Identification of Anther Length QTL and Construction of Chromosome Segment Substitution Lines of Oryza longistaminata


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
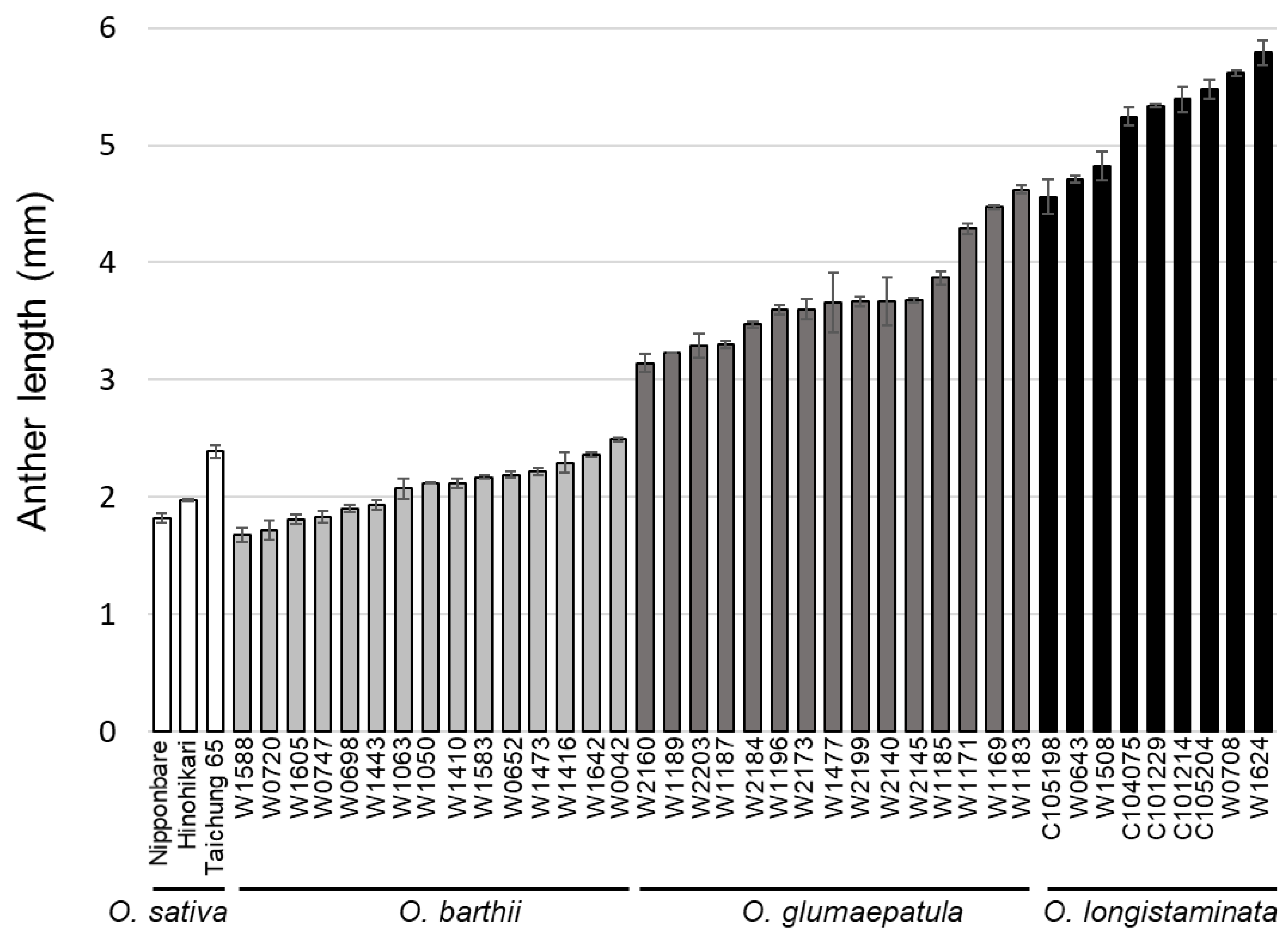
2. Results
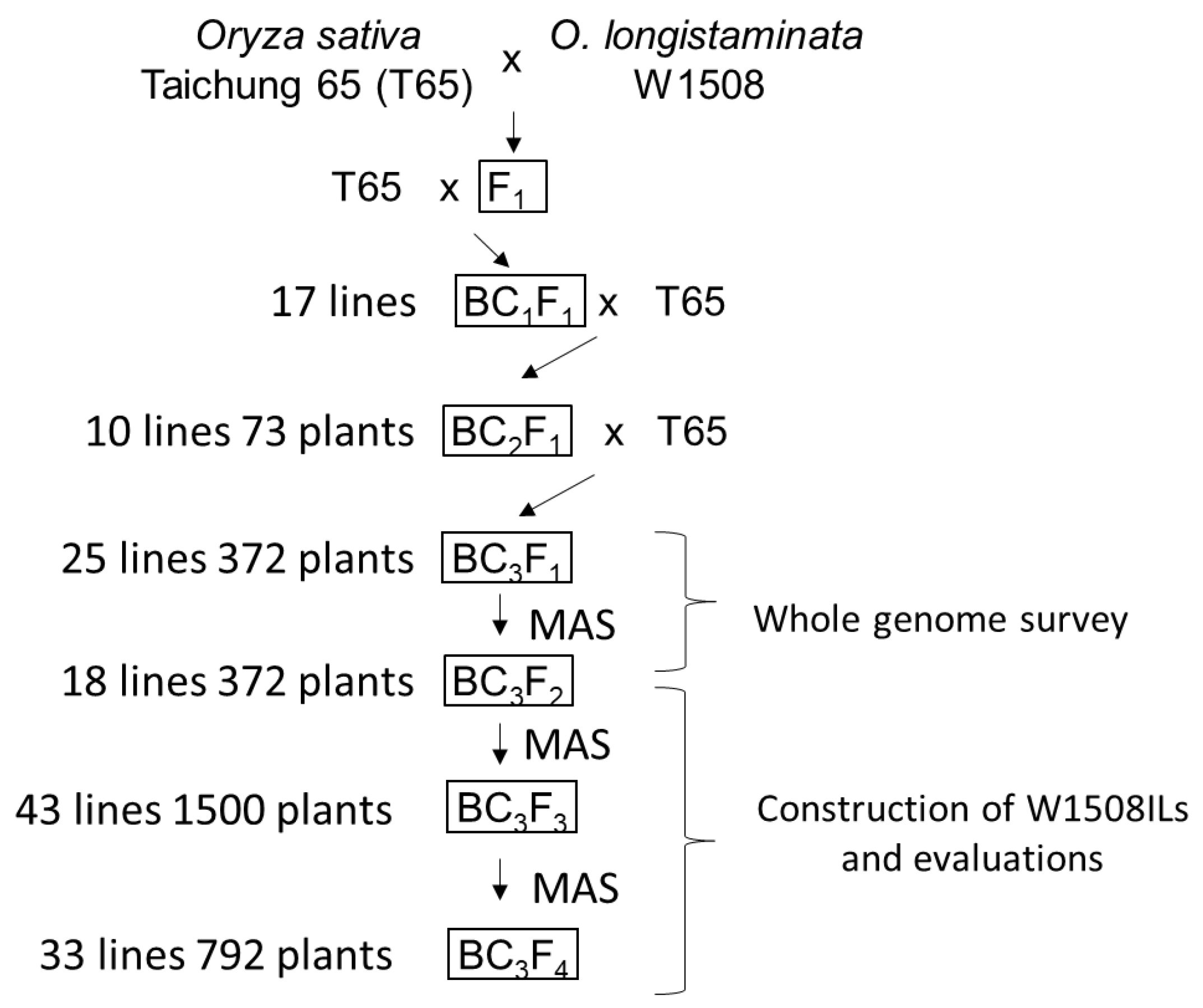
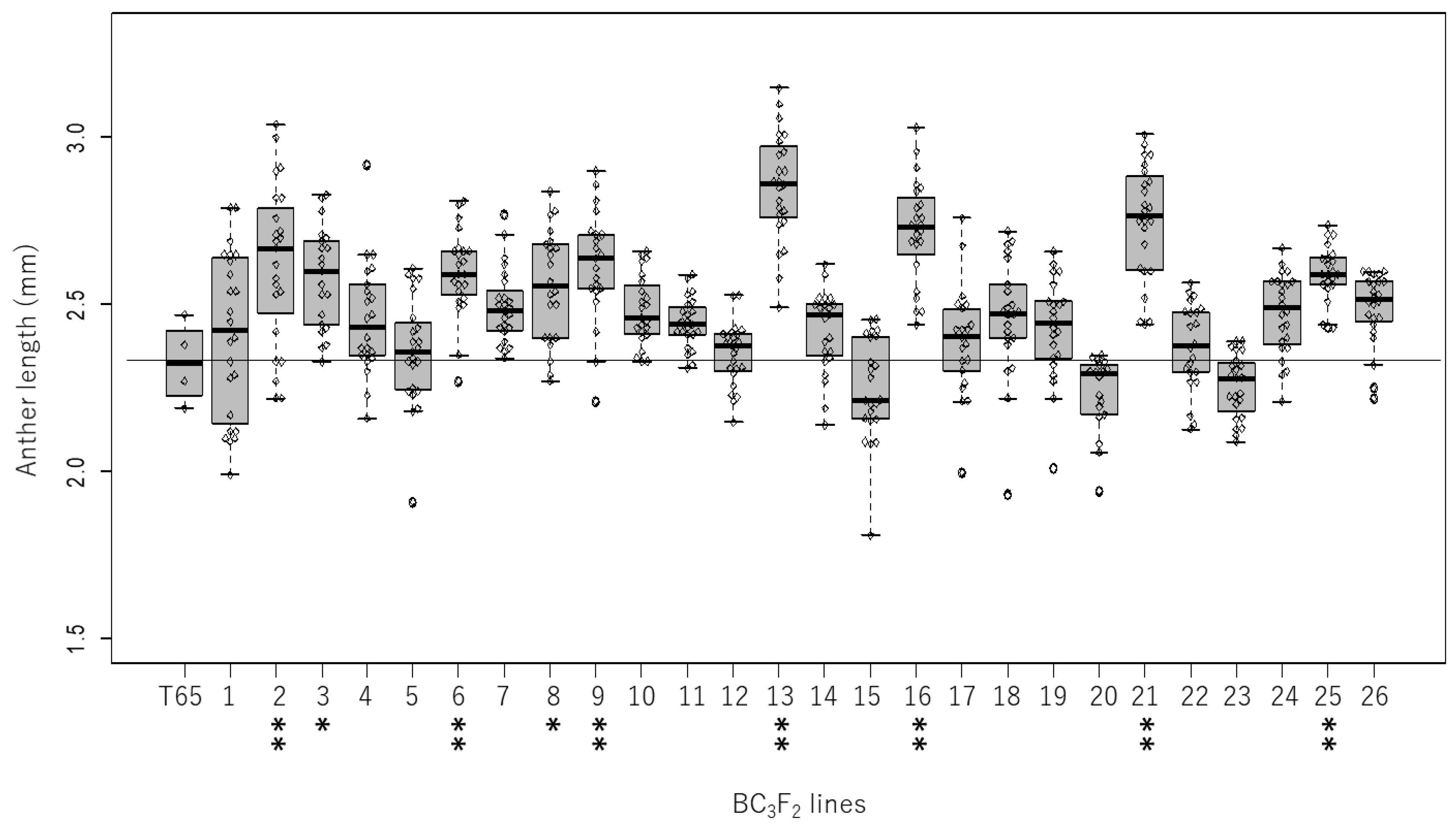
2.1. Genetic Variation in Anther Length in the Backcrossed Population
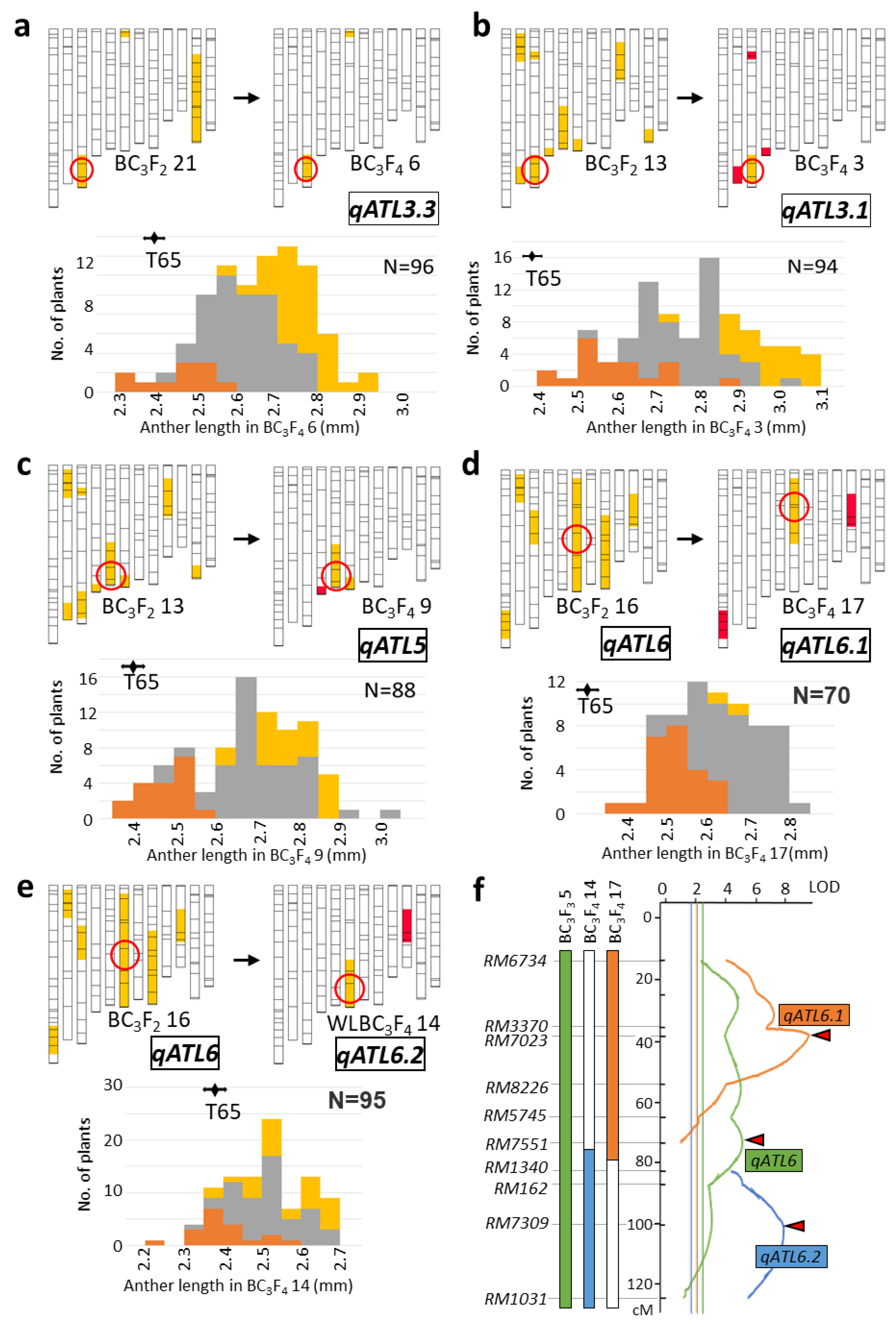
2.2. Validation of QTL
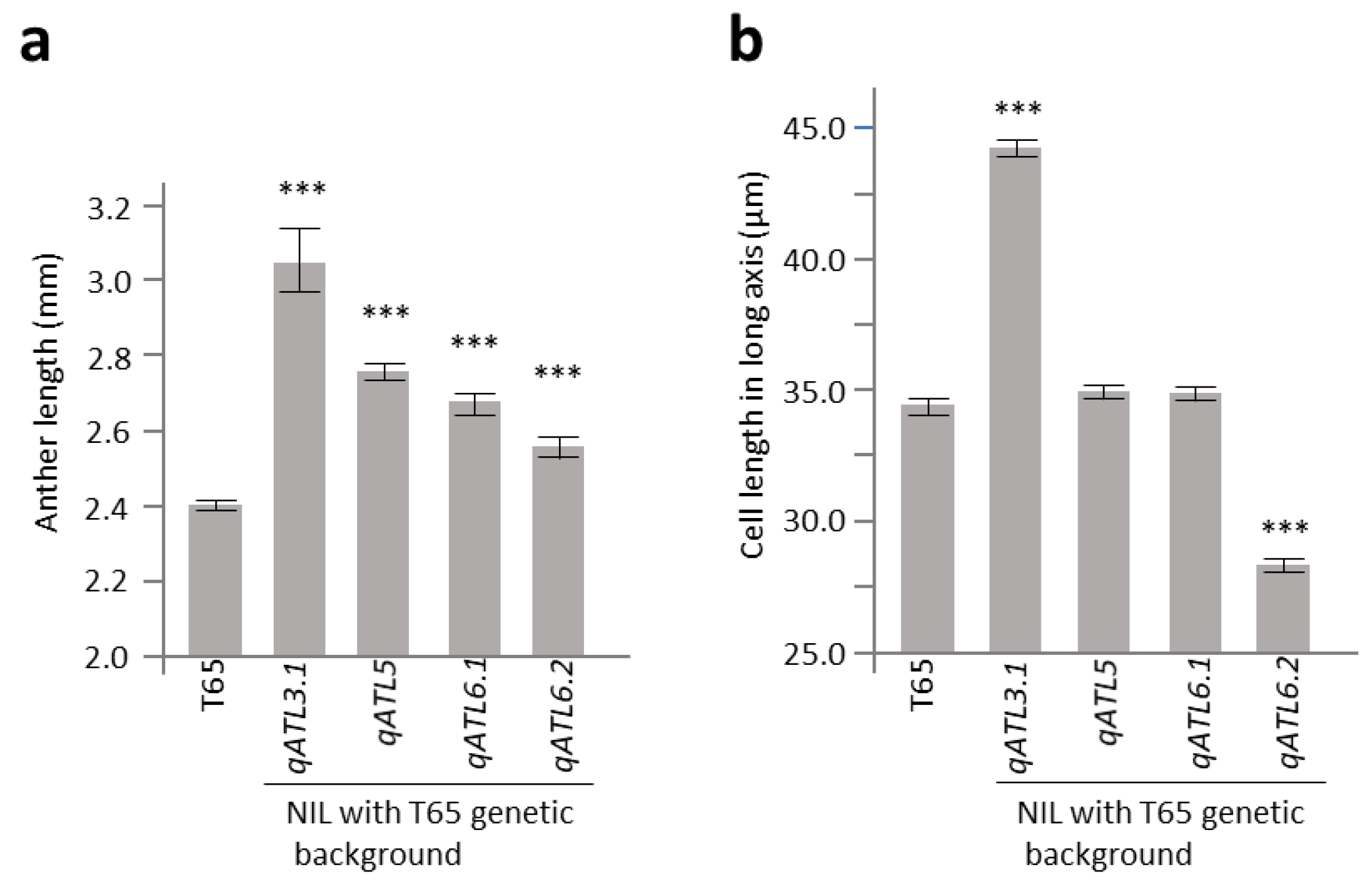
2.3. Characterization of Anther Length QTL Using Near-Isogenic Lines
2.4. Evaluation on W1508Ils
3. Discussion
4. Materials and Methods
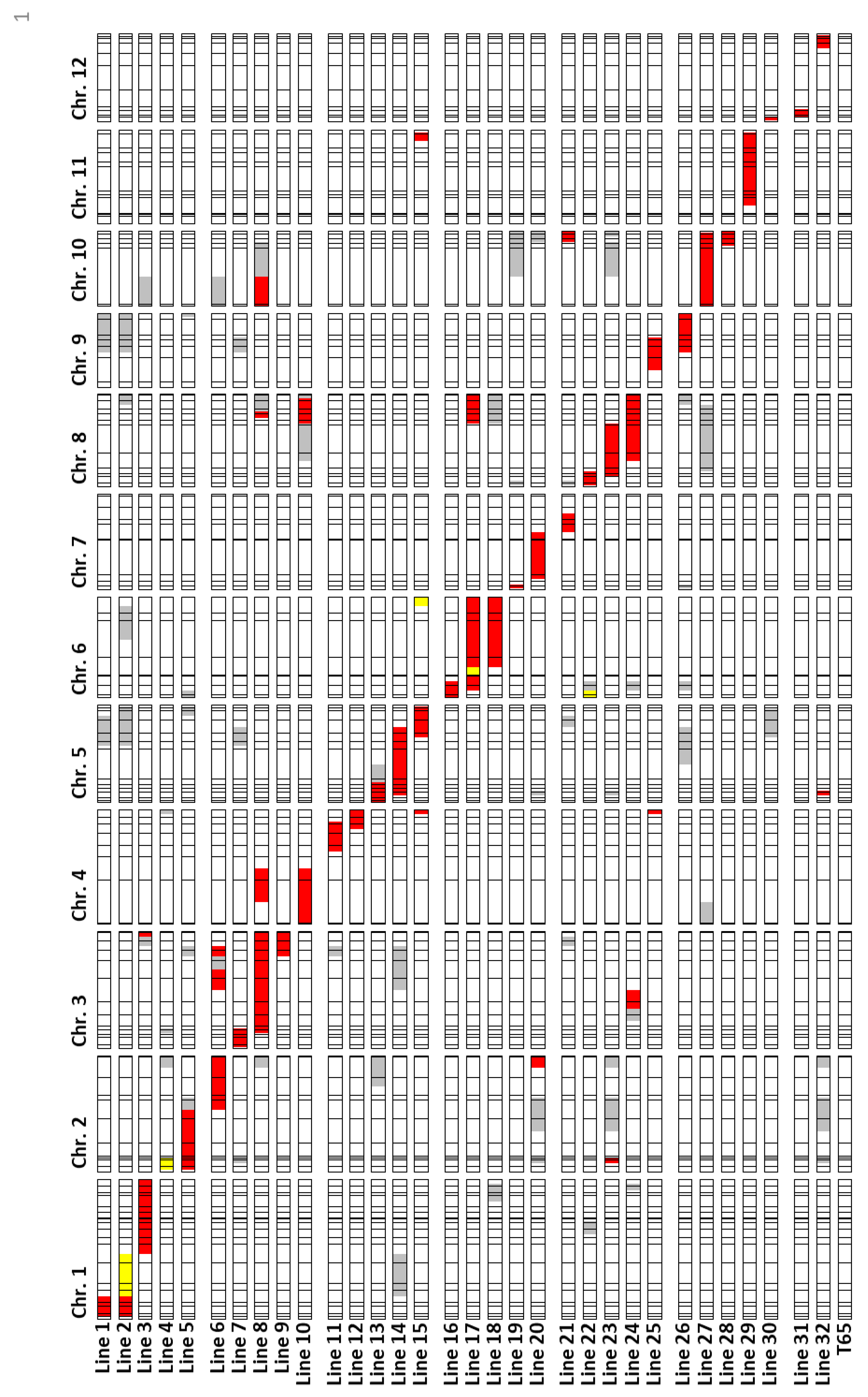
4.1. Plant Materials
4.2. Measurement of Anther Length
4.3. Genotyping
4.4. QTL Analysis
4.5. Measurement of Numbers of Pollen Grains
4.6. Observation of Epidermal Cells of Anthers
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Friedman, J.; Rubin, M.J. All in good time: Understanding annual and perennial strategies in plants. Am. J. Bot. 2015, 102, 497–499. [Google Scholar] [CrossRef] [PubMed]
- Kuroda, Y.; Urajrong, H.; Sato, Y.I. Population genetic structure of wild rice (Oryza rufipogon) in mainland southeast Asia as revealed by microsatellite polymorphisms. Topics 2002, 12, 159–170. [Google Scholar] [CrossRef][Green Version]
- Annavi, G.; Newman, C.; Buesching, C.D.; Macdonald, D.W.; Burke, T.; Dugdale, H.L. Heterozygosity-fitness correlations in a wild mammal population: Accounting for parental and environmental effects. Ecol. Evol. 2014, 4, 2594–2609. [Google Scholar] [CrossRef] [PubMed]
- Charpentier, M.J.E.; Williams, C.V.; Drea, C.M. Inbreeding depression in ring-tailed lemurs (Lemur catta): Genetic diversity predicts parasitism, immunocompetence, and survivorship. Conserv. Genet. 2008, 9, 1605–1615. [Google Scholar] [CrossRef]
- Slate, J.; Kruuk, L.E.B.; Marshall, T.C.; Pemberton, J.M.; Clutton-Brock, T.H. Inbreeding depression influences lifetime breeding success in a wild population of red deer (Cervus elaphus). Proc. R. Soc. Lond. B 2000, 267, 1657–1662. [Google Scholar] [CrossRef] [PubMed]
- Acevedo-Whitehouse, K.; Vicente, J.; Gortazar, C.; Hofle, U.; Fernandez-de-Mera, I.G.; Amos, W. Genetic resistance to bovine tuberculosis in the Iberian wild boar. Mol. Ecol. 2005, 14, 3209–3217. [Google Scholar] [CrossRef]
- David, P. Heterozygosity-fitness correlations: New perspectives on old problems. Heredity 1998, 80, 531–537. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, D. Evolution of plant breeding systems. Curr. Biol. 2006, 16, 726–735. [Google Scholar] [CrossRef]
- Hamrick, J.L.; Godt, M.J.W. Effects of life history traits on genetic diversity in plant species. Philos. Trans. R Soc. Biol. Sci. 1977, 351, 1291–1298. [Google Scholar] [CrossRef]
- Vaughan, D.A.; Lu, B.R.; Tomooka, N. The evolving story of rice evolution. Plant Sci. 2008, 174, 394–408. [Google Scholar] [CrossRef]
- Banaticla-Hilario, M.C.N.; Berg, R.G.; Hamilton, N.R.S.; McNally, K.L. Local differentiation amidst extensive allele sharing in Oryza nivara and O. rufipogon. Ecol. Evol. 2013, 3, 3047–3062. [Google Scholar] [CrossRef]
- Liu, R.; Zheng, X.M.; Zhou, L.; Zhou, H.F.; Ge, S. Population genetic structure of Oryza rufipogon and Oryza nivara: Implications for the origin of O. nivara. Mol. Ecol. 2015, 24, 5211–5228. [Google Scholar] [CrossRef] [PubMed]
- Reuscher, S.; Furuta, T.; Bessho-Uehara, K.; Cosi, M.; Jena, K.K.; Toyoda, A.; Fujiyama, A.; Kurata, N.; Ashikari, M. Assembling the genome of the African wild rice Oryza longistaminata by exploiting synteny in closely related Oryza species. Commun. Biol. 2018, 1, 162. [Google Scholar] [CrossRef] [PubMed]
- Hiroi, K.; Mamun, A.A.; Wada, T.; Takeoka, Y. A Study on the interspecific variation of spikelet structure in the genus Oryza. Jpn. J. Crop Sci. 1991, 60, 153–160. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, S.; Liu, H.; Fu, B.; Li, L.; Xie, M.; Song, Y.; Li, X.; Cai, J.; Wan, W.; et al. Genome and comparative transcriptomics of African wild rice Oryza longistaminata provide insights into molecular mechanism of rhizomatousness and self-incompatibility. Mol. Plant 2015, 8, 1683–1686. [Google Scholar] [CrossRef]
- Chu, Y.E.; Morishima, H.; Oka, H.I. Partial self-incompatibility found in Oryza perennis subsp. barthii. Gene Genet. Sys. 1969, 44, 225–229. [Google Scholar] [CrossRef][Green Version]
- Sahadevan, P.C.; Namboodiri, K.M.N. Natural crossing in rice. Proc. Indian Acad. Sci. Sect. B 1963, 58, 176–185. [Google Scholar]
- Oka, H.I.; Morishima, H. Variations in the breeding systems of wild rice, Oryza perennis. Evolution 1967, 21, 249–258. [Google Scholar] [CrossRef]
- Sakai, K.I.; Narise, T. Studies on the breeding behavior of wild rice. Annu. Rep. Natl. Inst. Genet. Jpn. 1959, 9, 64–65. [Google Scholar]
- Eshed, Y.; Zamir, D. A genomic library of Lycopersicon pennellii in L. esculentum: A tool for fine mapping genes. Euphytica 1994, 79, 175–179. [Google Scholar] [CrossRef]
- Eshed, Y.; Zamir, D. An introgression line population of Lycopersicon pennellii in the cultivated tomato enables the identification and fine mapping of yield associated QTL. Genetics 1995, 141, 1147–1162. [Google Scholar] [PubMed]
- Eshed, Y.; Zamir, D. Less-than-additive epistatic interactions of quantitative trait loci in tomato. Genetics 1996, 143, 1807–1817. [Google Scholar] [PubMed]
- Chetelat, R.T.; Meglic, V. Molecular mapping of chromosome segments introgressed from Solanum lycopersicoides into cultivated tomato (Lycopersicon esculentum). Theor. Appl. Genet. 2000, 100, 232–241. [Google Scholar] [CrossRef]
- Grandillo, S.; Ku, H.M.; Tanksley, S.D. Characterization of fs8.1, a major QTL influencing fruit shape in tomato. Mol. Breed. 1996, 2, 251–260. [Google Scholar] [CrossRef]
- Yamamoto, T.; Kuboki, Y.; Lin, S.Y.; Sasaki, T.; Yano, M. Fine mapping of quantitative trait loci Hd-1, Hd-2 and Hd-3, controlling heading date of rice, as single Mendelian factor. Theor. Appl. Genet. 1998, 97, 37–44. [Google Scholar] [CrossRef]
- Monforte, A.J.; Tanksley, S.D. Fine mapping of a quantitative trait locus (QTL) from Lycopersicon hirsutum chromosome 1 affecting fruit characteristics and agronomic traits: Breaking linkage among QTLs affecting different traits and dissection of heterosis for yield. Theor. Appl. Genet. 2000, 100, 471–479. [Google Scholar] [CrossRef]
- Xiong, L.Z.; Liu, K.D.; Dai, X.K.; Xu, C.G.; Zhang, Q.F. Identification of genetic factors controlling domestication related traits of rice using an F2 population of a cross between Oryza sativa and O. rufipogon. Theor. Appl. Genet. 1999, 98, 243–251. [Google Scholar] [CrossRef]
- Cai, W.; Morishima, H. (2002) QTL clusters reflect character associations in wild and cultivated rice. Theor. Appl. Genet. 2002, 104, 1217–1228. [Google Scholar] [CrossRef]
- Uga, Y.; Fukuta, Y.; Ohsawa, R.; Fujimura, T. Variations of floral traits in Asian cultivated rice (Oryza sativa L.) and its wild relatives (O rufipogon Griff.). Breed. Sci. 2003, 53, 345–352. [Google Scholar] [CrossRef]
- Uga, Y.; Siangliw, M.; Nagamine, T.; Ohsawa, R.; Fujimura, T.; Fukuta, Y. Comparative mapping of QTLs determining glume, pistil and stamen sizes in cultivated rice (Oryza sativa L.). Plant Breed. 2010, 129, 657–669. [Google Scholar] [CrossRef]
- Goldberg, R.B.; Beals, T.P.; Sanders, P.M. Anther development: Basic principles and practical applications. Plant Cell 1993, 5, 1217–1229. [Google Scholar] [CrossRef] [PubMed]
- McConn, M.; Browse, J. The critical requirement for linolenic acid is pollen development, not photosynthesis, in an Arabidopsis Mutant. Plant Cell 1996, 8, 403–416. [Google Scholar] [CrossRef] [PubMed]
- Cecchetti, V.; Altamura, M.M.; Falasca, G.; Costantino, P.; Cardarelli, M. Auxin regulates Arabidopsis anther dehiscence, pollen maturation, and filament elongation. Plant Cell 2008, 20, 1760–1774. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Dai, X.; Zhao, Y. Auxin biosynthesis by the YUCCA flavin monooxygenases controls the formation of floral organs and vascular tissues in Arabidopsis. Genes Dev. 2006, 20, 1790–1799. [Google Scholar] [CrossRef] [PubMed]
- Jewell, J.B.; Browse, J. Epidermal jasmonate perception is sufficient for all aspects of jasmonate-mediated male fertility in Arabidopsis. Plant J. 2016, 85, 634–647. [Google Scholar] [CrossRef] [PubMed]
- Plackett, A.R.; Thomas, S.G.; Wilson, Z.A.; Hedden, P. Gibberellin control of stamen development: A fertile field. Trends Plant Sci. 2011, 16, 568–578. [Google Scholar] [CrossRef] [PubMed]
- Murray, F.; Kalla, R.; Jacobsen, J.; Gubler, F. A role for HvGAMYB in anther development. Plant J. 2003, 33, 481–491. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Dellaporta, S.L.; Wood, J.; Hicks, J.B. A plant DNA minipreparation: Version II. Plant Mol. Biol. Rep. 1983, 1, 19–21. [Google Scholar] [CrossRef]
- Broman, K.W.; Wu, H.; Sen, S.; Churchill, G.A. R/qtl: QTL mapping in experimental crosses. Bioinformatics 2003, 19, 889–890. [Google Scholar] [CrossRef]






© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogami, T.; Yasui, H.; Yoshimura, A.; Yamagata, Y. Identification of Anther Length QTL and Construction of Chromosome Segment Substitution Lines of Oryza longistaminata. Plants 2019, 8, 388. https://doi.org/10.3390/plants8100388
Ogami T, Yasui H, Yoshimura A, Yamagata Y. Identification of Anther Length QTL and Construction of Chromosome Segment Substitution Lines of Oryza longistaminata. Plants. 2019; 8(10):388. https://doi.org/10.3390/plants8100388
Chicago/Turabian StyleOgami, Takayuki, Hideshi Yasui, Atsushi Yoshimura, and Yoshiyuki Yamagata. 2019. "Identification of Anther Length QTL and Construction of Chromosome Segment Substitution Lines of Oryza longistaminata" Plants 8, no. 10: 388. https://doi.org/10.3390/plants8100388
APA StyleOgami, T., Yasui, H., Yoshimura, A., & Yamagata, Y. (2019). Identification of Anther Length QTL and Construction of Chromosome Segment Substitution Lines of Oryza longistaminata. Plants, 8(10), 388. https://doi.org/10.3390/plants8100388