

Niches and Genotypes Determine the Diversity and Composition of Microbiomes After Herbicide Treatment in Beckmannia syzigachne
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Experimental Design for B. syzigachne Growth and Sampling
2.2. Comparison of Root Niches Bacterial in the S and R Populations
2.2.1. Sample Preparation and DNA Extraction
2.2.2. 16S rRNA Amplifying and Illumina MiSeq Sequencing
2.2.3. Bioinformatics Analysis
2.3. Comparison of Leaf Endophyte Fungi in the S and R Populations
2.4. Statistical Analyses
3. Results
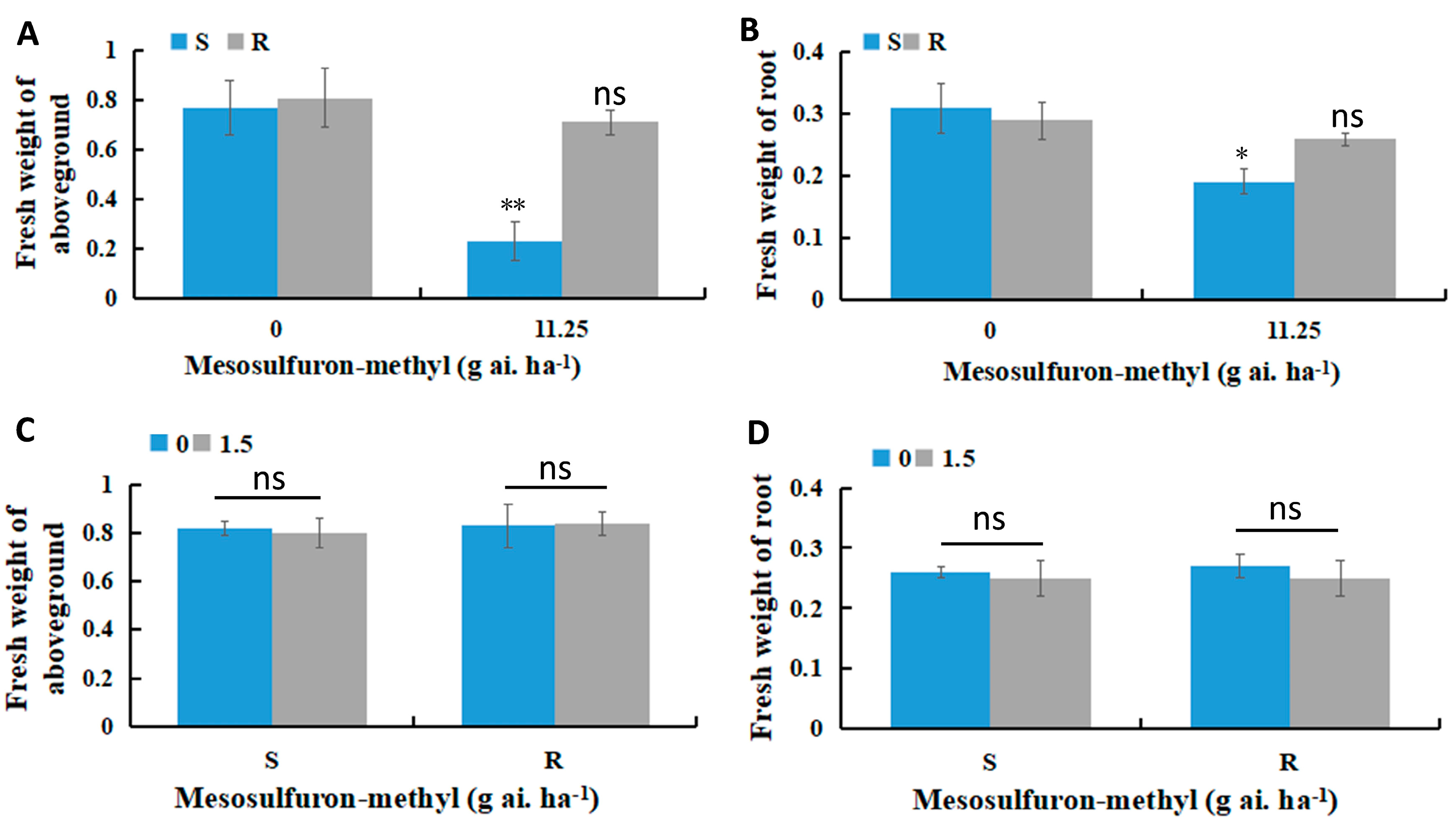
3.1. Mesosulfuron-Methyl Treatment Reduces Biomass in Sensitive B. syzigachne Populations
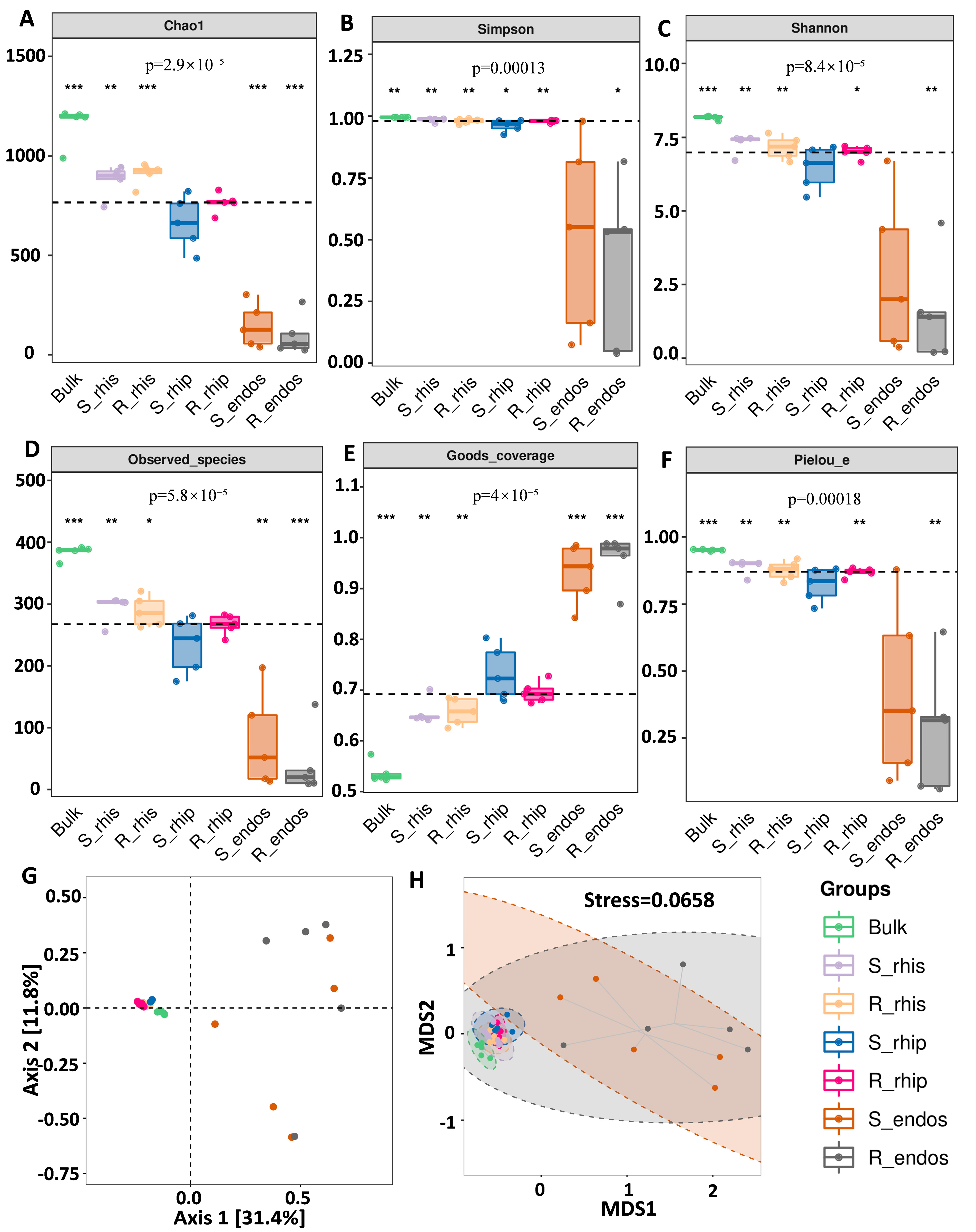
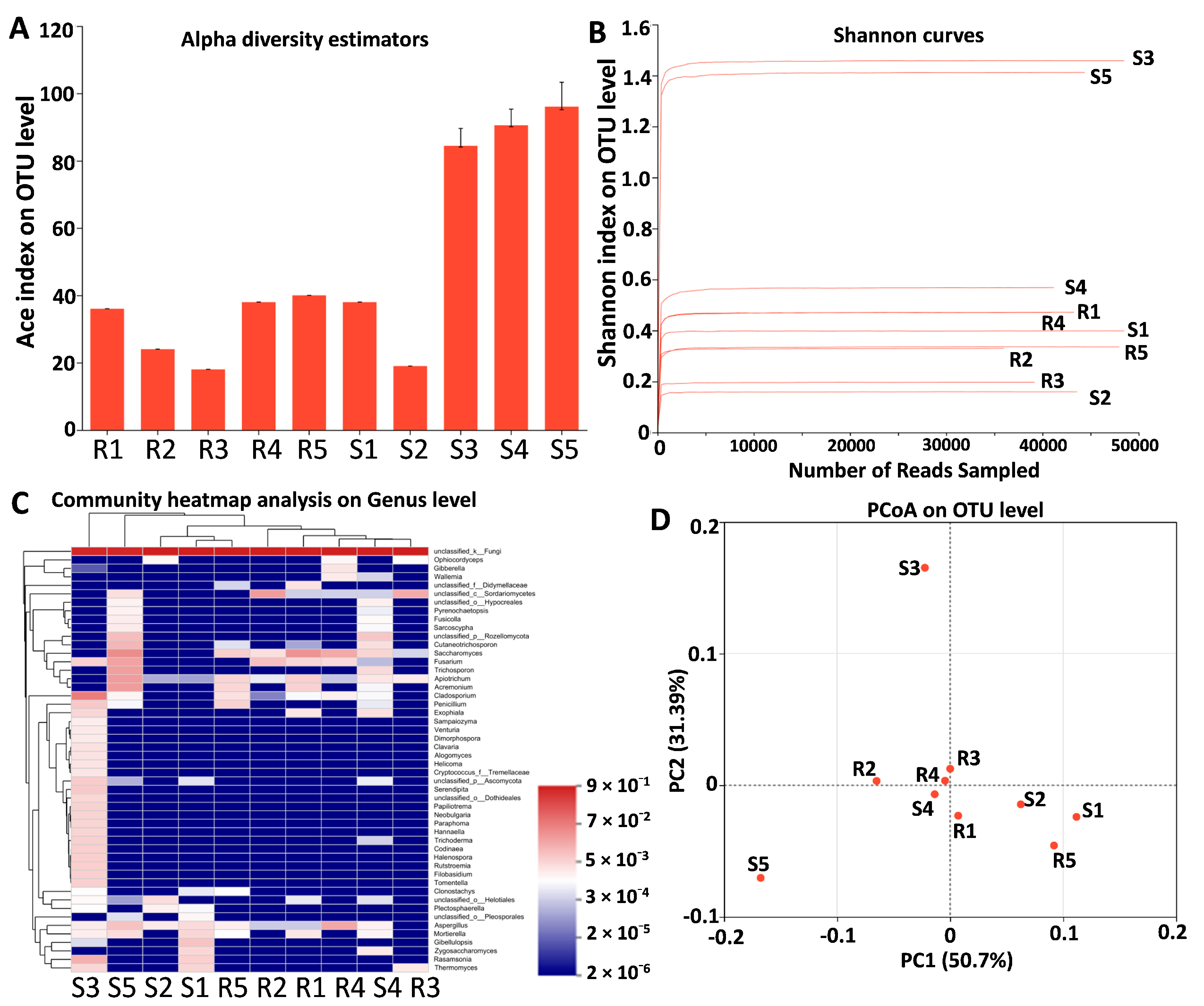
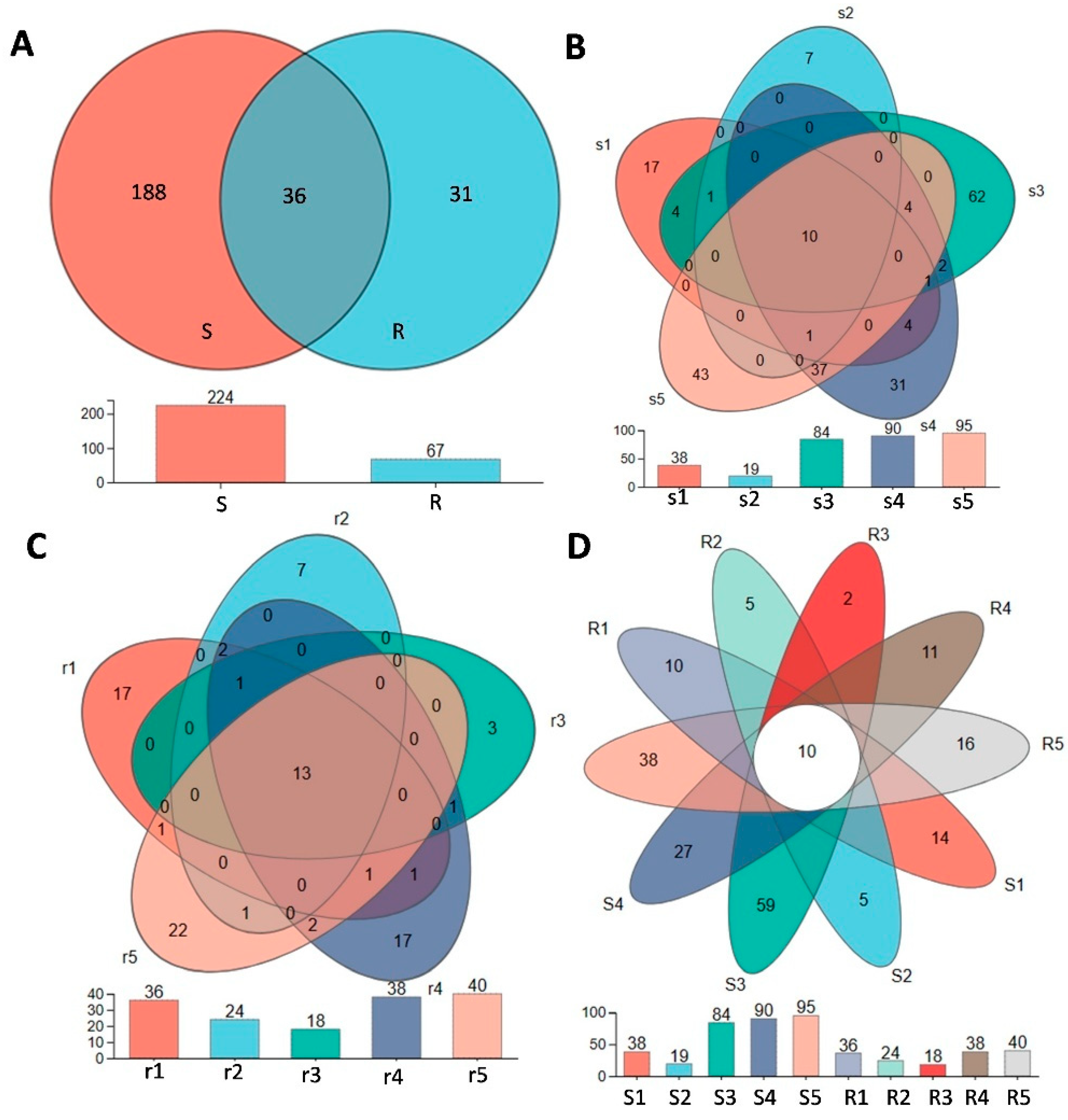
3.2. Niche and Genotype Influence the Diversity and Composition of B. syzigachne Root Microbiomes
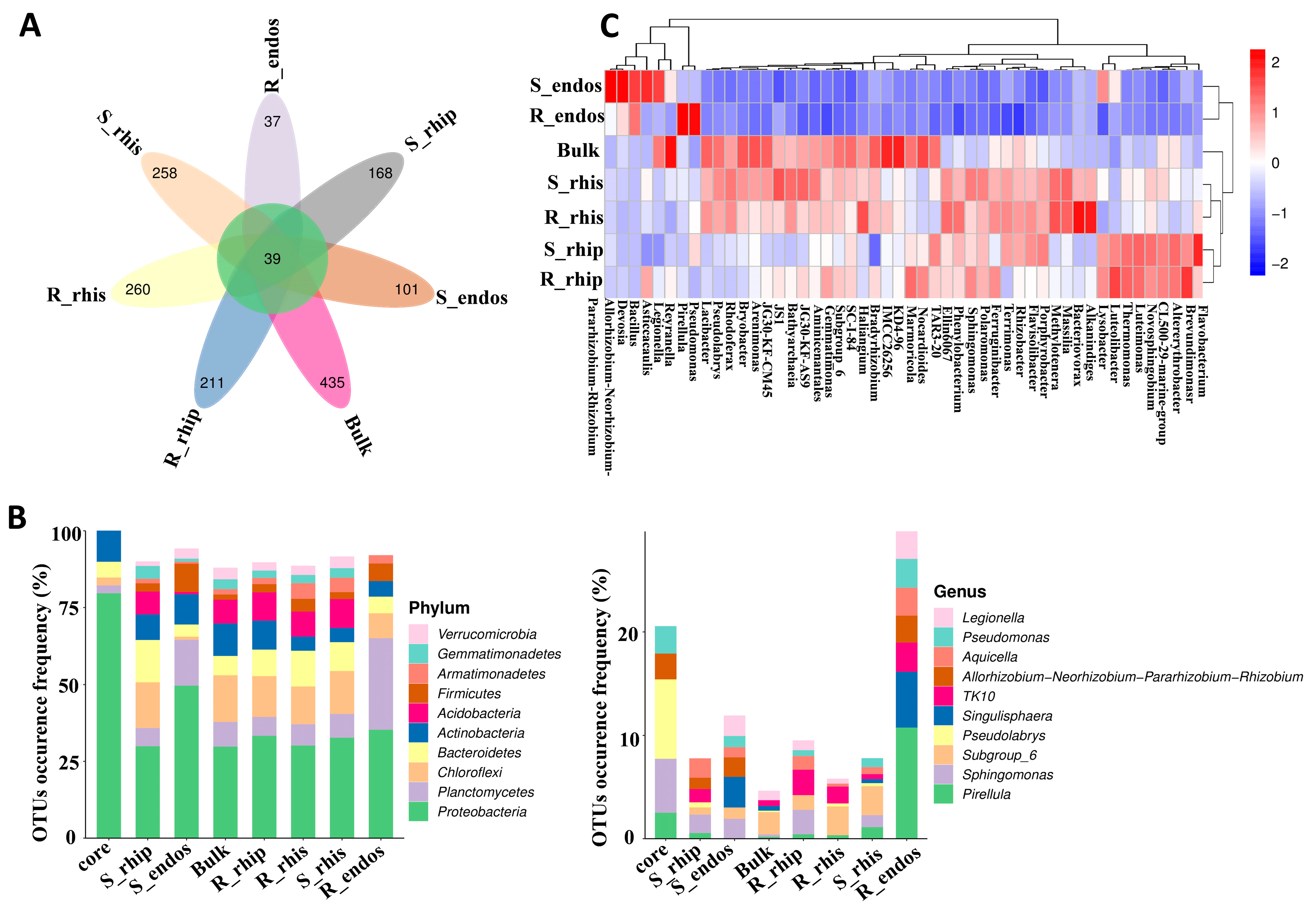
3.3. Genotypes Differentiation Analysis in Different Niches of B. syzigachne Roots
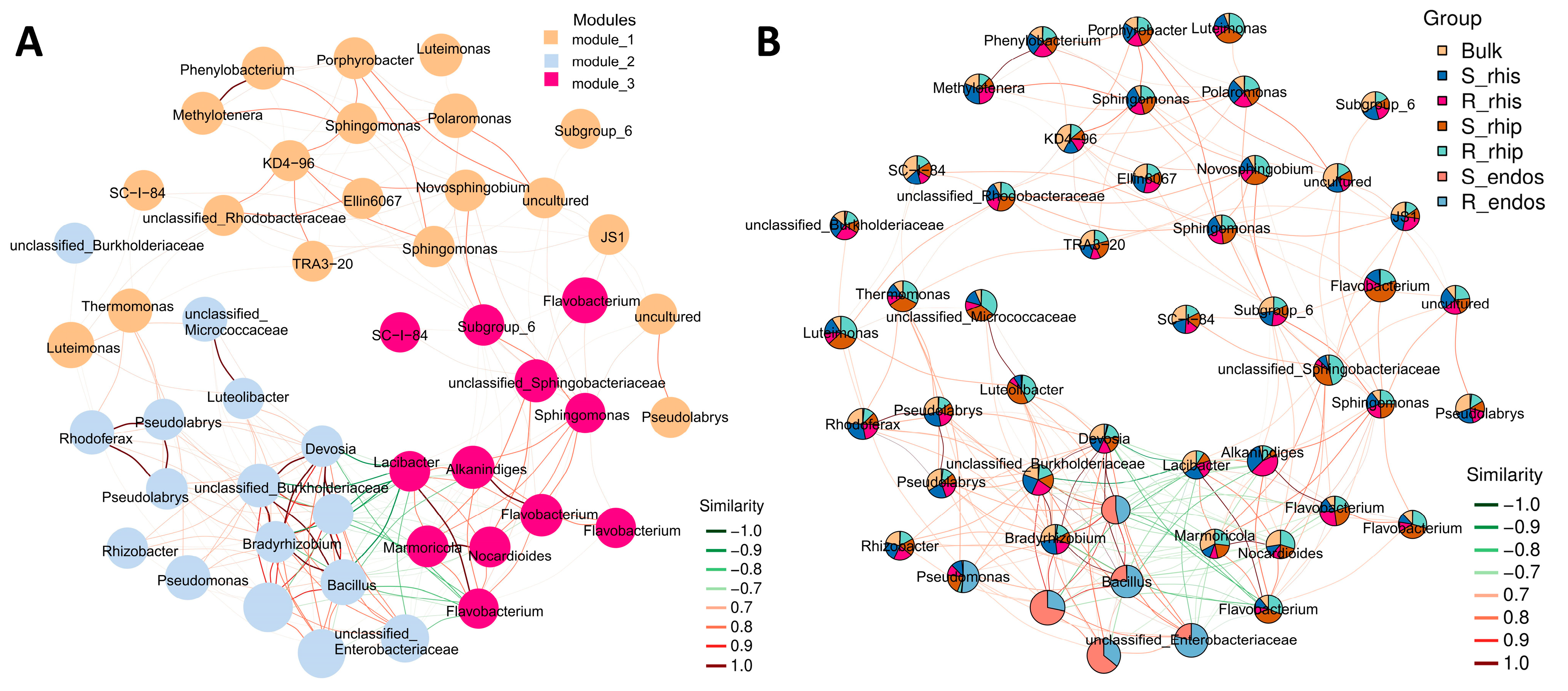
3.4. Potential Sources and Co-Occurrence Network of Root-Associated Microbiome
3.5. Functional Potential Prediction
3.6. Endophytic Fungal Microbiota in S and R Populations
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zheng, Y.; Luo, M.; Li, Y.; Wang, Y. The complete chloroplast genome sequence of Beckmannia syzigachne. Mitochondrial DNA B Resour. 2021, 6, 344–345. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Wu, J.; Zhu, Q.; Ye, C.; Li, X. Assembly of a reference-quality genome and resequencing diverse accessions of Beckmannia syzigachne provide insights into population structure and gene family evolution. Plant Commun. 2024, 6, 101174. [Google Scholar] [CrossRef]
- Li, L.; Du, L.; Liu, W.; Yuan, G.; Wang, J. Target-site mechanism of ACCase-inhibitors resistance in American sloughgrass (Beckmannia syzigachne Steud.) from China. Pestic. Biochem. Physiol. 2014, 110, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Li, J.; Zhang, W.; Dong, L. Detection of the I1781L mutation in fenoxaprop-p-ethyl-resistant American sloughgrass (Beckmannia syzigachne Steud.), based on the loop-mediated isothermal amplification method. Pest Manag. Sci. 2015, 71, 123–130. [Google Scholar] [CrossRef]
- Wang, J.; Peng, Y.; Chen, W.; Yu, Q.; Bai, L.; Pan, L. The Ile-2041-Val mutation in the ACCase gene confers resistance to clodinafop-propargyl in American sloughgrass (Beckmannia syzigachne Steud). Pest Manag. Sci. 2021, 77, 2425–2432. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lian, L.; Qi, J.; Fang, Y.; Nyporko, A.; Yu, Q.; Bai, L.; Pan, L. Metabolic resistance to acetolactate synthase inhibitors in Beckmannia syzigachne: Identification of CYP81Q32 and its transcription regulation. Plant J. 2023, 115, 317–334. [Google Scholar] [CrossRef]
- Délye, C.; Jasieniuk, M.; Le Corre, V. Deciphering the evolution of herbicide resistance in weeds. Trends Genet. 2013, 29, 649–658. [Google Scholar] [CrossRef]
- Bai, L.; Li, X.; Guo, X.; Chen, J.; Yu, H.; Cui, H. Distribution and Mechanism of Japanese Brome (Bromus japonicus) Resistance to ALS-Inhibiting Herbicides in China. Plants 2024, 13, 1139. [Google Scholar] [CrossRef]
- Yang, J.; Yu, H.; Cui, H.; Chen, J.; Li, X. High Antioxidant Ability Confer Resistance to Atrazine in Commelina communis L. Plants 2021, 10, 2685. [Google Scholar] [CrossRef]
- Yu, Q.; Han, H.; Vila-Aiub, M.M.; Powles, S.B. AHAS herbicide resistance endowing mutations: Effect on AHAS functionality and plant growth. J. Exp. Bot. 2010, 61, 3925–3934. [Google Scholar] [CrossRef]
- Li, Q.; Wang, H.; Yu, J.; Zhang, W.; Guo, W.; Liu, Y. Metabolism-Based Herbicide Resistance to Mesosulfuron-methyl and Identification of Candidate Genes in Bromus japonicus. Plants 2024, 13, 1751. [Google Scholar] [CrossRef] [PubMed]
- Bi, Y.; Liu, W.; Li, L.; Yuan, G.; Jin, T.; Wang, J. Molecular basis of resistance to mesosulfuron-methyl in Japanese foxtail, Alopecurus japonicus. J. Pestic. Sci. 2013, 38, 74–77. [Google Scholar] [CrossRef]
- Zhao, N.; Yan, Y.; Liu, W.; Wang, J. Cytochrome P450 CYP709C56 metabolizing mesosulfuron-methyl confers herbicide resistance in Alopecurus aequalis. Cell Mol. Life Sci. 2022, 79, 205. [Google Scholar] [CrossRef] [PubMed]
- Moretto, J.A.S.; Altarugio, L.M.; Andrade, P.A.; Fachin, A.L.; Andreote, F.D.; Stehling, E.G. Changes in bacterial community after application of three different herbicides. FEMS Microbiol. Lett. 2017, 364, 13. [Google Scholar] [CrossRef]
- Bending, G.D.; Rodríguez-Cruz, M.S.; Lincoln, S.D. Fungicide impacts on microbial communities in soils with contrasting management histories. Chemosphere 2007, 69, 82–88. [Google Scholar] [CrossRef]
- Liu, H.; Yu, Q.; Wang, G.; Ye, F.; Cong, Y. Biodegradation of phenol at high concentration by a novel yeast Trichosporon montevideense PHE1. Process Biochem. 2011, 46, 1678–1681. [Google Scholar] [CrossRef]
- Megharaj, M.; Ramakrishnan, B.; Venkateswarlu, K.; Sethunathan, N.; Naidu, R. Bioremediation approaches for organic pollutants: A critical perspective. Environ. Int. 2011, 37, 1362–1375. [Google Scholar] [CrossRef]
- Sarmah, A.K.; Sabadie, J. Hydrolysis of sulfonylurea herbicides in soils and aqueous solutions: A review. J. Agric. Food Chem. 2002, 50, 6253–6265. [Google Scholar] [CrossRef]
- Jiao, S.; Liu, Z.; Lin, Y.; Yang, J.; Chen, W.; Wei, G. Bacterial communities in oil contaminated soils: Biogeography and co-occurrence patterns. Soil Biol. Biochem. 2016, 98, 64–73. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Rio, T.G.D.; et al. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef]
- Lebeis, S.; Herrera Paredes, S.; Lundberg, D.; Breakfield, N.; Gehring, J.; Mcdonald, M.; Malfatti, S.; Rio, T.; Jones, C.; Tringe, S.; et al. Salicylic acid modulates colonization of the root microbiome by specific bacterial taxa. Science 2015, 349, 860–864. [Google Scholar] [CrossRef] [PubMed]
- Wagner, M.R.; Lundberg, D.S.; Del Rio, T.G.; Tringe, S.G.; Dangl, J.L.; Mitchell-Olds, T. Host genotype and age shape the leaf and root microbiomes of a wild perennial plant. Nat. Commun. 2016, 7, 12151. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef] [PubMed]
- Cardinale, M.; Grube, M.; Erlacher, A.; Quehenberger, J.; Berg, G. Bacterial networks and co-occurrence relationships in the lettuce root microbiota. Environ. Microbiol. 2014, 17, 239–252. [Google Scholar] [CrossRef]
- De Souza, R.; Okura, V.K.; Armanhi, J.S.L.; Jorrín, B.; Lozano, N.; Da Silva, M.J.; González-Guerrero, M.; De Araújo, L.M.; Verza, N.C.; Bagheri, H.C.; et al. Unlocking the bacterial and fungal communities assemblages of sugarcane microbiome. Sci. Rep. 2016, 6, 28774. [Google Scholar]
- Jimtha, J.C.; Smitha, P.V.; Anisha, C.; Deepthi, T.; Meekha, G.; Radhakrishnan, E.K.; Gayatri, G.P.; Remakanthan, A. Isolation of endophytic bacteria from embryogenic suspension culture of banana and assessment of their plant growth promoting properties. Plant Cell Tiss. Org. 2014, 118, 57–66. [Google Scholar] [CrossRef]
- Liu, K.; Luo, K.; Mao, A.; Pan, L.; Yan, B.; Wu, J.; Hu, L.; Liu, M.; Liu, X.; Bai, L. Endophytes enhance Asia minor bluegrass (Polypogon fugax) resistance to quizalofop-p-ethyl. Plant Soil 2020, 450, 373–384. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Jaccard, P. Nouvelles Recherches Sur la Distribution Florale. Bull. Soc. Vaudoise Sci. Nat. 1998, 44, 223–270. [Google Scholar]
- Beckers, B.; Beeck, M.; Thijs, S.; Truyens, S.; Weyens, N.; Boerjan, W.; Vangronsveld, J. Performance of 16s rDNA Primer Pairs in the Study of Rhizosphere and Endosphere Bacterial Microbiomes in Metabarcoding Studies. Front. Microbiol. 2016, 7, 650. [Google Scholar] [CrossRef]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef] [PubMed]
- Stackebrandt, E.; Brett, M. Taxonomic note: A place for DNA:DNA reassociation and 16s rRNA sequence analysis in the present Species Definition in Bacteriology. Int. J. Syst. Evol. Microbiol. 1994, 44, 846–849. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 2009, 75, 1098–5336. [Google Scholar] [CrossRef]
- Ondov, B.D.; Bergman, N.H.; Phillippy, A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinf. 2011, 12, 385. [Google Scholar] [CrossRef]
- Zhan, Q.; Qian, C. Microbial-induced synthesis of nanoparticles of zinc phosphate and basic zinc carbonate based on the degradation of glyphosate. Dig. J. Nanomater. Biostructures 2016, 11, 393–399. [Google Scholar]
- Andrade, I.; Figueiredo Carvalho, M.H.; Chaves, A.; Coelho, R.; Da Silva, F.; Zancopé Oliveira, R.; Frases, S.; Brito-Santos, F.; Paes, R. Metabolic and phenotypic plasticity may contribute for the higher virulence of Trichosporon asahii over other Trichosporonaceae members. Mycoses 2022, 66, 430–440. [Google Scholar] [CrossRef]
- Bissett, A.; Richardson, A.E.; Baker, G.; Wakelin, S.; Thrall, P.H. Life history determines biogeographical patterns of soil bacterial communities over multiple spatial scales. Mol. Ecol. 2020, 19, 4315–4327. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Dai, Y.; Huang, G.; Gu, Y.; Ni, J.; Wei, H.; Yuan, S. Soil microbial degradation of neonicotinoid insecticides imidacloprid, acetamiprid, thiacloprid and imidaclothiz and its effect on the persistence of bioefficacy against horsebean aphid Aphis craccivora Koch after soil application. Pest Manag. Sci. 2011, 67, 1245–1252. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, R.; Lee, S.M.; Ryu, C.M. Microbe-induced plant volatiles. New Phytol. 2018, 220, 684–691. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.; Lavoie, M.; Jin, Y.; Xu, J.; Fu, Z.; Qian, H. Diclofop-methyl affects microbial rhizosphere community and induces systemic acquired resistance in rice. J. Environ. Sci. 2017, 51, 352–360. [Google Scholar] [CrossRef]
- Gaiero, J.R.; Mccall, C.; Thompson, K.A.; Day, N.J.; Day, N.; Best, A.S.; Dunfield, K.E. Inside the root microbiome: Bacterial root endophytes and plant growth promotion. Am. J. Bot. 2013, 100, 1738–1750. [Google Scholar] [CrossRef] [PubMed]
- Bobrek, K.; Sokół, I.; Gaweł, A. Uncommon Non-Candida Yeasts in Healthy Turkeys-Antimicrobial Susceptibility and Biochemical Characteristic of Trichosporon Isolates. Pathogens 2021, 10, 538. [Google Scholar] [CrossRef]
- Sietmann, R.; Hammer, E.; Schauer, F. Biotransformation of Biarylic Compounds by Yeasts of the Genus Trichosporon. Syst. Appl. Microbiol. 2002, 25, 332–339. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bai, K.; Ouyang, Y.; Qi, J.; Zhan, Y.; Wang, J. Niches and Genotypes Determine the Diversity and Composition of Microbiomes After Herbicide Treatment in Beckmannia syzigachne. Plants 2025, 14, 876. https://doi.org/10.3390/plants14060876
Bai K, Ouyang Y, Qi J, Zhan Y, Wang J. Niches and Genotypes Determine the Diversity and Composition of Microbiomes After Herbicide Treatment in Beckmannia syzigachne. Plants. 2025; 14(6):876. https://doi.org/10.3390/plants14060876
Chicago/Turabian StyleBai, Kehan, Yulan Ouyang, Jiale Qi, You Zhan, and Junzhi Wang. 2025. "Niches and Genotypes Determine the Diversity and Composition of Microbiomes After Herbicide Treatment in Beckmannia syzigachne" Plants 14, no. 6: 876. https://doi.org/10.3390/plants14060876
APA StyleBai, K., Ouyang, Y., Qi, J., Zhan, Y., & Wang, J. (2025). Niches and Genotypes Determine the Diversity and Composition of Microbiomes After Herbicide Treatment in Beckmannia syzigachne. Plants, 14(6), 876. https://doi.org/10.3390/plants14060876