
Identification of Single Nucleotide Polymorphic Loci and Candidate Genes for Seed Germination Percentage in Okra under Salt and No-Salt Stresses by Genome-Wide Association Study

 ,
,
Abstract
1. Introduction
2. Results
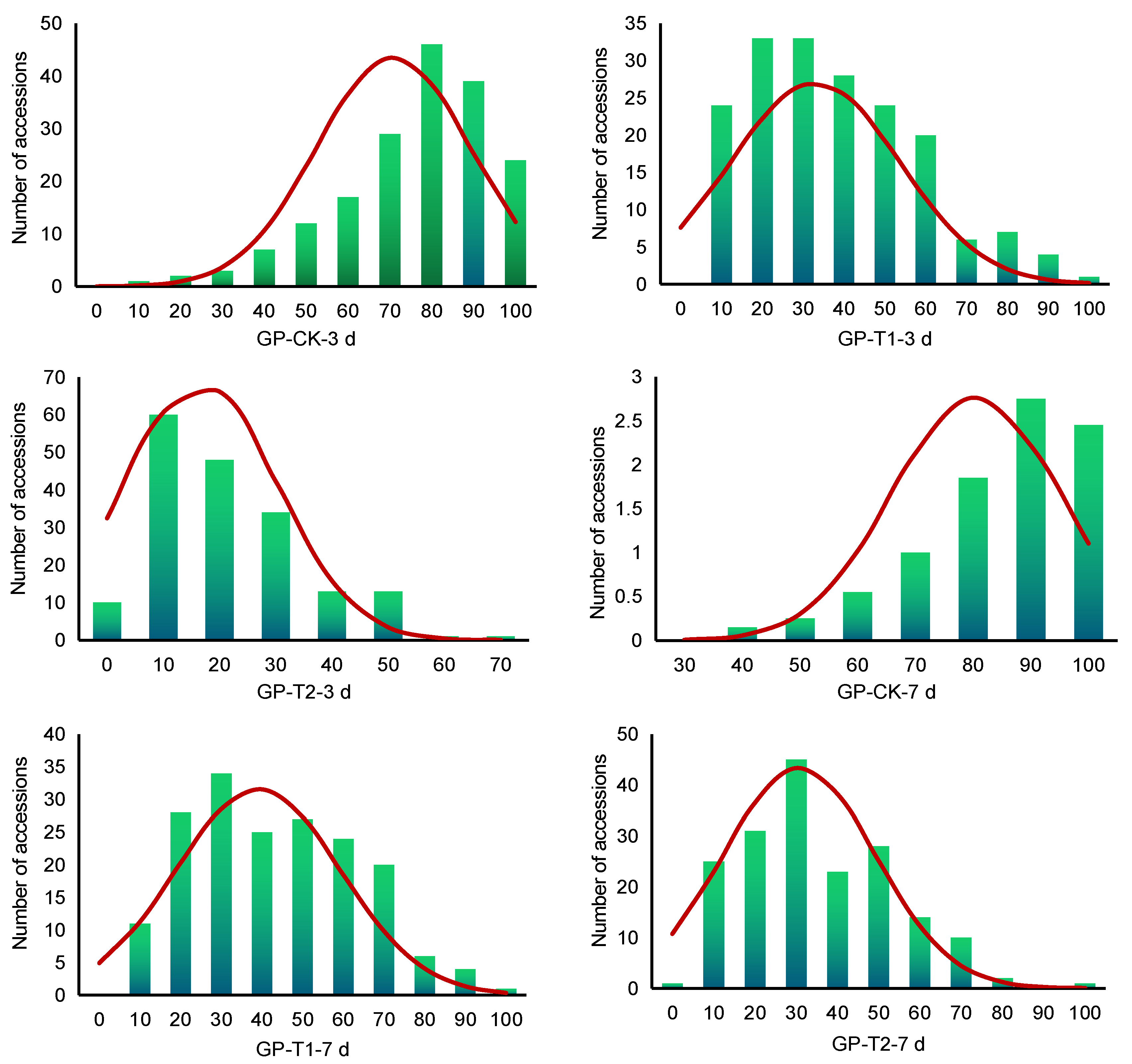
2.1. Phenotypic Variations
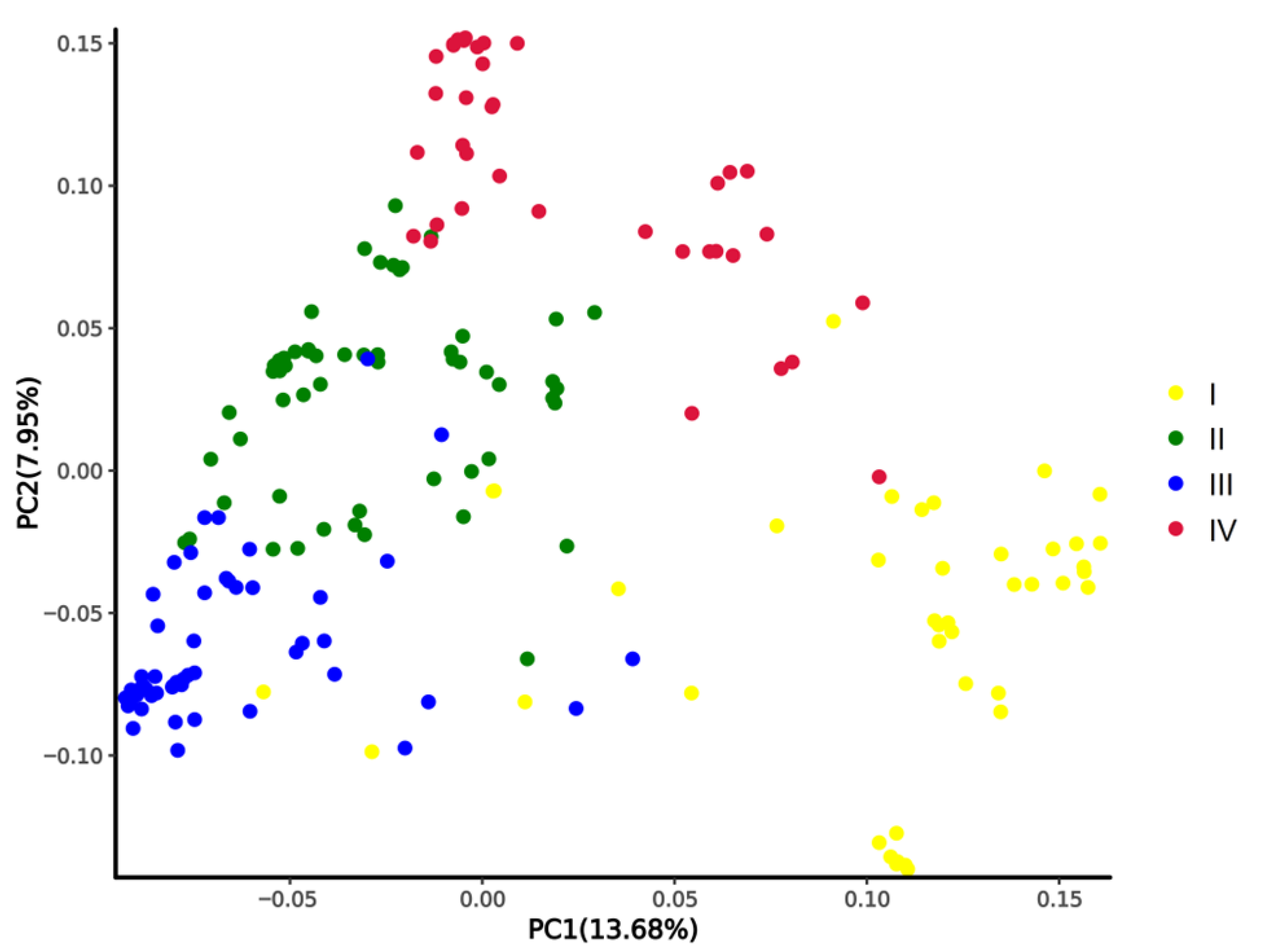
2.2. Genotyping and Population Structural Analysis
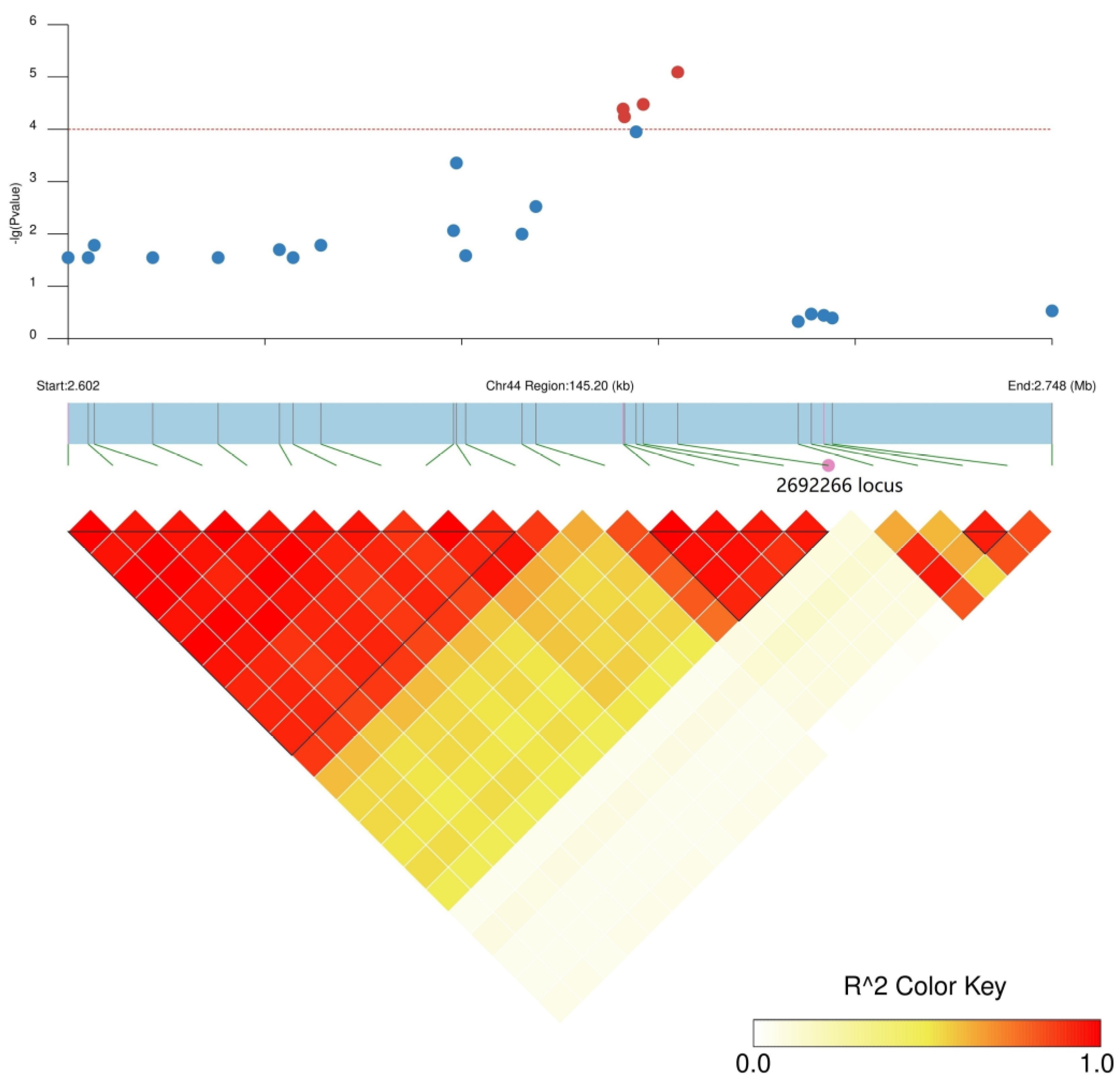
2.3. GWAS of GP under CK and Salt Stress Conditions
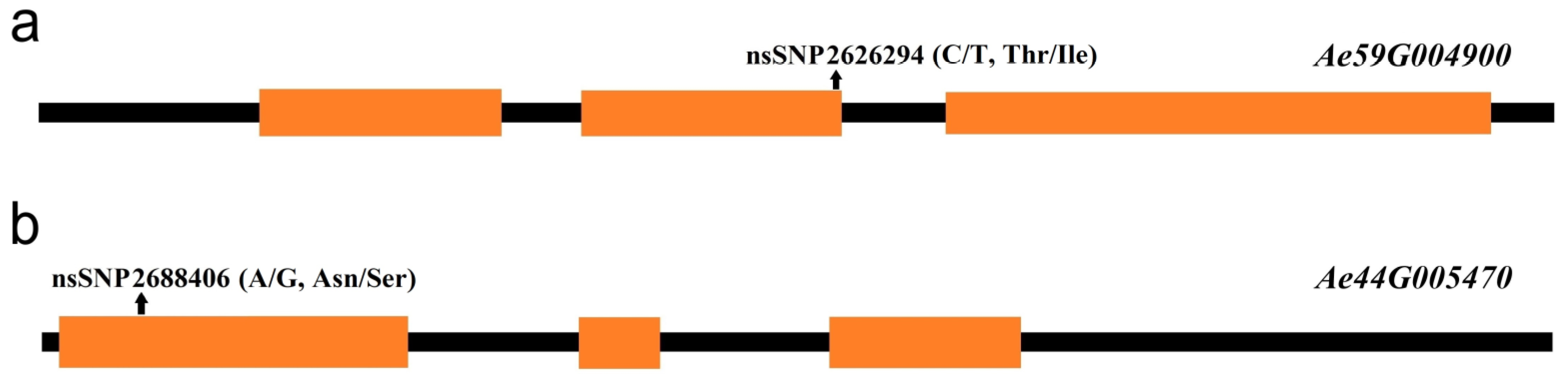
2.4. Candidate Gene Predictions
2.5. Candidate Gene Expression Analysis
3. Discussion
3.1. GP of Okra Seeds under Salt Stress
3.2. Mining the Candidate Genes Regulating the GP of Okra Seeds under Salt and No-Salt Stresses
3.3. Functions of PSK and ACF Genes under Salt Stress
4. Materials and Methods
4.1. Plant Materials
4.2. Seed Germination Experiment
4.3. DNA Extraction and Whole-Genome Resequencing
4.4. GWAS Analysis
4.5. Identification of Haplotype Blocks and Characterization of Candidate Genes
4.6. Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) Analysis of Candidate Genes
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Campbell, M.T.; Knecht, A.C.; Berger, B.; Brien, C.J.; Wang, D.; Walia, H. Integrating image-based phenomics and association analysis to dissect the genetic architecture of temporal salinity responses in rice. Plant Physiol. 2015, 168, 1476–1489. [Google Scholar] [CrossRef] [PubMed]
- Munns, R.; Tester, M. Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 2008, 59, 651–681. [Google Scholar] [CrossRef] [PubMed]
- Baloch, M.Y.J.; Zhang, W.J.; Sultana, T.; Akram, M.; Shoumik, B.A.A.; Khan, M.Z.; Farooq, M.A. Utilization of sewage sludge to manage saline-alkali soil and increase crop production: Is it safe or not? Environ. Technol. Inno. 2023, 32, 103266. [Google Scholar] [CrossRef]
- Yang, Y.Q.; Guo, Y. Elucidating the molecular mechanisms mediating plant salt-stress responses. New Phytol. 2018, 217, 523–539. [Google Scholar] [CrossRef] [PubMed]
- Lindberg, S.; Premkumar, A. Ion changes and signaling under salt stress in wheat and other important crops. Plants 2024, 13, 46. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.; Takeda, S.; Nick, P. Life and death under salt stress: Same players, different timing? J. Exp. Bot. 2014, 65, 2963–2979. [Google Scholar] [CrossRef] [PubMed]
- Zhu, J.K. Regulation of ion homeostasis under salt stress. Curr. Opin. Plant Biol. 2003, 6, 441–445. [Google Scholar] [CrossRef] [PubMed]
- Tarchoune, I.; Degl’ Innocenti, E.; Kaddour, R.; Guidi, L.; Lachaâl, M.; Navari-Izzo, F.; Ouerghi, Z. Effects of NaCl or Na2SO4 salinity on plant growth, ion content and photosynthetic activity in Ocimum basilicum L. Acta Physiol. Plant 2012, 34, 607–615. [Google Scholar] [CrossRef]
- Sewelam, N.; Kazan, K.; Schenk, P.M. Global plant stress signaling: Reactive oxygen species at the cross-road. Front. Plant Sci. 2016, 7, 187. [Google Scholar] [CrossRef]
- Cui, D.Z.; Wu, D.D.; Somarathna, Y.; Xu, C.Y.; Li, S.; Li, P.; Zhang, H.; Chen, H.B.; Zhao, L. QTL mapping for salt tolerance based on SNP markers at the seedling stage in maize (Zea mays L.). Euphytica 2014, 203, 273–283. [Google Scholar] [CrossRef]
- Xie, L.X.; Zheng, C.K.; Li, W.; Pu, M.L.; Zhou, G.H.; Sun, W.; Wu, X.; Zhao, X.Y.; Xie, X.Z. Mapping and identification a salt-tolerant QTL in a salt-resistant rice landrace, Haidao86. J. Plant Growth Regul. 2022, 41, 2347–2358. [Google Scholar] [CrossRef]
- Wang, Q.X.; Xie, W.B.; Xing, H.K.; Yan, J.; Meng, X.Z.; Li, X.L.; Fu, X.K.; Xu, J.Y.; Lian, X.M.; Yu, S.B.; et al. Genetic architecture of natural variation in rice chlorophyll content revealed by a genome-wide association study. Mol. Plant 2015, 8, 946–957. [Google Scholar] [CrossRef] [PubMed]
- Pang, Y.L.; Liu, C.X.; Wang, D.F.; Amand, P.S.; Bernardo, A.; Li, W.H.; He, F.; Li, L.Z.; Wang, L.M.; Yuan, X.F.; et al. High-resolution genome-wide association study identifies genomic regions and candidate genes for important agronomic traits in wheat. Mol. Plant 2020, 13, 1311–1327. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.C.; Xing, H.X.; Zeng, W.G.; Xu, J.L.; Mao, L.L.; Wang, L.Y.; Feng, W.; Tao, J.C.; Wang, H.R.; Zhang, H.J.; et al. Genome-wide association and differential expression analysis of salt tolerance in Gossypium hirsutum L. at the germination stage. BMC Plant Biol. 2019, 19, 394. [Google Scholar] [CrossRef]
- Cao, Y.C.; Jia, S.H.; Chen, L.X.; Zeng, S.N.; Zhao, T.J.; Karikari, B. Identification of major genomic regions for soybean seed weight by genome-wide association study. Mol. Breeding 2022, 42, 38. [Google Scholar] [CrossRef] [PubMed]
- Gong, C.S.; Zhao, S.J.; Yang, D.D.; Lu, X.Q.; Anees, M.; He, N.; Zhu, H.J.; Zhao, Y.; Liu, W.G. Genome-wide association analysis provides molecular insights into natural variation in watermelon seed size. Hortic. Res. 2022, 9, uhab074. [Google Scholar] [CrossRef] [PubMed]
- Holušová, K.; Čmejlová, J.; Suran, P.; Čmejla, R.; Sedlák, J.; Zelený, L.; Bartoš, J. High-resolution genome-wide association study of a large Czech collection of sweet cherry (Prunus avium L.) on fruit maturity and quality traits. Hortic. Res. 2023, 10, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Bewley, J.D. Seed germination and dormancy. Plant Cell 1997, 9, 1055–1066. [Google Scholar] [CrossRef]
- Zhu, G.H.; Liu, Y.G.; Ye, N.H.; Liu, R.; Zhang, J.H. Involvement of the abscisic acid catabolic gene CYP707A2 in the glucose-induced delay in seed germination and post-germination growth of Arabidopsis. Physiol. Plantarum 2011, 143, 375–384. [Google Scholar] [CrossRef]
- Li, W.H.; Zhang, H.Z.; Zeng, Y.L.; Xiang, L.J.; Lei, Z.H.; Huang, Q.X.; Li, T.Y.; Shen, F.; Cheng, Q. A salt tolerance evaluation method for sunflower (Helianthus annuus L.) at the seed germination stage. Sci. Rep. 2020, 10, 10626. [Google Scholar] [CrossRef]
- Sun, J.; Chen, J.Q.; Zhang, X.Y.; Xu, G.C.; Yu, Y.; Dai, Z.G.; Su, J.G. Genome-wide association study of salt tolerance at the germination stage in hemp. Euphytica 2022, 219, 5. [Google Scholar] [CrossRef]
- Shi, Y.Y.; Gao, L.L.; Wu, Z.C.; Zhang, X.J.; Wang, M.M.; Zhang, C.S.; Zhang, F.; Zhou, Y.L.; Li, Z.K. Genome-wide association study of salt tolerance at the seed germination stage in rice. BMC Plant Biol. 2017, 17, 92. [Google Scholar] [CrossRef] [PubMed]
- Wan, H.P.; Wei, Y.K.; Qian, J.L.; Gao, Y.L.; Wen, J.; Yi, B.; Ma, C.Z.; Tu, J.X.; Fu, T.D.; Shen, J.X. Association mapping of salt tolerance traits at germination stage of rapeseed (Brassica napus L.). Euphytica 2018, 214, 190. [Google Scholar] [CrossRef]
- Kan, G.Z.; Zhang, W.; Yang, W.M.; Ma, D.Y.; Zhang, D.; Hao, D.R.; Hu, Z.B.; Yu, D.Y. Association mapping of soybean seed germination under salt stress. Mol. Genet. Genom. 2015, 290, 2147–2162. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.X.; Liu, X.C.; Boge, W.; Liu, X.P. Genome-wide association study identifies loci for salt tolerance during germination in autotetraploid alfalfa (Medicago sativa L.) using genotyping-by-sequencing. Front. Plant Sci. 2016, 7, 956. [Google Scholar] [CrossRef] [PubMed]
- Ravelombola, W.; Shi, A.N.; Weng, Y.J.; Mou, B.Q.; Motes, D.; Clark, J.; Chen, P.Y.; Srivastava, V.; Qin, J.; Dong, L.D.; et al. Association analysis of salt tolerance in cowpea (Vigna unguiculata (L.) Walp) at germination and seedling stages. Theor. Appl. Genet. 2018, 131, 79–91. [Google Scholar] [CrossRef] [PubMed]
- Dhankhar, S.K.; Dhankhar, B.S.; Yadava, R.K. Inheritance of resistance to yellow vein mosaic virus in an interspecific cross of okra (Abelmoschus esculentus). Indian J. Agric. Sci. 2005, 75, 87–89. Available online: https://www.researchgate.net/publication/290956380 (accessed on 12 January 2024).
- Guebebia, S.; Gharsallaoui, A.; Dumas, E.; Baghi, F.; Zourgui, L.; Romdhane, M.; Agusti, G.; Ghnimi, S. Microencapsulation of phenolic compounds extracted from okra (Abelmoschus esculentus L.) leaves, fruits and seeds. Appl. Sci. 2023, 13, 12273. [Google Scholar] [CrossRef]
- Majd, N.E.; Tabandeh, M.R.; Shahriari, A.; Soleimani, Z. Okra (Abelmoscus esculentus) improved islets structure, and down-regulated PPARs gene expression in pancreas of high-fat diet and streptozotocin-induced diabetic rats. Cell J. 2018, 20, 31–40. [Google Scholar] [CrossRef]
- Tongjaroenbuangam, W.; Ruksee, N.; Chantiratikul, P.; Pakdeenarong, N.; Kongbuntad, W.; Govitrapong, P. Neuroprotective effects of quercetin, rutin and okra (Abelmoschus esculentus Linn.) in dexamethasone-treated mice. Neurochem. Int. 2011, 59, 677–685. [Google Scholar] [CrossRef]
- Alqasoumi, S.I. ‘Okra’ Hibiscus esculentus L.: A study of its hepatoprotective activity. Saudi Pharm. J. 2012, 20, 135–141. [Google Scholar] [CrossRef] [PubMed]
- Monte, L.G.; Santi-Gadelha, T.; Reis, L.B.; Braganhol, E.; Prietsch, R.F.; Dellagostin, O.A.; Lacerda, R.R.E.; Gadelha, C.A.A.; Conceição, F.R.; Pinto, L.S. Lectin of Abelmoschus esculentus (okra) promotes selective antitumor effects in human breast cancer cells. Biotechnol. Lett. 2014, 36, 461–469. [Google Scholar] [CrossRef] [PubMed]
- Sukanya, C.; Rajnarayan, S.; Parimal, P. Arsenic uptake and accumulation in okra (Abelmoschus esculentus) as affected by different arsenical speciation. Bull. Environ. Contam. Toxicol. 2016, 96, 395–400. [Google Scholar] [CrossRef]
- Haq, I.U.; Khan, A.A.; Khan, I.A.; Azmat, M.A. Comprehensive screening and selection of okra (Abelmoschus esculentus) germplasm for salinity tolerance at the seedling stage and during plant ontogeny. J. Zhejiang Univ.-Sci. B 2012, 13, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Habib, S.H.; Kausar, H.; Saud, H.M. Plant growth-promoting rhizobacteria enhance salinity stress tolerance in okra through ROS-scavenging enzymes. Biomed. Res. Int. 2016, 2016, 6284547. [Google Scholar] [CrossRef] [PubMed]
- Pandita, V.K.; Anand, A.; Nagarajan, S.; Seth, R.; Sinha, S.N. Solid matrix priming improves seed emergence and crop performance in okra. Seed Sci. Technol. 2010, 38, 665–674. [Google Scholar] [CrossRef]
- Mishra, G.P.; Singh, B.; Seth, T.; Singh, A.K.; Halder, J.; Krishnan, N.; Tiwari, S.K.; Singh, P.M. Biotechnological advancements and begomovirus management in okra (Abelmoschus esculentus L.): Status and perspectives. Front. Plant Sci. 2017, 8, 360. [Google Scholar] [CrossRef] [PubMed]
- Dkhil, B.B.; Issa, A.; Denden, M. Germination and seedling emergence of primed okra (Abelmoschus esculentus L.) seeds under salt stress and low temperature. Am. J. Plant Physiol. 2014, 9, 38–45. [Google Scholar] [CrossRef]
- Sharma, A.D.; Rathore, S.V.S.; Srinivasan, K.; Tyagi, R.K. Comparison of various seed priming methods for seed germination, seedling vigour and fruit yield in okra (Abelmoschus esculentus L. Moench). Sci. Hortic. 2014, 165, 75–81. [Google Scholar] [CrossRef]
- Rima, R. Seed priming significantly affects germination and post germination early seedling growth in okra. Malays. J. Halal Res. J. 2021, 4, 11–13. [Google Scholar] [CrossRef]
- Mohammadi, G.; Khah, E.M.; Honarmand, S.J.; Shirkhani, A.; Shabani, G. Effects of seed hardness breaking techniques on okra (Abelmoschus esculentus L.) germination. Int. J. Agric. Crop Sci. 2012, 4, 264–273. [Google Scholar]
- Musara, C.; Chitamba, J.; Nhuvira, C. Evaluation of different seed dormancy breaking techniques on okra (Abelmoschus esculentus L.) seed germination. Afr. J. Agric. Res. 2015, 10, 1952–1956. [Google Scholar] [CrossRef]
- Wu, H.; Guo, J.R.; Wang, C.F.; Li, K.L.; Zhang, X.W.; Yang, Z.; Li, M.T.; Wang, B.S. An effective screening method and a reliable screening trait for salt tolerance of Brassica napus at the germination stage. Front. Plant Sci. 2019, 10, 530. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Zhao, W.G.; Tong, W.; He, Q.; Yoon, M.Y.; Li, F.P.; Choi, B.; Heo, E.B.; Kim, K.W.; Park, Y.J. A Genome-wide association study reveals candidate genes related to salt tolerance in rice (Oryza sativa) at the germination stage. Int. J. Mol. Sci. 2018, 19, 3145. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.N.; Szczepanek, A.; Abdel-Haleem, H. Genome-wide association study (GWAS) analysis of camelina seedling germination under salt stress condition. Agronomy 2020, 10, 1444. [Google Scholar] [CrossRef]
- Tan, M.; Liao, F.; Hou, L.T.; Wang, J.; Wei, L.J.; Jian, H.J.; Xu, X.F.; Li, J.N.; Liu, L.Z. Genome-wide association analysis of seed germination percentage and germination index in Brassica napus L. under salt and drought stresses. Euphytica 2017, 213, 40. [Google Scholar] [CrossRef]
- Zhang, Z.W.; Ersoz, E.; Lai, C.Q.; Todhunter, R.J.; Tiwari, H.K.; Gore, M.A.; Bradbury, P.J.; Yu, J.M.; Arnett, D.K.; Ordovas, J.M.; et al. Mixed linear model approach adapted for genome-wide association studies. Nat. Genet. 2010, 42, 355–360. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.L.; Huang, M.; Fan, B.; Buckler, E.S.; Zhang, Z.W. Iterative usage of fixed and random effect models for powerful and efficient genome-wide association studies. PLoS Genet. 2016, 12, e1005767. [Google Scholar] [CrossRef]
- Kang, H.M.; Sul, J.H.; Service, S.K.; Zaitlen, N.A.; Kong, S.Y.; Freimer, N.B.; Sabatti, C.; Eskin, E. Variance component model to account for sample structure in genome-wide association studies. Nat. Genet. 2010, 42, 348–354. [Google Scholar] [CrossRef]
- Zhou, X.; Stephens, M. Genome-wide efficient mixed model analysis for association studies. Nat. Genet. 2012, 44, 821–824. [Google Scholar] [CrossRef]
- Smith, J.L.; Wilson, M.L.; Nilson, S.M.; Rowan, T.N.; Oldeschulte, D.L.; Schnabel, R.D.; Decker, J.E.; Seabury, C.M. Genome-wide association and genotype by environment interactions for growth traits in U.S. Gelbvieh cattle. BMC Genom. 2019, 20, 926. [Google Scholar] [CrossRef]
- Liu, J.Y.; Zhang, Y.W.; Han, X.; Zuo, J.F.; Zhang, Z.B.; Shang, H.H.; Song, Q.J.; Zhang, Y.M. An evolutionary population structure model reveals pleiotropic effects of GmPDAT for traits related to seed size and oil content in soybean. J. Exp. Bot. 2020, 71, 6988–7002. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Y.; Lin, Y.; Chen, J.B.; Yan, Q.; Xue, C.C.; Wu, R.R.; Chen, X.; Yuan, X.X. Genome-wide association studies provide genetic insights into natural variation of seed-size-related traits in mungbean. Front. Plant Sci. 2022, 13, 997988. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Kong, X.; Yang, Y.; Nie, X.; Lin, Z. Association mapping seed kernel oil content in upland cotton using genome-wide SSRs and SNPs. Mol. Breed. 2019, 39, 105. [Google Scholar] [CrossRef]
- Huang, H.; Zhao, S.; Chen, J.; Li, T.; Guo, G.; Xu, M.; Liao, S.; Wang, R.; Lan, J.; Su, Y.; et al. Genome-wide identification and functional analysis of cellulose synthase gene superfamily in Fragaria vesca. Front. Plant Sci. 2022, 13, 1044029. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.S.; Sun, L.; Dong, X.; Lu, S.J.; Tian, W.; Liu, J.X. Cellulose synthesis genes CESA6 and CSI1 are important for salt stress tolerance in Arabidopsis. J. Integr. Plant Biol. 2016, 58, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Li, S.T.; Zhang, L.; Wang, Y.; Xu, F.; Liu, M.; Lin, P.; Ren, S.; Ma, R.; Guo, Y.D. Knockdown of a cellulose synthase gene BoiCesA affects the leaf anatomy, cellulose content and salt tolerance in broccoli. Sci. Rep. 2017, 7, 41397. [Google Scholar] [CrossRef] [PubMed]
- Sharif, R.; Liu, P.; Wang, D.; Jin, Z.; Uzair, U.; Yadav, V.; Mujtaba, M.; Chen, P.; Li, Y. Genome-wide characterisation and expression analysis of cellulose synthase genes superfamily under various environmental stresses in Cucumis sativus L. New Zeal. J. Crop Hort. Sci. 2021, 49, 127–150. [Google Scholar] [CrossRef]
- Sauter, M. Phytosulfokine peptide signalling. J. Exp. Bot. 2015, 66, 5161–5169. [Google Scholar] [CrossRef]
- Rodiuc, N.; Barlet, X.; Hok, S.; Perfus-Barbeoch, L.; Allasia, V.; Engler, G.; Séassau, A.; Marteu, N.; Almeida-Engler, J.; Panabières, F.; et al. Evolutionarily distant pathogens require the Arabidopsis phytosulfokine signalling pathway to establish disease. Plant Cell Environ. 2016, 39, 1396–1407. [Google Scholar] [CrossRef]
- Nagar, P.; Sharma, N.; Jain, M.; Sharma, G.; Prasad, M.; Mustafiz, A. OsPSKR15, a phytosulfokine receptor from rice enhances abscisic acid response and drought stress tolerance. Physiol. Plant 2021, 174, e13569. [Google Scholar] [CrossRef] [PubMed]
- Stührwohldt, N.; Bühler, E.; Sauter, M.; Schaller, A. Phytosulfokine (PSK) precursor processing by subtilase SBT3.8 and PSK signaling improve drought stress tolerance in Arabidopsis. J. Exp. Bot. 2021, 72, 3427–3440. [Google Scholar] [CrossRef] [PubMed]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The genome analysis toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.Y.; Ruden, D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed]
- Price, A.L.; Patterson, N.J.; Plenge, R.M.; Weinblatt, M.E.; Shadick, N.A.; Reich, D. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 2006, 38, 904–909. [Google Scholar] [CrossRef]
- Alexander, D.H.; Novembre, J.; Lange, K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009, 19, 1655–1664. [Google Scholar] [CrossRef]
- Ginestet, C. ggplot2: Elegant graphics for data analysis. J. R. Stat. Soc. A Stat. 2011, 174, 245–246. [Google Scholar] [CrossRef]
- Turner, S.D. qqman: An R package for visualizing GWAS results using QQ and manhattan plots. bioRxiv 2014, 3, 731. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef]
- Zhan, Y.H.; Wu, T.T.; Zhao, X.; Wang, Z.Q.; Chen, Y. Comparative physiological and full-length transcriptome analyses reveal the molecular mechanism of melatonin-mediated salt tolerance in okra (Abelmoschus esculentus L.). BMC Plant Biol. 2021, 21, 180. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCt method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Trait | Mean | Max | Min | SD | CV (%) | Skew. | Kurt. |
|---|---|---|---|---|---|---|---|
| GP-CK-3 d | 0.7081 | 0.9800 | 0.1000 | 0.1836 | 25.92 | −0.9932 | 0.9420 |
| GP-T1-3 d | 0.3294 | 0.9333 | 0.0200 | 0.2069 | 62.82 | 0.6535 | −0.1401 |
| GP-T2-3 d | 0.1664 | 0.6333 | 0.0000 | 0.1366 | 82.07 | 0.9112 | 0.2628 |
| GP-CK-7 d | 0.8041 | 0.9933 | 0.3067 | 0.1445 | 17.96 | −1.0539 | 0.8917 |
| GP-T1-7 d | 0.3907 | 0.9400 | 0.0467 | 0.2022 | 51.76 | 0.3450 | −0.6176 |
| GP-T2-7 d | 0.3077 | 0.9200 | 0.0000 | 0.1841 | 59.82 | 0.5847 | −0.0123 |
| GP-CK-3 d | GP-CK-7 d | GP-T1-3 d | GP-T1-7 d | GP-T2-3 d | GP-T2-7 d | |
|---|---|---|---|---|---|---|
| GP-CK-3 d | ||||||
| GP-CK-7 d | 0.6287 ** | |||||
| GP-T1-3 d | 0.3863 ** | 0.2635 ** | ||||
| GP-T1-7 d | 0.3637 ** | 0.2879 ** | 0.6651 ** | |||
| GP-T2-3 d | 0.3082 ** | 0.1825 * | 0.6719 ** | 0.5297 ** | ||
| GP-T2-7 d | 0.3800 ** | 0.3164 ** | 0.5741 ** | 0.7069 ** | 0.6195 ** |
| SNP Name | Chromosome | Trait | Software | p Value | −log10 (p) | Allele | Count | MAF |
|---|---|---|---|---|---|---|---|---|
| SNP2619493 | Chr. 59 | CK-3 d | EMMAX | 1.71 × 10−5 | 4.77 | A/T | A: 178, T: 160 | 0.47 |
| GEMMA | 4.67 × 10−6 | 5.33 | A/T | A: 178, T: 160 | 0.47 | |||
| T1-3 d | EMMAX | 5.12 × 10−5 | 4.29 | A/T | A: 178, T: 160 | 0.47 | ||
| GEMMA | 1.69 × 10−5 | 4.77 | A/T | A: 178, T: 160 | 0.47 | |||
| T2-3 d | EMMAX | 7.51 × 10−5 | 4.12 | A/T | A: 178, T: 160 | 0.47 | ||
| GEMMA | 7.51 × 10−5 | 4.12 | A/T | A: 178, T: 160 | 0.47 | |||
| SNP2692266 | Chr. 44 | CK-7 d | EMMAX | 2.45 × 10−5 | 4.61 | A/G | A: 311, G: 47 | 0.13 |
| GEMMA | 1.08 × 10−5 | 4.97 | A/G | A: 311, G: 47 | 0.13 | |||
| T1-7 d | EMMAX | 4.81 × 10−5 | 4.32 | A/G | A: 311, G: 47 | 0.13 | ||
| GEMMA | 1.27 × 10−5 | 4.90 | A/G | A: 311, G: 47 | 0.13 | |||
| T2-7 d | EMMAX | 8.11 × 10−6 | 5.09 | A/G | A: 311, G: 47 | 0.13 | ||
| GEMMA | 4.36 × 10−6 | 5.36 | A/G | A: 311, G: 47 | 0.13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xu, G.; Cheng, Y.; Wang, X.; Dai, Z.; Kang, Z.; Ye, Z.; Pan, Y.; Zhou, L.; Xie, D.; Sun, J. Identification of Single Nucleotide Polymorphic Loci and Candidate Genes for Seed Germination Percentage in Okra under Salt and No-Salt Stresses by Genome-Wide Association Study. Plants 2024, 13, 588. https://doi.org/10.3390/plants13050588
Xu G, Cheng Y, Wang X, Dai Z, Kang Z, Ye Z, Pan Y, Zhou L, Xie D, Sun J. Identification of Single Nucleotide Polymorphic Loci and Candidate Genes for Seed Germination Percentage in Okra under Salt and No-Salt Stresses by Genome-Wide Association Study. Plants. 2024; 13(5):588. https://doi.org/10.3390/plants13050588
Chicago/Turabian StyleXu, Gaowen, Yujing Cheng, Xiaoqiu Wang, Zhigang Dai, Zepei Kang, Zhichao Ye, Yangyang Pan, Linkang Zhou, Dongwei Xie, and Jian Sun. 2024. "Identification of Single Nucleotide Polymorphic Loci and Candidate Genes for Seed Germination Percentage in Okra under Salt and No-Salt Stresses by Genome-Wide Association Study" Plants 13, no. 5: 588. https://doi.org/10.3390/plants13050588
APA StyleXu, G., Cheng, Y., Wang, X., Dai, Z., Kang, Z., Ye, Z., Pan, Y., Zhou, L., Xie, D., & Sun, J. (2024). Identification of Single Nucleotide Polymorphic Loci and Candidate Genes for Seed Germination Percentage in Okra under Salt and No-Salt Stresses by Genome-Wide Association Study. Plants, 13(5), 588. https://doi.org/10.3390/plants13050588