal PHE1 allele after fertilization. PHE1 expression in the fie and mea mutants was about 5–10 fold higher than wild type and remained at high levels until 4 days after pollination (DAP), whereas PHE1 expression levels decreased after 3 DAP in wild type. A decrease of PHE1 expression in the mea mutant seed partially rescues the mea seed-abortion phenotype, suggesting that PHE1 plays an important role in regulating MEA expression and is partially responsible for the lethal phenotype of fis-class mutants [111]. AGAMOUS-LIKE62 (AGL62), a type I MADS-box gene, is another example demonstrating that PcG complexes regulate MADS domain proteins [112,113]. AGL62 is specifically expressed in the Arabidopsis endosperm. During endosperm development, expression of AGL62 is high during the syncytial phase and then decreases dramatically before cellularization. Kang et al. showed AGL62 promotes nuclear proliferation and suppresses endosperm cellularization. The results suggest that the FIS-PRC2 complex is involved in silencing AGL62 at the late stage of endosperm development [113].
Increasing maternal genome dosage can bypass genes essential for imprinting control [114], whereas eliminating the paternal contribution has a similar effect as increasing maternal genome dosage [115]. Although FIS-class genes are required for endosperm development, increasing ratio of maternal-to-paternal genome in the endosperm could allow normal seed development in the fis mutant [114]. This suggests that one of main functions of FIS-PRC2 is to balance the ratio of parental genome in seeds of flowering plants and reduce parental dosage conflict. This study also found that AGL62 can reduce seed size in plants that have increased maternal genome dosage [114], presumably due to AGL62 involvement in controlling the timing of endosperm cellularization. Recently, the FIS-PRC2 complex has been found to regulate the type I MADS-box genes, including many AGL genes [112]. Interestingly, the data suggest that the FIS-PRC2 complex might play dual roles in regulating type I MADS-box genes—a general role in suppressing gene expression at both maternal and paternal alleles during endosperm cellularization and a specialized role in silencing the maternal allele of imprinted genes [112]. It has been shown that the FIS genes function to balance the contribution of the paternal genome. When the paternal parent is the cdka;1 mutant, the requirement of the maternal MEA and FIE alleles can be dispensable or the imprinted expression of MEA and FIE in developing seeds can be bypassed and viable homozygous mea or fie mutant seeds are developed [110,115]. This research showed that at certain conditions (i.e., in absence of the paternal contribution to the endosperm) the requirement of FIS genes in normal seed development can be dispensable [115]. Interestingly, CURLY LEAF (CLF), a H3K27me3 methyltransferase, has been shown to affect seed size [116]. The clf-28 mutant seed is larger, heavier, and contains higher oil content. Furthermore, transcriptomic analysis shows that expression of a set of genes is repressed by CLF, and 46% of CLF-repressed genes are associated with epigenetic modification of H3K27me3 [116].
Decreased expression of PcG gene OsFIE2 led to smaller seed in knockdown OsFIE2 rice lines, and enzymes involved in storage proteins and starch synthesis were reduced [117]. In a related study, analysis of rice plants with developing seeds under moderate (34 °C) and high (42 °C) heat stress showed that moderate heat stress caused increased endosperm cellularization but high heat stress caused failure of cellularization [117]. Endosperm cellularization in rice was regulated by OsFIE1, which was susceptible to temperature alteration and its expression was anticorrelated with heat stress. Transgenic plants overexpressing OsFIE1 had reduced seed size and premature cellularization. Furthermore, DNA methylation and histone modification were also altered in heat stress. This suggests that epigenetic regulation of endosperm development and thermal sensitivity of seed size could be linked through the PRC2 complex [118].
7. Effects of Histone Modifications on Seed Development
Histone modifications are involved in seed development. The PcG protein MEA is a histone H3 methyltransferase that methylates lysine 27 of histone H3 [119]. As described before, MEA is imprinted and regulates endosperm proliferation in seed development. The maternal MEA allele is expressed and antagonistically regulated directly or indirectly by MET1 and DME. The paternal MEA allele is silenced by DNA methylation and the silencing is maintained by the maternal FIS PRC2 (Figure 2). The maternal MEA allele expression silences the paternal MEA allele and also represses the transcription of imprinted genes such as PHE1 that are paternally active and maternally silenced [53,120,121]. MEA suppresses expression of its target genes by H3K27me3 trimethylation, a repressive histone modification [119], that is associated with DMRs of the maternal alleles of paternally expressed genes (PEGs) in Arabidopsis, rice, and maize [97,122,123].
Histone H1 is classically viewed to compact chromatin into a higher order structure [124]. Recent evidence has showed that histone H1 variants have complex functions in chromatin remodeling and epigenetic regulation of plant growth and reproduction [125]. Epigenetically, mutations in H1 can cause DNA hypomethylation and hypermethylation in different genomic loci and result in misregulation of gene expression [126]. Histone H1 and DME interact, and H1 is involved in DME-mediated DNA demethylation in the endosperm [127]. Genetic analysis of the histone h1 mutant indicated that the maternal histone H1 allele is involved in imprinted expression of MEA, FWA, and FIS2 in the endosperm of Arabidopsis [127]. The H1 mutations cause increased methylation in the promoter of the maternal MEA and FWA alleles in the endosperm [127]. Interestingly, it has been found that DNA sequences in the heterochromatic regions require DDM1 and CMT2 for methylation, and that this process involves histone H1 in seedlings [20]. Furthermore, it has been shown that DDM1 allows nucleosome-bound DNA methylation and the double mutant ddm1 h1 displays the strong linker-specific DNA methylation pattern in both Arabidopsis and mice [128]. These results suggest that naked DNA without nucleosomes is a preferred target of DNA methylation [128]. Recently, the STRUCTURE SPECIFIC RECOGNITION PROTEIN 1 (SSRP1), a subunit of the chromatin remodeler FACT (facilitates chromatin transactions), has been shown to co-localize with nuclear DME in vivo [129]. Furthermore, H1 mediates the requirement for FACT at some DME-target genes [129]. Interestingly, H1 is naturally depleted in the vegetative cell in pollen, and facilitates DME to access some heterochromatic TEs, demethylating and activating those TEs [130]. In summary, it seems that histone H1 variants might have subtly different roles in different tissues and cells at different developmental stages; for example, the egg cell versus the vegetative cells in regulating chromatin structure, DNA methylation, and expression of genes and TEs.
Histone H3 variants have been reported to participate in epigenetic reprogramming in plant reproduction. Although more than a dozen H3 variants are expressed in somatic cells, only a few H3 variants exist in male and female gametes in Arabidopsis [131]. After fertilization, H3 variants from male and female gametes are actively removed from the zygotic chromatin, and replaced with new somatic H3 variants in the embryo by de novo synthesis of H3 variants [131], suggesting reprogramming H3 variants in the zygote limit the inheritance of epigenetic information from parental genomes.
Histone acetylation is a vital chromatin modification for regulating gene expression during plant development [132]. Human SILENT INFORMATION REGULATOR6 (SIRT6) has been shown to deacetylate H3K9 and H3K56. Oryza sativa SIRTUIN1 (OsSRT1) is a histone deacetylase, closely related to the human SIRT6. Down-regulation of OsSRT1-induced expression of RICE STARCH REGULATOR1 (RSR1) and amylase genes in developing seeds, which resulted in decreased starch synthesis, increased starch degradation, and abnormal seed development [133]. OsSRT1 was required for reducing histone H3K9 acetylation on genes and transposons during starch metabolism in developing seeds [133]. These results indicate that seed development is regulated, in part, by OsSRT1-mediated histone deacetylation.
AtHDA7, an Arabidopsis HISTONE DEACETYLASE (HDAC), is required for the female gametophyte development and embryogenesis in Arabidopsis [134]. Knocking out AtHDA7 was shown to cause degeneration of micropylar nuclei at the four-nucleate embryo sac stage and delayed embryo development. Mutations in AtHDA7 induced histone hyperacetylation, and significantly increased the transcription of other HDACs including AtHDA6. In addition, loss of AtHDA7 expression affected expression of ARABIDOPSIS HOMOLOG OF SEPARASE (AESP), which was involved in female gametophyte and embryo development [135]. This suggests that maintaining appropriate histone acetylation patterns is necessary for plant reproductive development.
Studying MEIOTIC CONTROL OF CROSSOVERS1 (MCC1), a GENERAL CONTROL NON-REPRESSED PROTEIN 5 (GCN5)-related histone N-acetyltransferase, shows that histone hyperacetylation is important for meiosis in plants [136]. The MCC1 gene was needed for meiosis. The mcc1 siliques had 68% fewer seeds per silique than wild type and the embryo sacs failed to differentiate in 50% of mcc1 ovules [136]. The mcc1 mutant had defects in meiosis due to the mcc1 pollen mother cells showing 30% increase in histone H3 acetylation [136]. Interestingly, a recent study showed histone acetyltransferase GCN5 also affected the fatty acid composition in Arabidopsis seeds. This suggests that histone acetylation affects not only ovule development but also fatty acid biosynthesis during late stage of seed maturation.
8. Small RNA Regulation is Critical in Seed Development
sRNAs are important regulators during plant growth and development [60], and especially important for reproductive development [137]. The homozygous male gametophytic kokopelli (kpl) mutant shows a high proportion of single fertilization event and 70% reduced seed set due to undeveloped ovules and aborted seeds [64]. ARIADNE14 (ARI14), encoding a putative ubiquitin E3 ligase, and KPL generate sperm-specific natural cis-antisense siRNAs (cis-nat-siRNAs). In the kpl mutant, ARI14 transcripts increased in sperm and fertilization was impaired. Furthermore, expression of ARI14 was increased in the siRNA biogenesis mutants dcl1, hen1, hyl1, and rdr2, suggesting that the siRNA pathway is involved in generating cis-nat-siRNAs from the KPL locus [64].
The 21 nt and 24 nt phased siRNAs (phasiRNAs) are generated by RDR6 and DCL4/DCL5 [138,139,140]. Although premeiotic 21 nt phasiRNAs lack known targets, loss of MEIOSIS ARRESTED AT LEPTOTENE 1 (MEL1), an AGO protein that interacts with 21 nt phasiRNAs, results in abnormalities in tapetum formation and early arrest in pollen mother cells in rice [141,142]. The closest homolog of MEL1 in maize is AGO5, and the mutant ago5-4 plants show defects in initiation of megagametogenesis and are unfertile in females. AGO4, a component involved in RdDM, is also involved in seed development. The ago4-1 mutant has been shown to have reduced fertility, which was only associated with floral organ defects [143]. However, pollen grain viability varies in the mutant, suggesting that the RdDM pathway can be important for meiosis [144]. Interestingly, Walker et al. showed that RdDM affected meiosis in the male meiocyte [145]. They found hypermethylated loci in the male sexual lineage called sexual-lineage-hypermethylated loci (SLHs), which were caused by RdDM. Loss of methylation at the sexual-lineage-specific methylated (SLM) loci by interrupting RdDM affected splicing of MULTIPOLAR SPINDLE1 (MPS1), a gene required for meiosis in Arabidopsis. Retention of the last intron of MPS1 produces aberrant MPS1 protein that interferes with meiosis, resulting in abnormal meiosis and impaired tetrad formation [145].
siRNAs also provide important insights into the mechanism of parental genome dosage imbalance in seeds [146]. Seed size is affected by maternal and paternal genome ratio. Paternal-excess crosses (2n × 4n) delay endosperm cellularization and generate larger seeds, whereas maternal-excess crosses (4n × 2n) promote early cellularization and generate smaller seeds [147]. Although a mechanism to explain an imbalance of parental genome dosage could involve parent-of-origin-specific factors and imprinted genes, an alternative explanation could involve 24 nt siRNAs, which are also called p4-siRNAs, as RNA Pol IV is responsible for generating these RNAs [148,149,150]. Genetic analyses of reciprocal crosses between 2n and 4n showed that p4-siRNA production depends on the maternal genome dosage [146]. In the maternal-excess endosperm, maternal p4-siRNA levels increased and p4-siRNAs-associated AGL genes were repressed, causing early cellularization and smaller seeds. In the paternal-excess endosperm, there was a lower amount of maternal p4-siRNAs and upregulation of AGLs, resulting in nuclear proliferation and larger seeds [146]. There are inconsistent results as to whether p4-siRNAs are from the maternal [148] or from both maternal and paternal genomes [151,152]. Genome-wide sRNAs in whole seeds of three different Arabidopsis strains and their reciprocal crosses were analyzed and genes involved in RdDM were found to be expressed in both endosperm and embryo at 6 DAP, suggesting that both endosperm and embryo can potentially produce sRNAs [153]. On the contrary, 6%–24% sRNAs were from the paternal genome, which was similar to the fraction of the paternal mRNA reads in seeds. Furthermore, NRPD1a is primarily paternally expressed in all the reciprocal crosses [153]. These studies suggest that siRNAs produced by Pol IV are involved in regulating seed size mediated by RdDM.
Interestingly, the reciprocal crosses between Arabidopsis thaliana and Arabidopsis lyrata revealed that allelic dosage is regulated by a sRNA pathway in the endosperm [154]. The ratio between maternal and paternal transcripts is actively maintained by the Pol IV-mediated pathway. Mutations in Pol IV suppress negative effects of extra paternal genome dosage. Inheritance of a paternal Pol IV mutation can rescue the seed abortion phenotype caused by excess paternal genome dosage [154]. In a following up study, RdDM activity from the paternal parent was shown to be sufficient to determine seed viability in the cross with paternal excess in Arabidopsis [155]. Misregulation in TEs and imprinted genes was found to be unlikely to cause seed abortion in the cross with paternal excess, and a model of a transcriptional buffering system of balanced gene expression between parental alleles was proposed [155]. The above results were not consistent with the role of easiRNAs in dosage effects on seed development discussed in the previous paragraph. As discussed before, the recent research shows that production of paternal easiRNAs regulates parental genome dosage [74,75]. Martinez et al. showed that Pol IV produced TE-derived 21–22 nt easiRNAs in pollen and loss of paternal Pol IV suppressed easiRNA biogenesis and restored CHH methylation of TEs mediated by RdDM [75]. One big difference was that they showed that these 21–22 nt easiRNAs depend on Pol IV-DCL2/4-RDR6 non-canonical RdDM pathway, whereas the paternal mutations in DCL3, RDR2, and Pol V cannot rescue the seed abortion caused by paternal excess [74,75]. However, Satyaki and Gehring showed that mutations in canonical RdDM pathway genes (Pol V and DRM2) are sufficient for suppressing seed abortion with paternal excess [155]. One of the reasons to explain this discrepancy is that Martinez et al. used the omission of second division (osd1) mutant to generate conditions of paternal excess [75], whereas the other study used the true tetraploid wild type and mutants generated by treatment with colchicine [155]. One explanation is that different paternal parents were used in these two studies. Furthermore, seed viability is a combinational consequence of genetic/epigenetic status of seeds, and physiological and environmental interaction. Nevertheless, considering the fact that these studies all show that the paternal mutation of Pol IV can suppress seed abortion of paternal excess, it is quite puzzling that contradictory results were obtained.
9. Summary and Discussion
Seed development is controlled by programmed gene expression. To date, many genes have been identified and characterized as being important for seed development [156]. The research in the last decade has shown epigenetics is important for seed development, but it remains largely unclear how different epigenetic mechanisms interact to regulate seed development. Since the first report that DNA methylation regulates embryogenesis and seed development in plants [76,157], we now know much more about the role of DNA methylation, histone modifications, and sRNAs in regulating seed development. Furthermore, DNA methylomes of gametes, embryo, endosperm, and seeds at different developmental stages have been sequenced in Arabidopsis and other organisms, which has greatly increased our understanding of how epigenetics affects reproductive development [158]. Most results discussed in this review have been obtained from studying Arabidopsis thaliana, which is very resilient to epigenetic perturbations, differing it from other plant species or mammals. These studies have shown a significant variation of DNA methylation among gametes, different seed compartments, and seeds at different stages (Figure 1). It is not clear what exact mechanisms cause dynamic DNA methylation during reproduction and how it evolved.
DNA methylation occurs in CG, CHG, CHH contexts of genes, repeats, and TEs, and its levels are quite diverse. In flowering plants, one of the major functions of DNA methylation is to silence TEs and to maintain genome stability during plant growth and development, including gametogenesis and seed development. In addition to silencing TEs and repeats, DNA methylation, as an epigenetic modification, regulates gene expression during seed development. Low levels of methylation in the gene promoter have been shown to correlate with high expression for some genes in Arabidopsis, rice, and soybean [65,85,101]. Although DNA methylome has been sequenced in many plant species, it remains to be determined how many genes are directly regulated by DNA methylation during seed development.
Besides the general role in gene regulation, DNA methylation has been demonstrated to regulate expression of a specific set of genes, including PcG and imprinted genes in seed development. PcG genes MEA and FIS2 are imprinted, and their maternal alleles are antagonistically regulated by MET1 and DME in the endosperm. The paternal MEA allele is silenced by DNA methylation and this silenced state is maintained by its maternal FIS PRC2 complex in the endosperm. To date, many genes have been revealed to show imprinted expression [46,51,55,57,58], and it is very likely that they are regulated by methylation and other epigenetic mechanisms. It remains to be experimentally proven how many genes are actually imprinted and their specific function in seed development [46,58].
DNA methylation reprogramming occurs in gametogenesis prior to fertilization and embryogenesis. In the endosperm, hypomethylated maternal alleles can cause expression of 24 nt sRNA that might move to the embryo to methylate the homologous genes and/or TEs through RdDM, fortifying the silencing of TEs in the embryo (Figure 2). Similarly, in the male gametophyte, DME demethylates CG sites of DME-targeted genes or nearby TEs. It is hypothesized that expressed siRNA from hypomethylated TEs in the vegetative nucleus can move to adjacent sperm cells to further silence TEs in the sperm cell that passes its genetic information to the zygote [78,84,91]. It seems that both canonical (DCL3, RDR2, Pol IV, and Pol V) and non-canonical (DCL2/4, RDR6, Pol IV, and Pol II) RdDM pathways exist during reproduction. Epigenetic reprogramming of methylation in female and male gametophytes and during seed development is to ensure overall genome stability and inheritance of traits to the next generation. It still needs to be experimentally demonstrated how sRNAs are moved from microspores and/or the companion cell to the gametes to silence targeted genes through the RdDM pathway. In addition, exact mechanisms of hybridization barriers between different ploidy parents remain elusive—it can be due to misregulation of TEs and imprinted genes by Pol IV-mediated sRNAs, transcriptional buffering of parental alleles [155], or subtle indirect effects of misregulation of genes and TEs caused by collective epigenetic modifications.
Histone modifications are critical for development. Multiple lines of evidence have shown that histone H3 methyltransferases (MEA), histone acetyltransferases (AtHDA7 and GCN5), and deacetylases (OsSRT1) are involved in regulating seed development. It is apparent that DNA methylation, histone modifications (methylation, acetylation, etc.), PcG proteins, parent-of-origin and parental dosage effects, RdDM, and dynamic sRNA homeostasis are, in part, an integral epigenetic system during seed development. It remains to be elucidated how these different epigenetic pathways intertwine to affect dynamic epigenetic status in sexual lineage, zygote, and embryogenesis, and maintain transgenerational epigenetic inheritance and variations in reproduction.
Author Contributions
Conceptualization, W.X.; investigation, Q.H., A.B., X.C., and A.M.; writing—original draft preparation, Q.H., A.B., and W.X.; writing—review and editing, Q.H., A.B., Y.-Q.C.A., T.-F.H., and W.X.
Funding
This project was funded by the U.S. National Science Foundation grant (project #: 1715115) to T.-F.H. and W.X.
Acknowledgments
The authors apologize for not being able to include many important publications due to limited space, and thank R. Keith Slotkin for helpful discussion and comments on Figure 2.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Drews, G.N.; Wang, D.; Steffen, J.G.; Schumaker, K.S.; Yadegari, R. Identification of genes expressed in the angiosperm female gametophyte. J. Exp. Bot. 2011, 62, 1593–1599. [Google Scholar] [CrossRef] [PubMed]
- Drews, G.N.; Yadegari, R. Development and function of the Angiosperm female gametophyte. Annu. Rev. Genet. 2002, 36, 99–124. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.C.; Lemmon, B.E.; Nguyen, H.; Olsen, O.-A. Development of endosperm in Arabidopsis thaliana. Sex. Plant Reprod. 1999, 12, 32–42. [Google Scholar] [CrossRef]
- Gehring, M.; Choi, Y.; Fischer, R.L. Imprinting and seed development. Plant Cell 2004, 16. [Google Scholar] [CrossRef] [PubMed]
- Goldberg, R.B.; de Paiva, G.; Yadegari, R. Plant embryogenesis: Zygote to seed. Science 1994, 266, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Jurgens, G. Apical-basal pattern formation in Arabidopsis embryogenesis. EMBO J. 2001, 20, 3609–3616. [Google Scholar] [CrossRef]
- Pelletier, J.M.; Kwong, R.W.; Park, S.; Le, B.H.; Baden, R.; Cagliari, A.; Hashimoto, M.; Munoz, M.D.; Fischer, R.L.; Goldberg, R.B.; et al. LEC1 sequentially regulates the transcription of genes involved in diverse developmental processes during seed development. Proc. Natl. Acad. Sci. USA 2017, 114, E6710–E6719. [Google Scholar] [CrossRef]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef]
- Martienssen, R.A.; Colot, V. DNA methylation and epigenetic inheritance in plants and filamentous fungi. Science 2001, 293, 1070–1074. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. DNA methylation, a key regulator of plant development and other processes. Curr. Opin. Genet. Dev. 2000, 10, 217–223. [Google Scholar] [CrossRef]
- Richards, E.J. Inherited epigenetic variation—Revisiting soft inheritance. Nat. Rev. Genet. 2006, 7, 395–401. [Google Scholar] [CrossRef] [PubMed]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Bestor, T.H.; Jaenisch, R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell 1992, 69, 915–926. [Google Scholar] [CrossRef]
- Vongs, A.; Kakutani, T.; Martienssen, R.A.; Richards, E.J. Arabidopsis thaliana DNA methylation mutants. Science 1993, 260, 1926–1928. [Google Scholar] [CrossRef] [PubMed]
- Finnegan, E.J.; Dennis, E.S. Isolation and identification by sequence homology of a putative cytosine methyltransferase from Arabidopsis thaliana. Nucl. Acids Res. 1993, 21, 2383–2388. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Kovac, K.A. Plant DNA Methyltransferases. Plant Mol. Biol. 2000, 43, 189–201. [Google Scholar] [CrossRef] [PubMed]
- Kankel, M.W.; Ramsey, D.E.; Stokes, T.L.; Flowers, S.K.; Haag, J.R.; Jeddeloh, J.A.; Riddle, N.C.; Verbsky, M.L.; Richards, E.J. Arabidopsis MET1 Cytosine Methyltransferase Mutants. Genetics 2003, 163, 1109–1122. [Google Scholar]
- Bartee, L.; Malagnac, F.; Bender, J. Arabidopsis cmt3 chromomethylase mutations block non-CG methylation and silencing of an endogenous gene. Genes Dev. 2001, 15, 1753–1758. [Google Scholar] [CrossRef]
- Lindroth, A.M.; Cao, X.; Jackson, J.P.; Zilberman, D.; McCallum, C.M.; Henikoff, S.; Jacobsen, S.E. Requirement of CHROMOMETHYLASE3 for maintenance of CpXpG methylation. Science 2001, 292, 2077–2080. [Google Scholar] [CrossRef]
- Zemach, A.; Kim, M.Y.; Hsieh, P.H.; Coleman-Derr, D.; Eshed-Williams, L.; Thao, K.; Harmer, S.L.; Zilberman, D. The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1-containing heterochromatin. Cell 2013, 153, 193–205. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-CG methylation patterns shape the epigenetic landscape in Arabidopsis. Nat. Struct. Mol. Biol. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Du, J.; Hale, C.J.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.A.; Chory, J.; Wohlschlegel, J.A.; Patel, D.J.; Jacobsen, S.E. Molecular mechanism of action of plant DRM de novo DNA methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.B. A gene complex controlling segmentation in Drosophila. Nature 1978, 276, 565–570. [Google Scholar] [CrossRef] [PubMed]
- Lewis, E.B.; Mislove, R.F. New mutants report. Drosoph. Inf. Serv. 1947, 21, 69. [Google Scholar]
- Pirrotta, V. Polycombing the genome: PcG, trxG, and chromatin silencing. Cell 1998, 93, 333–336. [Google Scholar] [CrossRef]
- Xiao, J.; Jin, R.; Yu, X.; Shen, M.; Wagner, J.D.; Pai, A.; Song, C.; Zhuang, M.; Klasfeld, S.; He, C.; et al. Cis and trans determinants of epigenetic silencing by Polycomb repressive complex 2 in Arabidopsis. Nat. Genet. 2017, 49, 1546–1552. [Google Scholar] [CrossRef]
- Grossniklaus, U.; Vielle-Calzada, J.P.; Hoeppner, M.A.; Gagliano, W.B. Maternal control of embryogenesis by MEDEA, a polycomb group gene in Arabidopsis. Science 1998, 280, 446–450. [Google Scholar] [CrossRef]
- Luo, M.; Bilodeau, P.; Koltunow, A.; Dennis, E.S.; Peacock, W.J.; Chaudhury, A.M. Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 1999, 96, 296–301. [Google Scholar] [CrossRef]
- Ohad, N.; Margossian, L.; Hsu, Y.C.; Williams, C.; Repetti, P.; Fischer, R.L. A mutation that allows endosperm development without fertilization. Proc. Natl. Acad. Sci. USA 1996, 93, 5319–5324. [Google Scholar] [CrossRef]
- Hennig, L.; Taranto, P.; Walser, M.; Schonrock, N.; Gruissem, W. Arabidopsis MSI1 is required for epigenetic maintenance of reproductive development. Development 2003, 130, 2555–2565. [Google Scholar] [CrossRef] [PubMed]
- Chaudhury, A.M.; Ming, L.; Miller, C.; Craig, S.; Dennis, E.S.; Peacock, W.J. Fertilization-independent seed development in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 1997, 94, 4223–4228. [Google Scholar] [CrossRef] [PubMed]
- Gates, L.A.; Foulds, C.E.; O’Malley, B.W. Histone Marks in the ‘Driver’s Seat’: Functional Roles in Steering the Transcription Cycle. Trends Biochem. Sci. 2017, 42, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef]
- Zhang, X.; Bernatavichute, Y.V.; Cokus, S.; Pellegrini, M.; Jacobsen, S.E. Genome-wide analysis of mono-, di- and trimethylation of histone H3 lysine 4 in Arabidopsis thaliana. Genome Biol. 2009, 10, R62. [Google Scholar] [CrossRef]
- Guo, L.; Yu, Y.; Law, J.A.; Zhang, X. SET DOMAIN GROUP2 is the major histone H3 lysine [corrected] 4 trimethyltransferase in Arabidopsis. Proc. Natl. Acad. Sci. USA 2010, 107, 18557–18562. [Google Scholar] [CrossRef]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef]
- Tan, M.; Luo, H.; Lee, S.; Jin, F.; Yang, J.S.; Montellier, E.; Buchou, T.; Cheng, Z.; Rousseaux, S.; Rajagopal, N.; et al. Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 2011, 146, 1016–1028. [Google Scholar] [CrossRef]
- Roudier, F.; Ahmed, I.; Berard, C.; Sarazin, A.; Mary-Huard, T.; Cortijo, S.; Bouyer, D.; Caillieux, E.; Duvernois-Berthet, E.; Al-Shikhley, L.; et al. Integrative epigenomic mapping defines four main chromatin states in Arabidopsis. EMBO J. 2011, 30, 1928–1938. [Google Scholar] [CrossRef]
- Liu, C.L.; Kaplan, T.; Kim, M.; Buratowski, S.; Schreiber, S.L.; Friedman, N.; Rando, O.J. Single-nucleosome mapping of histone modifications in S. cerevisiae. PLoS Biol. 2005, 3, e328. [Google Scholar] [CrossRef]
- Zhou, M.; Palanca, A.M.S.; Law, J.A. Locus-specific control of the de novo DNA methylation pathway in Arabidopsis by the CLASSY family. Nat. Genet. 2018, 50, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Mikkelsen, T.S.; Ku, M.; Jaffe, D.B.; Issac, B.; Lieberman, E.; Giannoukos, G.; Alvarez, P.; Brockman, W.; Kim, T.K.; Koche, R.P.; et al. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 2007, 448, 553–560. [Google Scholar] [CrossRef] [PubMed]
- Kharchenko, P.V.; Alekseyenko, A.A.; Schwartz, Y.B.; Minoda, A.; Riddle, N.C.; Ernst, J.; Sabo, P.J.; Larschan, E.; Gorchakov, A.A.; Gu, T.; et al. Comprehensive analysis of the chromatin landscape in Drosophila melanogaster. Nature 2011, 471, 480–485. [Google Scholar] [CrossRef] [PubMed]
- Ferguson-Smith, A.C.; Surani, M.A. Imprinting and the epigenetic asymmetry between parental genomes. Science 2001, 293, 1086–1089. [Google Scholar] [CrossRef]
- Reik, W.; Walter, J. Genomic imprinting: Parental influence on the genome. Nature Rev. Genet. 2001, 2, 21–32. [Google Scholar] [CrossRef]
- Hsieh, T.F.; Shin, J.; Uzawa, R.; Silva, P.; Cohen, S.; Bauer, M.J.; Hashimoto, M.; Kirkbride, R.C.; Harada, J.J.; Zilberman, D.; et al. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc. Natl. Acad. Sci. USA 2011, 108, 1755–1762. [Google Scholar] [CrossRef]
- Jahnke, S.; Scholten, S. Epigenetic resetting of a gene imprinted in plant embryos. Curr. Biol. 2009, 19, 1677–1681. [Google Scholar] [CrossRef]
- Huh, J.H.; Bauer, M.J.; Hsieh, T.F.; Fischer, R.L. Cellular programming of plant gene imprinting. Cell 2008, 132, 735–744. [Google Scholar] [CrossRef]
- Gregg, C.; Zhang, J.; Weissbourd, B.; Luo, S.; Schroth, G.P.; Haig, D.; Dulac, C. High-resolution analysis of parent-of-origin allelic expression in the mouse brain. Science 2010, 329, 643–648. [Google Scholar] [CrossRef]
- Haig, D.; Westoby, M. Genomic imprinting in endosperm: Its effect on seed development in crosses between species, and between different ploidies of the same species, and its implications for the evolution of apomixis. Phil. Trans. R. Soc. Lond. B 1991, 333, 1–14. [Google Scholar]
- Kohler, C.; Page, D.R.; Gagliardini, V.; Grossniklaus, U. The Arabidopsis thaliana MEDEA Polycomb group protein controls expression of PHERES1 by parental imprinting. Nat. Genet. 2005, 37, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Baroux, C.; Gagliardini, V.; Page, D.R.; Grossniklaus, U. Dynamic regulatory interactions of Polycomb group genes: MEDEA autoregulation is required for imprinted gene expression in Arabidopsis. Genes Dev. 2006, 20, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Gehring, M.; Huh, J.H.; Hsieh, T.F.; Penterman, J.; Choi, Y.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DEMETER DNA glycosylase establishes MEDEA polycomb gene self-imprinting by allele-specific demethylation. Cell 2006, 124, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Jullien, P.E.; Kinoshita, T.; Ohad, N.; Berger, F. Maintenance of DNA methylation during the Arabidopsis life cycle is essential for parental imprinting. Plant Cell 2006, 18, 1360–1372. [Google Scholar] [CrossRef]
- Jullien, P.E.; Katz, A.; Oliva, M.; Ohad, N.; Berger, F. Polycomb group complexes self-regulate imprinting of the Polycomb group gene MEDEA in Arabidopsis. Curr. Biol. 2006, 16, 486–492. [Google Scholar] [CrossRef]
- Raissig, M.T.; Bemer, M.; Baroux, C.; Grossniklaus, U. Genomic imprinting in the Arabidopsis embryo is partly regulated by PRC2. PLoS Genet. 2013, 9, e1003862. [Google Scholar] [CrossRef]
- Kinoshita, T.; Yadegari, R.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. Imprinting of the MEDEA polycomb gene in the Arabidopsis endosperm. Plant Cell 1999, 11, 1945–1952. [Google Scholar] [CrossRef]
- Gehring, M.; Missirian, V.; Henikoff, S. Genomic analysis of parent-of-origin allelic expression in Arabidopsis thaliana seeds. PLoS ONE 2011, 6, e23687. [Google Scholar] [CrossRef]
- Vu, T.M.; Nakamura, M.; Calarco, J.P.; Susaki, D.; Lim, P.Q.; Kinoshita, T.; Higashiyama, T.; Martienssen, R.A.; Berger, F. RNA-directed DNA methylation regulates parental genomic imprinting at several loci in Arabidopsis. Development 2013, 140, 2953–2960. [Google Scholar] [CrossRef]
- Chen, X. Small RNAs in development—Insights from plants. Curr. Opin. Genet. Dev. 2012, 22, 361–367. [Google Scholar] [CrossRef]
- Mallory, A.C.; Elmayan, T.; Vaucheret, H. MicroRNA maturation and action—The expanding roles of ARGONAUTEs. Curr. Opin. Plant Biol. 2008, 11, 560–566. [Google Scholar] [CrossRef] [PubMed]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef] [PubMed]
- Axtell, M.J. Classification and comparison of small RNAs from plants. Annu. Rev. Plant Biol. 2013, 64, 137–159. [Google Scholar] [CrossRef] [PubMed]
- Ron, M.; Alandete Saez, M.; Eshed Williams, L.; Fletcher, J.C.; McCormick, S. Proper regulation of a sperm-specific cis-nat-siRNA is essential for double fertilization in Arabidopsis. Genes Dev. 2010, 24, 1010–1021. [Google Scholar] [CrossRef] [PubMed]
- Zemach, A.; Kim, M.Y.; Silva, P.; Rodrigues, J.A.; Dotson, B.; Brooks, M.D.; Zilberman, D. Local DNA hypomethylation activates genes in rice endosperm. Proc. Natl. Acad. Sci. USA 2010, 107, 18729–18734. [Google Scholar] [CrossRef] [PubMed]
- Ji, L.; Chen, X. Regulation of small RNA stability: Methylation and beyond. Cell Res. 2012, 22, 624–636. [Google Scholar] [CrossRef]
- He, X.J.; Hsu, Y.F.; Zhu, S.; Wierzbicki, A.T.; Pontes, O.; Pikaard, C.S.; Liu, H.L.; Wang, C.S.; Jin, H.; Zhu, J.K. An effector of RNA-directed DNA methylation in Arabidopsis is an ARGONAUTE 4- and RNA-binding protein. Cell 2009, 137, 498–508. [Google Scholar] [CrossRef]
- Bies-Etheve, N.; Pontier, D.; Lahmy, S.; Picart, C.; Vega, D.; Cooke, R.; Lagrange, T. RNA-directed DNA methylation requires an AGO4-interacting member of the SPT5 elongation factor family. EMBO Rep. 2009, 10, 649–654. [Google Scholar] [CrossRef]
- Huang, Y.; Ji, L.; Huang, Q.; Vassylyev, D.G.; Chen, X.; Ma, J.B. Structural insights into mechanisms of the small RNA methyltransferase HEN1. Nature 2009, 461, 823–827. [Google Scholar] [CrossRef]
- Nuthikattu, S.; McCue, A.D.; Panda, K.; Fultz, D.; DeFraia, C.; Thomas, E.N.; Slotkin, R.K. The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21-22 nucleotide small interfering RNAs. Plant Physiol. 2013, 162, 116–131. [Google Scholar] [CrossRef]
- Wu, L.; Mao, L.; Qi, Y. Roles of dicer-like and argonaute proteins in TAS-derived small interfering RNA-triggered DNA methylation. Plant Physiol. 2012, 160, 990–999. [Google Scholar] [CrossRef]
- Garcia, D.; Garcia, S.; Pontier, D.; Marchais, A.; Renou, J.P.; Lagrange, T.; Voinnet, O. Ago hook and RNA helicase motifs underpin dual roles for SDE3 in antiviral defense and silencing of nonconserved intergenic regions. Mol. Cell 2012, 48, 109–120. [Google Scholar] [CrossRef]
- Pontier, D.; Picart, C.; Roudier, F.; Garcia, D.; Lahmy, S.; Azevedo, J.; Alart, E.; Laudie, M.; Karlowski, W.M.; Cooke, R.; et al. NERD, a plant-specific GW protein, defines an additional RNAi-dependent chromatin-based pathway in Arabidopsis. Mol. Cell 2012, 48, 121–132. [Google Scholar] [CrossRef]
- Borges, F.; Parent, J.S.; van Ex, F.; Wolff, P.; Martinez, G.; Kohler, C.; Martienssen, R.A. Transposon-derived small RNAs triggered by miR845 mediate genome dosage response in Arabidopsis. Nat. Genet. 2018, 50, 186–192. [Google Scholar] [CrossRef]
- Martinez, G.; Wolff, P.; Wang, Z.; Moreno-Romero, J.; Santos-Gonzalez, J.; Conze, L.L.; DeFraia, C.; Slotkin, R.K.; Kohler, C. Paternal easiRNAs regulate parental genome dosage in Arabidopsis. Nat. Genet. 2018, 50, 193–198. [Google Scholar] [CrossRef]
- Xiao, W.; Custard, K.D.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. DNA methylation is critical for Arabidopsis embryogenesis and seed viability. Plant Cell 2006, 18, 805–814. [Google Scholar] [CrossRef]
- Park, K.; Kim, M.Y.; Vickers, M.; Park, J.S.; Hyun, Y.; Okamoto, T.; Zilberman, D.; Fischer, R.L.; Feng, X.; Choi, Y.; et al. DNA demethylation is initiated in the central cells of Arabidopsis and rice. Proc. Natl. Acad. Sci. USA 2016, 113, 15138–15143. [Google Scholar] [CrossRef]
- Ibarra, C.A.; Feng, X.; Schoft, V.K.; Hsieh, T.F.; Uzawa, R.; Rodrigues, J.A.; Zemach, A.; Chumak, N.; Machlicova, A.; Nishimura, T.; et al. Active DNA demethylation in plant companion cells reinforces transposon methylation in gametes. Science 2012, 337, 1360–1364. [Google Scholar] [CrossRef]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef]
- Widman, N.; Feng, S.; Jacobsen, S.E.; Pellegrini, M. Epigenetic differences between shoots and roots in Arabidopsis reveals tissue-specific regulation. Epigenetics 2014, 9, 236–242. [Google Scholar] [CrossRef]
- Feng, S.; Cokus, S.J.; Zhang, X.; Chen, P.Y.; Bostick, M.; Goll, M.G.; Hetzel, J.; Jain, J.; Strauss, S.H.; Halpern, M.E.; et al. Conservation and divergence of methylation patterning in plants and animals. Proc. Natl. Acad. Sci. USA 2010, 107, 8689–8694. [Google Scholar] [CrossRef]
- Ganguly, D.R.; Crisp, P.A.; Eichten, S.R.; Pogson, B.J. The Arabidopsis DNA methylome is stable under transgenerational drought stress. Plant Physiol. 2017, 175, 1893–1912. [Google Scholar] [CrossRef]
- Becker, C.; Hagmann, J.; Muller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef]
- Calarco, J.P.; Borges, F.; Donoghue, M.T.; Van Ex, F.; Jullien, P.E.; Lopes, T.; Gardner, R.; Berger, F.; Feijo, J.A.; Becker, J.D.; et al. Reprogramming of DNA methylation in pollen guides epigenetic inheritance via small RNA. Cell 2012, 151, 194–205. [Google Scholar] [CrossRef]
- Hsieh, T.F.; Ibarra, C.A.; Silva, P.; Zemach, A.; Eshed-Williams, L.; Fischer, R.L.; Zilberman, D. Genome-wide demethylation of Arabidopsis endosperm. Science 2009, 324, 1451–1454. [Google Scholar] [CrossRef]
- Lin, J.Y.; Le, B.H.; Chen, M.; Henry, K.F.; Hur, J.; Hsieh, T.F.; Chen, P.Y.; Pelletier, J.M.; Pellegrini, M.; Fischer, R.L.; et al. Similarity between soybean and Arabidopsis seed methylomes and loss of non-CG methylation does not affect seed development. Proc. Natl. Acad. Sci. USA 2017, 114, E9730–E9739. [Google Scholar] [CrossRef]
- Choi, Y.; Gehring, M.; Johnson, L.; Hannon, M.; Harada, J.J.; Goldberg, R.B.; Jacobsen, S.E.; Fischer, R.L. DEMETER, a DNA glycosylase domain protein, is required for endosperm gene imprinting and seed viability in Arabidopsis. Cell 2002, 110, 33–42. [Google Scholar] [CrossRef]
- Xiao, W.; Gehring, M.; Choi, Y.; Margossian, L.; Pu, H.; Harada, J.J.; Goldberg, R.B.; Pennell, R.I.; Fischer, R.L. Imprinting of the MEA Polycomb gene is controlled by antagonism between MET1 methyltransferase and DME glycosylase. Develop. Cell 2003, 5, 891–901. [Google Scholar] [CrossRef]
- Kinoshita, T.; Miura, A.; Choi, Y.; Kinoshita, Y.; Cao, X.; Jacobsen, S.E.; Fischer, R.L.; Kakutani, T. One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science 2004, 303, 521–523. [Google Scholar] [CrossRef]
- Schmidt, A.; Wohrmann, H.J.; Raissig, M.T.; Arand, J.; Gheyselinck, J.; Gagliardini, V.; Heichinger, C.; Walter, J.; Grossniklaus, U. The Polycomb group protein MEDEA and the DNA methyltransferase MET1 interact to repress autonomous endosperm development in Arabidopsis. Plant J. 2013, 73, 776–787. [Google Scholar] [CrossRef]
- Slotkin, R.K.; Vaughn, M.; Borges, F.; Tanurdzic, M.; Becker, J.D.; Feijo, J.A.; Martienssen, R.A. Epigenetic reprogramming and small RNA silencing of transposable elements in pollen. Cell 2009, 136, 461–472. [Google Scholar] [CrossRef]
- Schoft, V.K.; Chumak, N.; Choi, Y.; Hannon, M.; Garcia-Aguilar, M.; Machlicova, A.; Slusarz, L.; Mosiolek, M.; Park, J.S.; Park, G.T.; et al. Function of the DEMETER DNA glycosylase in the Arabidopsis thaliana male gametophyte. Proc. Natl. Acad. Sci. USA 2011, 108, 8042–8047. [Google Scholar] [CrossRef]
- Martinez, G.; Panda, K.; Kohler, C.; Slotkin, R.K. Silencing in sperm cells is directed by RNA movement from the surrounding nurse cell. Nat. Plants 2016, 2, 16030. [Google Scholar] [CrossRef]
- Gehring, M.; Bubb, K.L.; Henikoff, S. Extensive demethylation of repetitive elements during seed development underlies gene imprinting. Science 2009, 324, 1447–1451. [Google Scholar] [CrossRef]
- Wang, G.; Jiang, H.; Del Toro de Leon, G.; Martinez, G.; Kohler, C. Sequestration of a transposon-derived siRNA by a target mimic imprinted gene induces postzygotic reproductive isolation in Arabidopsis. Dev. Cell 2018, 46, 696–705. [Google Scholar] [CrossRef]
- Ten Hove, C.A.; Lu, K.J.; Weijers, D. Building a plant: Cell fate specification in the early Arabidopsis embryo. Development 2015, 142, 420–430. [Google Scholar] [CrossRef]
- Zhang, M.; Xie, S.; Dong, X.; Zhao, X.; Zeng, B.; Chen, J.; Li, H.; Yang, W.; Zhao, H.; Wang, G.; et al. Genome-wide high resolution parental-specific DNA and histone methylation maps uncover patterns of imprinting regulation in maize. Genome Res. 2014, 24, 167–176. [Google Scholar] [CrossRef]
- Bouyer, D.; Kramdi, A.; Kassam, M.; Heese, M.; Schnittger, A.; Roudier, F.; Colot, V. DNA methylation dynamics during early plant life. Genome Biol. 2017, 18, 179. [Google Scholar] [CrossRef]
- Kawakatsu, T.; Nery, J.R.; Castanon, R.; Ecker, J.R. Dynamic DNA methylation reconfiguration during seed development and germination. Genome Biol. 2017, 18, 171. [Google Scholar] [CrossRef]
- Narsai, R.; Gouil, Q.; Secco, D.; Srivastava, A.; Karpievitch, Y.V.; Liew, L.C.; Lister, R.; Lewsey, M.G.; Whelan, J. Extensive transcriptomic and epigenomic remodelling occurs during Arabidopsis thaliana germination. Genome Biol. 2017, 18, 172. [Google Scholar] [CrossRef]
- An, Y.C.; Goettel, W.; Han, Q.; Bartels, A.; Liu, Z.; Xiao, W. Dynamic Changes of Genome-Wide DNA Methylation during Soybean Seed Development. Sci. Rep. 2017, 7, 12263. [Google Scholar] [CrossRef]
- Stroud, H.; Greenberg, M.V.; Feng, S.; Bernatavichute, Y.V.; Jacobsen, S.E. Comprehensive analysis of silencing mutants reveals complex regulation of the Arabidopsis methylome. Cell 2013, 152, 352–364. [Google Scholar] [CrossRef]
- Li, Q.; Eichten, S.R.; Hermanson, P.J.; Zaunbrecher, V.M.; Song, J.; Wendt, J.; Rosenbaum, H.; Madzima, T.F.; Sloan, A.E.; Huang, J.; et al. Genetic perturbation of the maize methylome. Plant Cell 2014, 26, 4602–4616. [Google Scholar] [CrossRef]
- Grover, J.W.; Kendall, T.; Baten, A.; Burgess, D.; Freeling, M.; King, G.J.; Mosher, R.A. Maternal components of RNA-directed DNA methylation are required for seed development in Brassica rapa. Plant J. 2018, 94, 575–582. [Google Scholar] [CrossRef]
- Mirouze, M.; Reinders, J.; Bucher, E.; Nishimura, T.; Schneeberger, K.; Ossowski, S.; Cao, J.; Weigel, D.; Paszkowski, J.; Mathieu, O. Selective epigenetic control of retrotransposition in Arabidopsis. Nature 2009, 461, 427–430. [Google Scholar] [CrossRef]
- Lotan, L.; Ohto, M.-A.; Yee, K.M.; West, M.A.L.; Lo, R.; Kwong, R.W.; Yamagishi, K.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. Arabidopsis LEAFY COTYLEDON1 is sufficient to induce embryo development in vegetative cells. Cell 1998, 93, 1195–1205. [Google Scholar] [CrossRef]
- Lee, H.; Fischer, R.L.; Goldberg, R.B.; Harada, J.J. Arabidopsis LEAFY COTYLEDON1 represents a functionally specialized subunit of the CCAAT binding transcription factor. Proc. Natl. Acad. Sci. USA 2003, 100, 2152–2156. [Google Scholar] [CrossRef]
- Tao, Z.; Shen, L.; Gu, X.; Wang, Y.; Yu, H.; He, Y. Embryonic epigenetic reprogramming by a pioneer transcription factor in plants. Nature 2017, 551, 124–128. [Google Scholar] [CrossRef]
- Crevillen, P.; Yang, H.; Cui, X.; Greeff, C.; Trick, M.; Qiu, Q.; Cao, X.; Dean, C. Epigenetic reprogramming that prevents transgenerational inheritance of the vernalized state. Nature 2014, 515, 587–590. [Google Scholar] [CrossRef]
- Bouyer, D.; Roudier, F.; Heese, M.; Andersen, E.D.; Gey, D.; Nowack, M.K.; Goodrich, J.; Renou, J.P.; Grini, P.E.; Colot, V.; et al. Polycomb repressive complex 2 controls the embryo-to-seedling phase transition. PLoS Genet. 2011, 7, e1002014. [Google Scholar] [CrossRef]
- Kohler, C.; Hennig, L.; Spillane, C.; Pien, S.; Gruissem, W.; Grossniklaus, U. The Polycomb-group protein MEDEA regulates seed development by controlling expression of the MADS-box gene PHERES1. Genes Dev. 2003, 17, 1540–1553. [Google Scholar] [CrossRef]
- Zhang, S.; Wang, D.; Zhang, H.; Skaggs, M.I.; Lloyd, A.; Ran, D.; An, L.; Schumaker, K.S.; Drews, G.N.; Yadegari, R. FERTILIZATION-INDEPENDENT SEED-Polycomb Repressive Complex 2 Plays a Dual Role in Regulating Type I MADS-Box Genes in Early Endosperm Development. Plant Physiol. 2018, 177, 285–299. [Google Scholar] [CrossRef]
- Kang, I.H.; Steffen, J.G.; Portereiko, M.F.; Lloyd, A.; Drews, G.N. The AGL62 MADS domain protein regulates cellularization during endosperm development in Arabidopsis. Plant Cell 2008, 20, 635–647. [Google Scholar] [CrossRef]
- Kradolfer, D.; Hennig, L.; Kohler, C. Increased maternal genome dosage bypasses the requirement of the FIS polycomb repressive complex 2 in Arabidopsis seed development. PLoS Genet. 2013, 9, e1003163. [Google Scholar] [CrossRef]
- Nowack, M.K.; Shirzadi, R.; Dissmeyer, N.; Dolf, A.; Endl, E.; Grini, P.E.; Schnittger, A. Bypassing genomic imprinting allows seed development. Nature 2007, 447, 312–315. [Google Scholar] [CrossRef]
- Liu, J.; Deng, S.; Wang, H.; Ye, J.; Wu, H.W.; Sun, H.X.; Chua, N.H. CURLY LEAF Regulates Gene Sets Coordinating Seed Size and Lipid Biosynthesis. Plant Physiol. 2016, 171, 424–436. [Google Scholar] [CrossRef]
- Nallamilli, B.R.; Zhang, J.; Mujahid, H.; Malone, B.M.; Bridges, S.M.; Peng, Z. Polycomb group gene OsFIE2 regulates rice (Oryza sativa) seed development and grain filling via a mechanism distinct from Arabidopsis. PLoS Genet. 2013, 9, e1003322. [Google Scholar] [CrossRef]
- Folsom, J.J.; Begcy, K.; Hao, X.; Wang, D.; Walia, H. Rice fertilization-Independent Endosperm1 regulates seed size under heat stress by controlling early endosperm development. Plant Physiol. 2014, 165, 238–248. [Google Scholar] [CrossRef]
- Jacob, Y.; Bergamin, E.; Donoghue, M.T.; Mongeon, V.; LeBlanc, C.; Voigt, P.; Underwood, C.J.; Brunzelle, J.S.; Michaels, S.D.; Reinberg, D.; et al. Selective methylation of histone H3 variant H3.1 regulates heterochromatin replication. Science 2014, 343, 1249–1253. [Google Scholar] [CrossRef]
- Villar, C.B.; Erilova, A.; Makarevich, G.; Trosch, R.; Kohler, C. Control of PHERES1 imprinting in Arabidopsis by direct tandem repeats. Mol. Plant 2009, 2, 654–660. [Google Scholar] [CrossRef]
- Wohrmann, H.J.; Gagliardini, V.; Raissig, M.T.; Wehrle, W.; Arand, J.; Schmidt, A.; Tierling, S.; Page, D.R.; Schob, H.; Walter, J.; et al. Identification of a DNA methylation-independent imprinting control region at the Arabidopsis MEDEA locus. Genes Dev. 2012, 26, 1837–1850. [Google Scholar] [CrossRef]
- Moreno-Romero, J.; Jiang, H.; Santos-Gonzalez, J.; Kohler, C. Parental epigenetic asymmetry of PRC2-mediated histone modifications in the Arabidopsis endosperm. EMBO J. 2016, 35, 1298–1311. [Google Scholar] [CrossRef]
- Makarevitch, I.; Eichten, S.R.; Briskine, R.; Waters, A.J.; Danilevskaya, O.N.; Meeley, R.B.; Myers, C.L.; Vaughn, M.W.; Springer, N.M. Genomic distribution of maize facultative heterochromatin marked by trimethylation of H3K27. Plant Cell 2013, 25, 780–793. [Google Scholar] [CrossRef]
- Lever, M.A.; Th’ng, J.P.; Sun, X.; Hendzel, M.J. Rapid exchange of histone H1.1 on chromatin in living human cells. Nature 2000, 408, 873–876. [Google Scholar] [CrossRef]
- Rutowicz, K.; Lirski, M.; Mermaz, B.; Teano, G.; Schubert, J.; Mestiri, I.; Kroten, M.A.; Fabrice, T.N.; Fritz, S.; Grob, S.; et al. Linker histones are fine-scale chromatin architects modulating developmental decisions in Arabidopsis. Genome Biol. 2019, 20, 157. [Google Scholar] [CrossRef]
- Fan, Y.; Nikitina, T.; Zhao, J.; Fleury, T.J.; Bhattacharyya, R.; Bouhassira, E.E.; Stein, A.; Woodcock, C.L.; Skoultchi, A.I. Histone H1 depletion in mammals alters global chromatin structure but causes specific changes in gene regulation. Cell 2005, 123, 1199–1212. [Google Scholar] [CrossRef]
- Rea, M.; Zheng, W.; Chen, M.; Braud, C.; Bhangu, D.; Rognan, T.N.; Xiao, W. Histone H1 affects gene imprinting and DNA methylation in Arabidopsis. Plant J. 2012, 71, 776–786. [Google Scholar] [CrossRef]
- Lyons, D.B.; Zilberman, D. DDM1 and Lsh remodelers allow methylation of DNA wrapped in nucleosomes. Elife 2017, 6, e30674. [Google Scholar] [CrossRef]
- Frost, J.M.; Kim, M.Y.; Park, G.T.; Hsieh, P.H.; Nakamura, M.; Lin, S.J.H.; Yoo, H.; Choi, J.; Ikeda, Y.; Kinoshita, T.; et al. FACT complex is required for DNA demethylation at heterochromatin during reproduction in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E4720–E4729. [Google Scholar] [CrossRef]
- He, S.; Vickers, M.; Zhang, J.; Feng, X. Natural depletion of histone H1 in sex cells causes DNA demethylation, heterochromatin decondensation and transposon activation. Elife 2019, 8. [Google Scholar] [CrossRef]
- Ingouff, M.; Rademacher, S.; Holec, S.; Soljic, L.; Xin, N.; Readshaw, A.; Foo, S.H.; Lahouze, B.; Sprunck, S.; Berger, F. Zygotic resetting of the HISTONE 3 variant repertoire participates in epigenetic reprogramming in Arabidopsis. Curr. Biol. 2010, 20, 2137–2143. [Google Scholar] [CrossRef]
- Wang, Z.; Cao, H.; Chen, F.; Liu, Y. The roles of histone acetylation in seed performance and plant development. Plant Physiol. Biochem. 2014, 84, 125–133. [Google Scholar] [CrossRef]
- Zhong, X.; Zhang, H.; Zhao, Y.; Sun, Q.; Hu, Y.; Peng, H.; Zhou, D.X. The rice NAD(+)-dependent histone deacetylase OsSRT1 targets preferentially to stress- and metabolism-related genes and transposable elements. PLoS ONE 2013, 8, e66807. [Google Scholar] [CrossRef]
- Cigliano, R.A.; Cremona, G.; Paparo, R.; Termolino, P.; Perrella, G.; Gutzat, R.; Consiglio, M.F.; Conicella, C. Histone deacetylase AtHDA7 is required for female gametophyte and embryo development in Arabidopsis. Plant Physiol. 2013, 163, 431–440. [Google Scholar] [CrossRef]
- Liu, Z.; Makaroff, C.A. Arabidopsis separase AESP is essential for embryo development and the release of cohesin during meiosis. Plant Cell 2006, 18, 1213–1225. [Google Scholar] [CrossRef]
- Perrella, G.; Consiglio, M.F.; Aiese-Cigliano, R.; Cremona, G.; Sanchez-Moran, E.; Barra, L.; Errico, A.; Bressan, R.A.; Franklin, F.C.; Conicella, C. Histone hyperacetylation affects meiotic recombination and chromosome segregation in Arabidopsis. Plant J. 2010, 62, 796–806. [Google Scholar] [CrossRef]
- Van Ex, F.; Jacob, Y.; Martienssen, R.A. Multiple roles for small RNAs during plant reproduction. Curr. Opin. Plant Biol. 2011, 14, 588–593. [Google Scholar] [CrossRef]
- Johnson, C.; Kasprzewska, A.; Tennessen, K.; Fernandes, J.; Nan, G.L.; Walbot, V.; Sundaresan, V.; Vance, V.; Bowman, L.H. Clusters and superclusters of phased small RNAs in the developing inflorescence of rice. Genome Res. 2009, 19, 1429–1440. [Google Scholar] [CrossRef]
- Song, X.; Li, P.; Zhai, J.; Zhou, M.; Ma, L.; Liu, B.; Jeong, D.H.; Nakano, M.; Cao, S.; Liu, C.; et al. Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J. 2012, 69, 462–474. [Google Scholar] [CrossRef]
- Zhai, J.; Zhang, H.; Arikit, S.; Huang, K.; Nan, G.L.; Walbot, V.; Meyers, B.C. Spatiotemporally dynamic, cell-type-dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc. Natl. Acad. Sci. USA 2015, 112, 3146–3151. [Google Scholar] [CrossRef]
- Nonomura, K.; Morohoshi, A.; Nakano, M.; Eiguchi, M.; Miyao, A.; Hirochika, H.; Kurata, N. A germ cell specific gene of the ARGONAUTE family is essential for the progression of premeiotic mitosis and meiosis during sporogenesis in rice. Plant Cell 2007, 19, 2583–2594. [Google Scholar] [CrossRef] [PubMed]
- Komiya, R.; Ohyanagi, H.; Niihama, M.; Watanabe, T.; Nakano, M.; Kurata, N.; Nonomura, K. Rice germline-specific Argonaute MEL1 protein binds to phasiRNAs generated from more than 700 lincRNAs. Plant J. 2014, 78, 385–397. [Google Scholar] [CrossRef] [PubMed]
- Zilberman, D. Role of Arabidopsis ARGONAUTE4 in RNA-directed DNA methylation triggered by inverted repeats. Curr. Biol. 2004, 14, 1214–1220. [Google Scholar] [CrossRef] [PubMed]
- Oliver, C.; Santos, J.L.; Pradillo, M. Accurate Chromosome Segregation at First Meiotic Division Requires AGO4, a Protein Involved in RNA-Dependent DNA Methylation in Arabidopsis thaliana. Genetics 2016, 204, 543–553. [Google Scholar] [CrossRef]
- Walker, J.; Gao, H.; Zhang, J.; Aldridge, B.; Vickers, M.; Higgins, J.D.; Feng, X. Sexual-lineage-specific DNA methylation regulates meiosis in Arabidopsis. Nat. Genet. 2018, 50, 130–137. [Google Scholar] [CrossRef]
- Lu, J.; Zhang, C.; Baulcombe, D.C.; Chen, Z.J. Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc. Natl. Acad. Sci. USA 2012, 109, 5529–5534. [Google Scholar] [CrossRef]
- Scott, R.J.; Spielman, M.; Bailey, J.; Dickinson, H.G. Parent-of-origin effects on seed development in Arabidopsis thaliana. Development 1998, 125, 3329–3341. [Google Scholar]
- Mosher, R.A.; Melnyk, C.W.; Kelly, K.A.; Dunn, R.M.; Studholme, D.J.; Baulcombe, D.C. Uniparental expression of PolIV-dependent siRNAs in developing endosperm of Arabidopsis. Nature 2009, 460, 283–286. [Google Scholar] [CrossRef]
- Herr, A.J.; Jensen, M.B.; Dalmay, T.; Baulcombe, D.C. RNA polymerase IV directs silencing of endogenous DNA. Science 2005, 308, 118–120. [Google Scholar] [CrossRef]
- Onodera, Y.; Haag, J.R.; Ream, T.; Costa Nunes, P.; Pontes, O.; Pikaard, C.S. Plant nuclear RNA polymerase IV mediates siRNA and DNA methylation-dependent heterochromatin formation. Cell 2005, 120, 613–622. [Google Scholar] [CrossRef]
- Rodrigues, J.A.; Ruan, R.; Nishimura, T.; Sharma, M.K.; Sharma, R.; Ronald, P.C.; Fischer, R.L.; Zilberman, D. Imprinted expression of genes and small RNA is associated with localized hypomethylation of the maternal genome in rice endosperm. Proc. Natl. Acad. Sci. USA 2013, 110, 7934–7939. [Google Scholar] [CrossRef]
- Xin, M.; Yang, R.; Yao, Y.; Ma, C.; Peng, H.; Sun, Q.; Wang, X.; Ni, Z. Dynamic parent-of-origin effects on small interfering RNA expression in the developing maize endosperm. BMC Plant Biol. 2014, 14, 192. [Google Scholar] [CrossRef]
- Pignatta, D.; Erdmann, R.M.; Scheer, E.; Picard, C.L.; Bell, G.W.; Gehring, M. Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. Elife 2014, 3, e03198. [Google Scholar] [CrossRef]
- Erdmann, R.M.; Satyaki, P.R.V.; Klosinska, M.; Gehring, M. A Small RNA Pathway Mediates Allelic Dosage in Endosperm. Cell Rep. 2017, 21, 3364–3372. [Google Scholar] [CrossRef]
- Satyaki, P.R.V.; Gehring, M. Paternally Acting Canonical RNA-Directed DNA Methylation Pathway Genes Sensitize Arabidopsis Endosperm to Paternal Genome Dosage. Plant Cell 2019, 31, 1563–1578. [Google Scholar] [CrossRef]
- Meinke, D.W. Genome-wide identification of EMBRYO-DEFECTIVE (EMB) genes required for growth and development in Arabidopsis. N. Phytol. 2019. [Google Scholar] [CrossRef]
- Xiao, W.; Brown, R.C.; Lemmon, B.E.; Harada, J.J.; Goldberg, R.B.; Fischer, R.L. Regulation of seed size by hypomethylation of maternal and paternal genomes. Plant Physiol. 2006, 142, 1160–1168. [Google Scholar] [CrossRef]
- Bartels, A.; Han, Q.; Nair, P.; Stacey, L.; Gaynier, H.; Mosley, M.; Huang, Q.Q.; Pearson, J.K.; Hsieh, T.F.; An, Y.C.; et al. Dynamic DNA methylation in plant growth and development. Int. J. Mol. Sci. 2018, 19, 2144. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).