
Transcriptome Analyses Throughout Chili Pepper Fruit Development Reveal Novel Insights into the Domestication Process

 ,
,  , , , , and
, , , , and 
Abstract
1. Introduction
2. Results
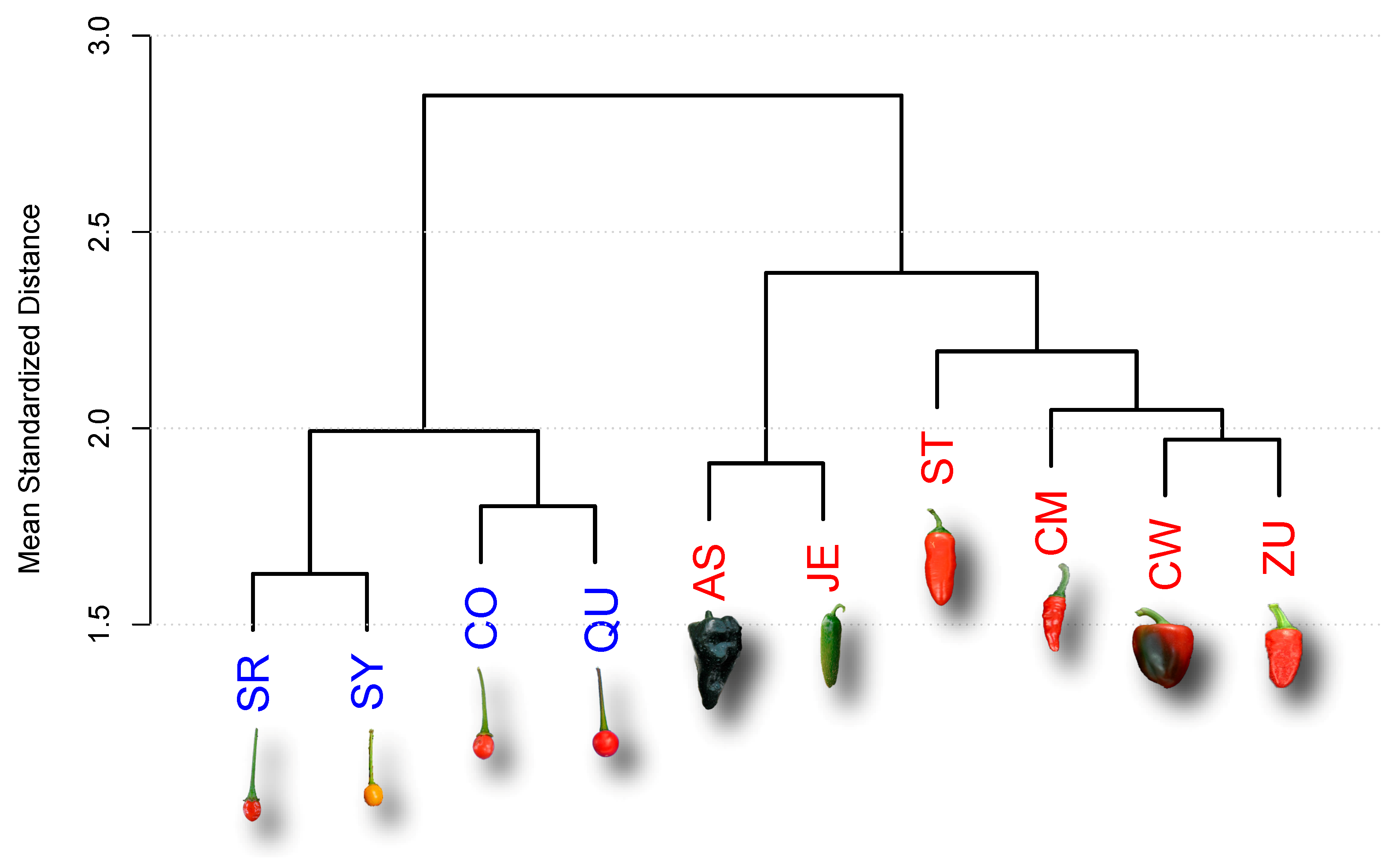
2.1. Analysis of Late Maturing Times
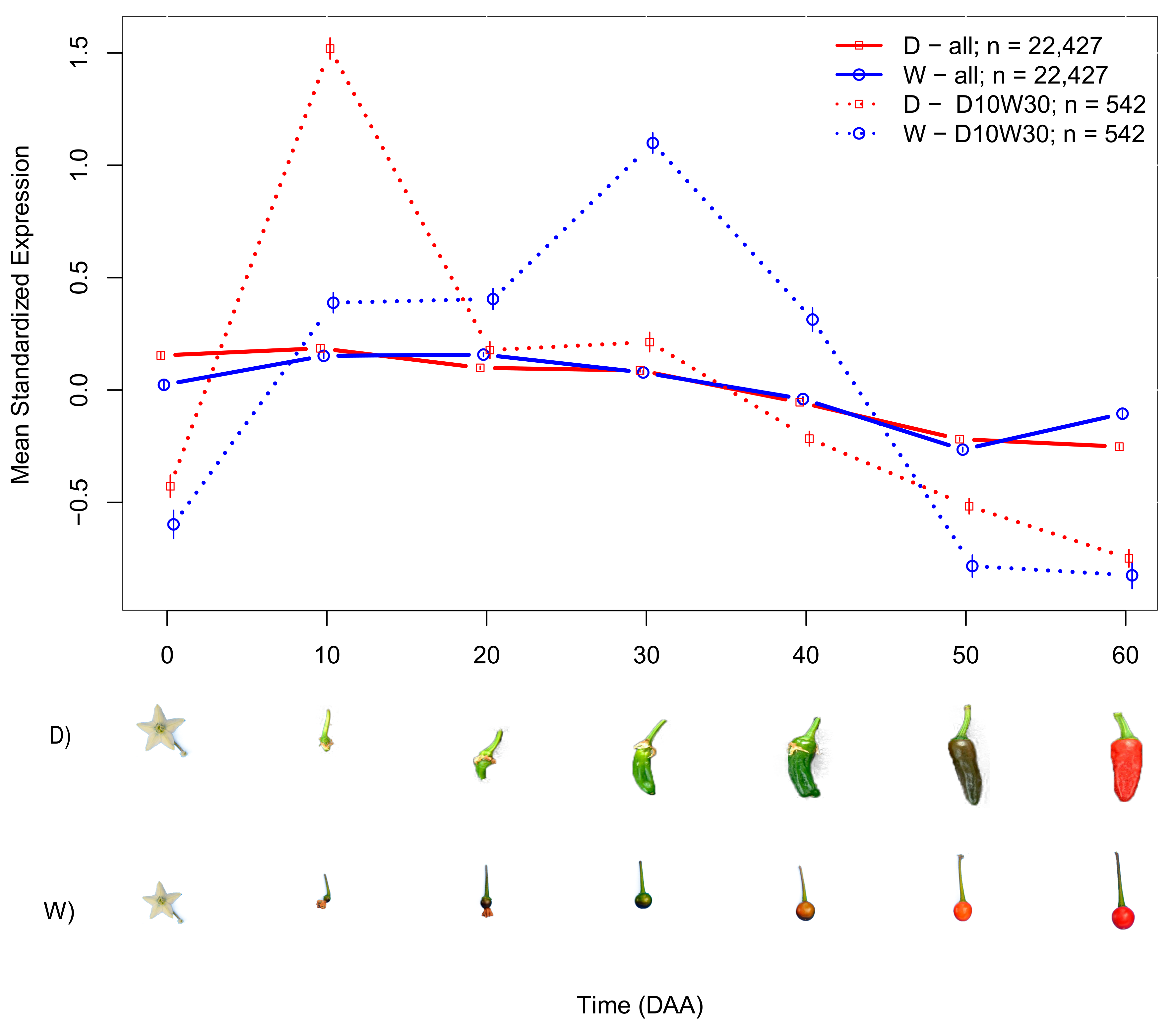
2.2. Domesticated (D) and Wild (W) Accessions Have Different Mean SEPs
2.3. Differences in SEPs of Individual Genes Varied between D and W
2.4. Differences in Expression of Genes Related to Cell Reproduction Appear Earlier and Are Larger in Domesticated Than in Wild Genotypes
2.5. Biological Processes Were Enriched in Genes That Are Expressed Earlier in Domesticated Genotypes
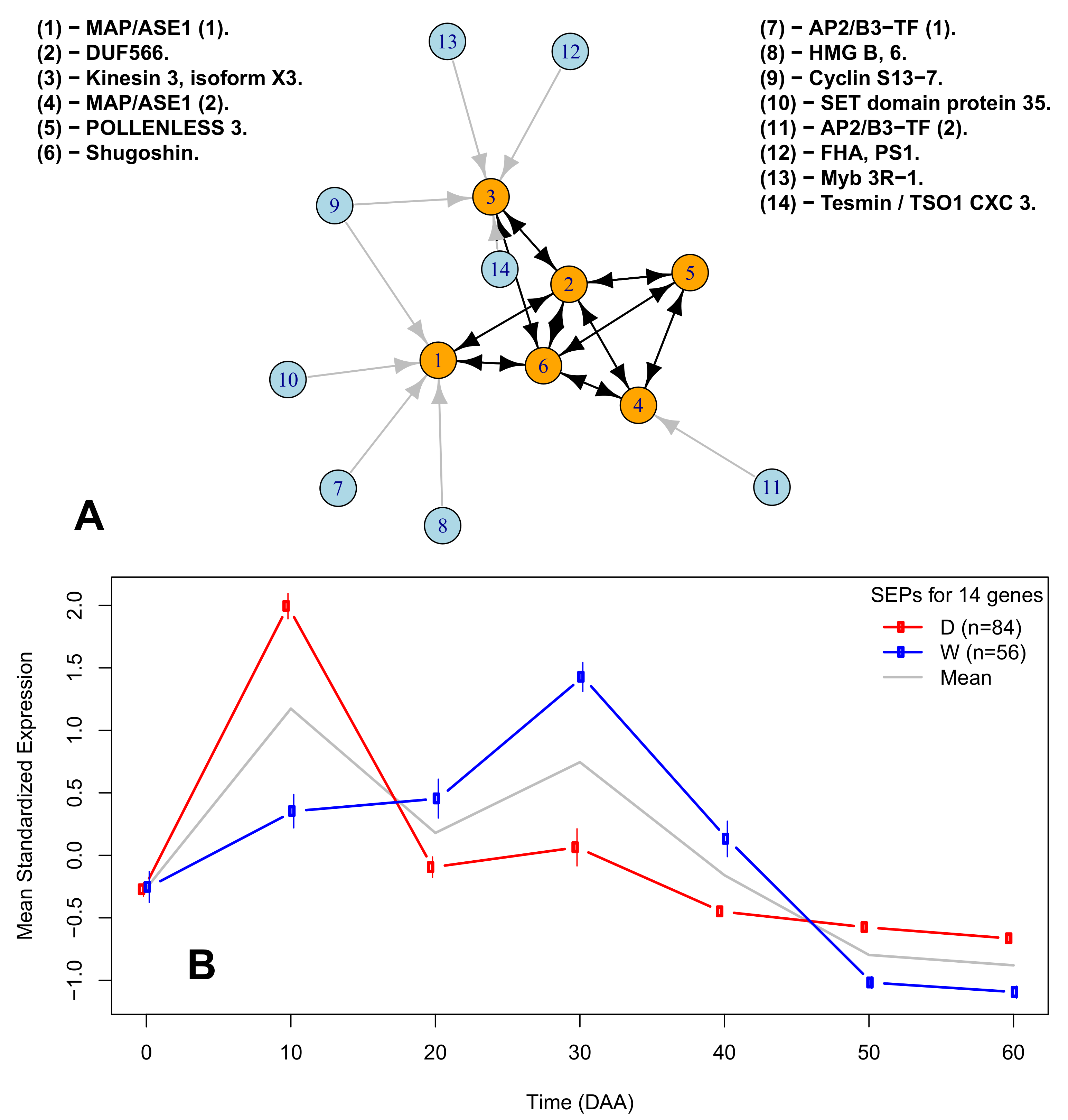
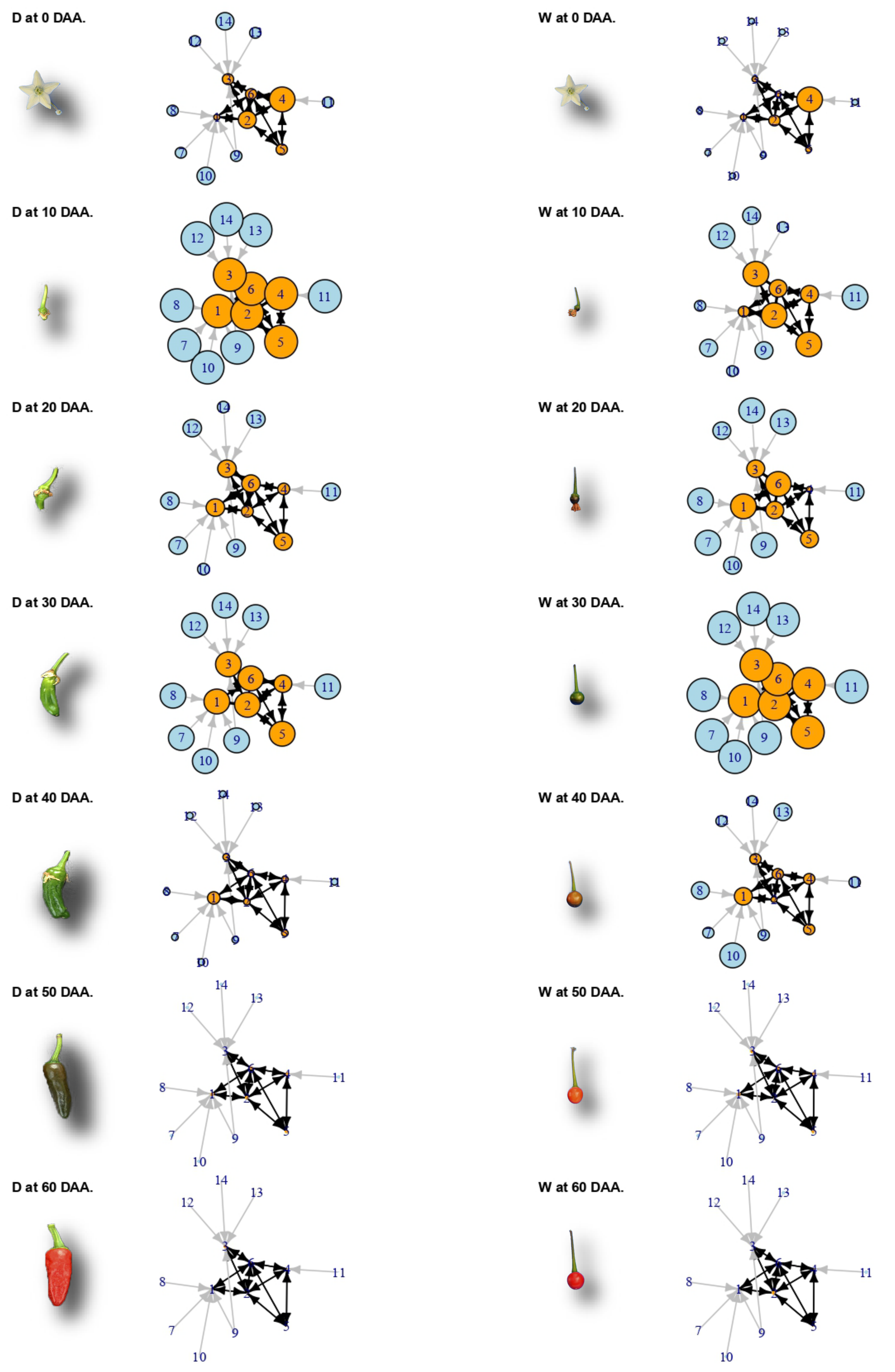
2.6. A Network of Cell Cycle Genes with the D10W30 Expression Pattern
3. Discussion
4. Materials and Methods
4.1. Statistical Design
4.2. Plant Materials and Cultivation
4.3. RNA-Seq Library Construction and Processing
4.4. Estimations and Analyses of Standardized Expression Profiles (SEPs)
4.5. Network Estimation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Pickersgill, B. Relationships between weedy and cultivated forms in some species of chili peppers (genus Capsicum). Evolution 1971, 25, 683–691. [Google Scholar] [PubMed]
- Jarret, R.L.; Barboza, G.E.; da Costa Batista, F.R.; Berke, T.; Chou, Y.Y.; Hulse-Kemp, A.; Ochoa-Alejo, N.; Tripodi, P.; Veres, A.; Garcia, C.C.; et al. Capsicum—An abbreviated compendium. J. Am. Soc. Hortic. Sci. 2019, 144, 3–22. [Google Scholar] [CrossRef]
- Badia, A.D.; Spina, A.A.; Vassalotti, G. Capsicum annuum L.: An Overview of Biological Activities and Potential Nutraceutical Properties in Humans and Animals. J. Nutr. Ecol. Food Res. 2017, 4, 167–177. [Google Scholar] [CrossRef]
- Cervantes-Hernández, F.; Alcalá-González, P.; Martínez, O.; Ordaz-Ortiz, J.J. Placenta, Pericarp, and Seeds of Tabasco Chili Pepper Fruits Show a Contrasting Diversity of Bioactive Metabolites. Metabolites 2019, 9, 206. [Google Scholar] [CrossRef]
- Gómez-García, M.d.R.; Ochoa-Alejo, N. Biochemistry and molecular biology of carotenoid biosynthesis in chili peppers (Capsicum spp.). Int. J. Mol. Sci. 2013, 14, 19025–19053. [Google Scholar] [CrossRef]
- Gómez-García, M.d.R.; Ochoa-Alejo, N. Predominant role of the l-galactose pathway in l-ascorbic acid biosynthesis in fruits and leaves of the Capsicum annuum L. chili pepper. Braz. J. Bot. 2016, 39, 157–168. [Google Scholar] [CrossRef]
- Martínez-López, L.A.; Ochoa-Alejo, N.; Martínez, O. Dynamics of the chili pepper transcriptome during fruit development. BMC Genom. 2014, 15, 143. [Google Scholar] [CrossRef]
- Arce-Rodríguez, M.L.; Ochoa-Alejo, N. An R2R3-MYB transcription factor regulates capsaicinoid biosynthesis. Plant Physiol. 2017, 174, 1359–1370. [Google Scholar] [CrossRef]
- Tanaka, Y.; Nakashima, F.; Kirii, E.; Goto, T.; Yoshida, Y.; Yasuba, K.I. Difference in capsaicinoid biosynthesis gene expression in the pericarp reveals elevation of capsaicinoid contents in chili peppers (Capsicum chinense). Plant Cell Rep. 2017, 36, 267–279. [Google Scholar] [CrossRef]
- Fayos, O.; Ochoa-Alejo, N.; Octavio, M.; Savirǿn, M.; Orduna, J.; Mallor, C.; Barbero, G.F.; Garces-Claver, A. Assessment of Capsaicinoid and Capsinoid Accumulation Patterns during Fruit Development in Three Chili Pepper Genotypes (Capsicum spp.) Carrying Pun1 and pAMT Alleles Related to Pungency. J. Agric. Food Chem. 2019, 67, 12219–12227. [Google Scholar] [CrossRef]
- Osorio, S.; Alba, R.; Nikoloski, Z.; Kochevenko, A.; Fernie, A.R.; Giovannoni, J.J. Integrative comparative analyses of transcript and metabolite profiles from pepper and tomato ripening and development stages uncovers species-specific patterns of network regulatory behavior. Plant Physiol. 2012, 159, 1713–1729. [Google Scholar] [CrossRef] [PubMed]
- Jang, Y.K.; Jung, E.S.; Lee, H.A.; Choi, D.; Lee, C.H. Metabolomic Characterization of Hot Pepper (Capsicum annuum ‘CM334’) during Fruit Development. J. Agric. Food Chem. 2015, 63, 9452–9460. [Google Scholar] [CrossRef] [PubMed]
- Dubey, M.; Jaiswal, V.; Rawoof, A.; Kumar, A.; Nitin, M.; Chhapekar, S.S.; Kumar, N.; Ahmad, I.; Islam, K.; Brahma, V.; et al. Identification of genes involved in fruit development/ripening in Capsicum and development of functional markers. Genomics 2019, 111, 1913–1922. [Google Scholar] [CrossRef] [PubMed]
- Hayano-Kanashiro, C.; Gámez-Meza, N.; Medina-Juárez, L.Á. Wild Pepper Capsicum annuum L. var. glabriusculum: Taxonomy, Plant Morphology, Distribution, Genetic Diversity, Genome Sequencing, and Phytochemical Compounds. Crop Sci. 2016, 56, 1–11. [Google Scholar]
- Kraft, K.H.; Brown, C.H.; Nabhan, G.P.; Luedeling, E.; Ruiz, J.D.J.L.; d’Eeckenbrugge, G.C.; Hijmans, R.J.; Gepts, P. Multiple lines of evidence for the origin of domesticated chili pepper, Capsicum annuum, in Mexico. Proc. Natl. Acad. Sci. USA 2014, 111, 6165–6170. [Google Scholar] [CrossRef] [PubMed]
- McClung de Tapia, E. The origins of agriculture in Mesoamerica and Central America. Orig. Agric. Int. Perspect. 1992, 1, 143–171. [Google Scholar]
- Pickersgill, B. Chile peppers (Capsicum spp.). In Ethnobotany of Mexico; Springer: Berlin/Heidelberg, Germany, 2016; pp. 417–437. [Google Scholar]
- Perry, L.; Dickau, R.; Zarrillo, S.; Holst, I.; Pearsall, D.M.; Piperno, D.R.; Berman, M.J.; Cooke, R.G.; Rademaker, K.; Ranere, A.J.; et al. Starch fossils and the domestication and dispersal of chili peppers (Capsicum spp. L.) in the Americas. Science 2007, 315, 986–988. [Google Scholar] [CrossRef]
- Doebley, J.F.; Gaut, B.S.; Smith, B.D. The molecular genetics of crop domestication. Cell 2006, 127, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Zeder, M.A. Core questions in domestication research. Proc. Natl. Acad. Sci. USA 2015, 112, 3191–3198. [Google Scholar] [CrossRef]
- Ross-Ibarra, J.; Morrell, P.L.; Gaut, B.S. Plant domestication, a unique opportunity to identify the genetic basis of adaptation. Proc. Natl. Acad. Sci. USA 2007, 104, 8641–8648. [Google Scholar] [CrossRef]
- Doebley, J.; Stec, A.; Wendel, J.; Edwards, M. Genetic and morphological analysis of a maize-teosinte F2 population: Implications for the origin of maize. Proc. Natl. Acad. Sci. USA 1990, 87, 9888–9892. [Google Scholar] [CrossRef] [PubMed]
- Tian, F.; Stevens, N.M.; Buckler, E.S. Tracking footprints of maize domestication and evidence for a massive selective sweep on chromosome 10. Proc. Natl. Acad. Sci. USA 2009, 106, 9979–9986. [Google Scholar] [CrossRef]
- Studer, A.; Zhao, Q.; Ross-Ibarra, J.; Doebley, J. Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 2011, 43, 1160. [Google Scholar] [CrossRef] [PubMed]
- Hufford, M.B.; Xu, X.; Van Heerwaarden, J.; Pyhäjärvi, T.; Chia, J.M.; Cartwright, R.A.; Elshire, R.J.; Glaubitz, J.C.; Guill, K.E.; Kaeppler, S.M.; et al. Comparative population genomics of maize domestication and improvement. Nat. Genet. 2012, 44, 808–811. [Google Scholar] [CrossRef]
- Bellucci, E.; Bitocchi, E.; Ferrarini, A.; Benazzo, A.; Biagetti, E.; Klie, S.; Minio, A.; Rau, D.; Rodriguez, M.; Panziera, A.; et al. Decreased nucleotide and expression diversity and modified coexpression patterns characterize domestication in the common bean. Plant Cell 2014, 26, 1901–1912. [Google Scholar] [CrossRef] [PubMed]
- Singh, J.; Zhao, J.; Vallejos, C.E. Differential transcriptome patterns associated with early seedling development in a wild and a domesticated common bean (Phaseolus vulgaris L.) accession. Plant Sci. 2018, 274, 153–162. [Google Scholar] [CrossRef]
- Lippman, Z.; Tanksley, S.D. Dissecting the genetic pathway to extreme fruit size in tomato using a cross between the small-fruited wild species Lycopersicon pimpinellifolium and L. esculentum var. Giant Heirloom. Genetics 2001, 158, 413–422. [Google Scholar]
- Müller, N.A.; Wijnen, C.L.; Srinivasan, A.; Ryngajllo, M.; Ofner, I.; Lin, T.; Ranjan, A.; West, D.; Maloof, J.N.; Sinha, N.R.; et al. Domestication selected for deceleration of the circadian clock in cultivated tomato. Nat. Genet. 2016, 48, 89–93. [Google Scholar] [CrossRef] [PubMed]
- Sauvage, C.; Rau, A.; Aichholz, C.; Chadoeuf, J.; Sarah, G.; Ruiz, M.; Santoni, S.; Causse, M.; David, J.; Glémin, S. Domestication rewired gene expression and nucleotide diversity patterns in tomato. Plant J. 2017, 91, 631–645. [Google Scholar] [CrossRef]
- Razifard, H.; Ramos, A.; Della Valle, A.L.; Bodary, C.; Goetz, E.; Manser, E.J.; Li, X.; Zhang, L.; Visa, S.; Tieman, D.; et al. Genomic evidence for complex domestication history of the cultivated tomato in Latin America. Mol. Biol. Evol. 2020, 37, 1118–1132. [Google Scholar] [CrossRef]
- Hernández-Verdugo, S.; Luna-Reyes, R.; Oyama, K. Genetic structure and differentiation of wild and domesticated populations of Capsicum annuum (Solanaceae) from Mexico. Plant Syst. Evol. 2001, 226, 129–142. [Google Scholar] [CrossRef]
- Paran, I.; Van Der Knaap, E. Genetic and molecular regulation of fruit and plant domestication traits in tomato and pepper. J. Exp. Bot. 2007, 58, 3841–3852. [Google Scholar] [CrossRef]
- Carvalho, S.; Ragassi, C.; Bianchetti, L.; Reifschneider, F.; Buso, G.; Faleiro, F. Morphological and genetic relationships between wild and domesticated forms of peppers (Capsicum frutescens L. and C. chinense Jacquin). Genet. Mol. Res. 2014, 13, 7447–7464. [Google Scholar] [CrossRef] [PubMed]
- Taitano, N.; Bernau, V.; Jardón-Barbolla, L.; Leckie, B.; Mazourek, M.; Mercer, K.; McHale, L.; Michel, A.; Baumler, D.; Kantar, M.; et al. Genome-wide genotyping of a novel Mexican Chile Pepper collection illuminates the history of landrace differentiation after Capsicum annuum L. domestication. Evol. Appl. 2019, 12, 78–92. [Google Scholar] [CrossRef]
- Qin, C.; Yu, C.; Shen, Y.; Fang, X.; Chen, L.; Min, J.; Cheng, J.; Zhao, S.; Xu, M.; Luo, Y.; et al. Whole-genome sequencing of cultivated and wild peppers provides insights into Capsicum domestication and specialization. Proc. Natl. Acad. Sci. USA 2014, 111, 5135–5140. [Google Scholar] [CrossRef]
- Kim, S.; Park, M.; Yeom, S.I.; Kim, Y.M.; Lee, J.M.; Lee, H.A.; Seo, E.; Choi, J.; Cheong, K.; Kim, K.T.; et al. Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 2014, 46, 270–278. [Google Scholar] [CrossRef]
- Ahn, Y.K.; Karna, S.; Jun, T.H.; Yang, E.Y.; Lee, H.E.; Kim, J.H.; Kim, J.H. Complete genome sequencing and analysis of Capsicum annuum varieties. Mol. Breed. 2016, 36, 140. [Google Scholar] [CrossRef]
- Hulse-Kemp, A.M.; Maheshwari, S.; Stoffel, K.; Hill, T.A.; Jaffe, D.; Williams, S.R.; Weisenfeld, N.; Ramakrishnan, S.; Kumar, V.; Shah, P.; et al. Reference quality assembly of the 3.5-Gb genome of Capsicum annuum from a single linked-read library. Hortic. D 2018, 5, 4. [Google Scholar] [CrossRef] [PubMed]
- Albert, V.A.; Chang, T.H. Evolution of a hot genome. Proc. Natl. Acad. Sci. USA 2014, 111, 5069–5070. [Google Scholar] [CrossRef] [PubMed]
- Díaz-Valenzuela, E.; Sawers, R.H.; Cibrián-Jaramillo, A. Cis-and Trans-Regulatory Variations in the Domestication of the Chili Pepper Fruit. Mol. Biol. Evol. 2020, 37, 1593–1603. [Google Scholar] [CrossRef]
- Holland, M.J. Transcript abundance in yeast varies over six orders of magnitude. J. Biol. Chem. 2002, 277, 14363–14366. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Liu, W.; Jin, X.; Ji, H.; Wang, H.; Glusman, G.; Robinson, M.; Liu, L.; Ruan, J.; Gao, S. NormExpression: An R package to normalize gene expression data using evaluated methods. Front. Genet. 2019, 10, 400. [Google Scholar] [CrossRef] [PubMed]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Kothari, S.; Joshi, A.; Kachhwaha, S.; Ochoa-Alejo, N. Chilli peppers? A review on tissue culture and transgenesis. Biotechnol. Adv. 2010, 28, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.L.; Li, L.; Shah, S.; Gong, Z.H. The relationship between red fruit colour formation and key genes of capsanthin biosynthesis pathway in Capsicum annuum. Biol. Plant. 2015, 59, 507–513. [Google Scholar] [CrossRef]
- Liu, W.; Chen, L.; Zhang, S.; Hu, F.; Wang, Z.; Lyu, J.; Wang, B.; Xiang, H.; Zhao, R.; Tian, Z.; et al. Decrease of gene expression diversity during domestication of animals and plants. BMC Evol. Biol. 2019, 19, 19. [Google Scholar] [CrossRef]
- Santaguida, S.; Musacchio, A. The life and miracles of kinetochores. EMBO J. 2009, 28, 2511–2531. [Google Scholar] [CrossRef]
- D?Archivio, S.; Wickstead, B. Trypanosome outer kinetochore proteins suggest conservation of chromosome segregation machinery across eukaryotes. J. Cell Biol. 2017, 216, 379–391. [Google Scholar] [CrossRef]
- Filkov, V. Identifying gene regulatory networks from gene expression data. In Handbook of Computational Molecular Biology; CRC Press: Boca Raton, FL, USA, 2005; pp. 101–132. [Google Scholar]
- Allocco, D.J.; Kohane, I.S.; Butte, A.J. Quantifying the relationship between co-expression, co-regulation and gene function. BMC Bioinform. 2004, 5, 18. [Google Scholar] [CrossRef]
- Spies, D.; Ciaudo, C. Dynamics in transcriptomics: Advancements in RNA-seq time course and downstream analysis. Comput. Struct. Biotechnol. J. 2015, 13, 469–477. [Google Scholar] [CrossRef]
- Darwin, C. The Variation of Animals and Plants Under Domestication; Cambridge University Press: Cambridge, UK, 1868; Volume 2. [Google Scholar]
- Swain, P.S.; Elowitz, M.B.; Siggia, E.D. Intrinsic and extrinsic contributions to stochasticity in gene expression. Proc. Natl. Acad. Sci. USA 2002, 99, 12795–12800. [Google Scholar] [CrossRef]
- Chapman, M.A.; Pashley, C.H.; Wenzler, J.; Hvala, J.; Tang, S.; Knapp, S.J.; Burke, J.M. A genomic scan for selection reveals candidates for genes involved in the evolution of cultivated sunflower (Helianthus annuus). Plant Cell 2008, 20, 2931–2945. [Google Scholar] [CrossRef]
- Li, Y.h.; Zhao, S.c.; Ma, J.x.; Li, D.; Yan, L.; Li, J.; Qi, X.t.; Guo, X.s.; Zhang, L.; He, W.m.; et al. Molecular footprints of domestication and improvement in soybean revealed by whole genome re-sequencing. BMC Genom. 2013, 14, 579. [Google Scholar] [CrossRef]
- Huang, C.L.; Hung, C.Y.; Chiang, Y.C.; Hwang, C.C.; Hsu, T.W.; Huang, C.C.; Hung, K.H.; Tsai, K.C.; Wang, K.H.; Osada, N.; et al. Footprints of natural and artificial selection for photoperiod pathway genes in Oryza. Plant J. 2012, 70, 769–782. [Google Scholar] [CrossRef] [PubMed]
- Pollinger, J.P.; Bustamante, C.D.; Fledel-Alon, A.; Schmutz, S.; Gray, M.M.; Wayne, R.K. Selective sweep mapping of genes with large phenotypic effects. Genome Res. 2005, 15, 1809–1819. [Google Scholar] [CrossRef]
- Rothammer, S.; Seichter, D.; Förster, M.; Medugorac, I. A genome-wide scan for signatures of differential artificial selection in ten cattle breeds. BMC Genom. 2013, 14, 908. [Google Scholar] [CrossRef]
- Dai, Q.; Geng, L.; Lu, M.; Jin, W.; Nan, X.; He, P.a.; Yao, Y. Comparative transcriptome analysis of the different tissues between the cultivated and wild tomato. PLoS ONE 2017, 12, e0172411. [Google Scholar] [CrossRef]
- Rong, J.; Lammers, Y.; Strasburg, J.L.; Schidlo, N.S.; Ariyurek, Y.; De Jong, T.J.; Klinkhamer, P.G.; Smulders, M.J.; Vrieling, K. New insights into domestication of carrot from root transcriptome analyses. BMC Genom. 2014, 15, 895. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Tang, S.; Zhu, S.; Tang, Q.; Zheng, X. Transcriptome comparison reveals the patterns of selection in domesticated and wild ramie (Boehmeria nivea L. Gaud). Plant Mol. Biol. 2014, 86, 85–92. [Google Scholar] [CrossRef]
- Chaudhary, B.; Hovav, R.; Rapp, R.; Verma, N.; Udall, J.A.; Wendel, J.F. Global analysis of gene expression in cotton fibers from wild and domesticated Gossypium barbadense. Evol. Dev. 2008, 10, 567–582. [Google Scholar] [CrossRef] [PubMed]
- Monson, M.S.; Cardona, C.J.; Coulombe, R.A.; Reed, K.M. Hepatic transcriptome responses of domesticated and wild turkey embryos to aflatoxin B1. Toxins 2016, 8, 16. [Google Scholar] [CrossRef]
- Christie, M.R.; Marine, M.L.; Fox, S.E.; French, R.A.; Blouin, M.S. A single generation of domestication heritably alters the expression of hundreds of genes. Nat. Commun. 2016, 7, 10676. [Google Scholar] [CrossRef]
- Albert, F.W.; Somel, M.; Carneiro, M.; Aximu-Petri, A.; Halbwax, M.; Thalmann, O.; Blanco-Aguiar, J.A.; Plyusnina, I.Z.; Trut, L.; Villafuerte, R.; et al. A comparison of brain gene expression levels in domesticated and wild animals. PLoS Genet. 2012, 8, e1002962. [Google Scholar] [CrossRef]
- Azzi, L.; Deluche, C.; Gévaudant, F.; Frangne, N.; Delmas, F.; Hernould, M.; Chevalier, C. Fruit growth-related genes in tomato. J. Exp. Bot. 2015, 66, 1075–1086. [Google Scholar] [CrossRef]
- Bourdon, M.; Frangne, N.; Mathieu-Rivet, E.; Nafati, M.; Cheniclet, C.; Renaudin, J.P.; Chevalier, C. Endoreduplication and growth of fleshy fruits. In Progress in Botany 71; Springer: Berlin/Heidelberg, Germany, 2010; pp. 101–132. [Google Scholar]
- Chevalier, C. Cell cycle control and fruit development. Annu. Plant Rev. Online 2018, 32, 269–293. [Google Scholar]
- Ogawa, D.; Ishikawa, K.; Nunomura, O.; Mii, M. Correlation between fruit characters and degree of polysomaty in fruit tissues of Capsicum. J. Jpn. Soc. Hortic. Sci. 2010, 79, 168–173. [Google Scholar] [CrossRef]
- Ogawa, D.; Ishikawa, K.; Mii, M. Difference in the polysomaty degree during fruit development among plants with different ploidy levels produced by artificial chromosome doubling of a pepper (Capsicum annuum) cultivar ‘Shishitou No. 562’. Sci. Hortic. 2012, 134, 121–126. [Google Scholar] [CrossRef]
- Vanneste, S.; Coppens, F.; Lee, E.; Donner, T.J.; Xie, Z.; Van Isterdael, G.; Dhondt, S.; De Winter, F.; De Rybel, B.; Vuylsteke, M.; et al. Developmental regulation of CYCA2s contributes to tissue-specific proliferation in Arabidopsis. EMBO J. 2011, 30, 3430–3441. [Google Scholar] [CrossRef]
- Guo, M.; Simmons, C.R. Cell number counts—The fw2. 2 and CNR genes and implications for controlling plant fruit and organ size. Plant Sci. 2011, 181, 1–7. [Google Scholar] [CrossRef]
- Gonzalez, N.; Gévaudant, F.; Hernould, M.; Chevalier, C.; Mouras, A. The cell cycle-associated protein kinase WEE1 regulates cell size in relation to endoreduplication in developing tomato fruit. Plant J. 2007, 51, 642–655. [Google Scholar] [CrossRef] [PubMed]
- Ariizumi, T.; Shinozaki, Y.; Ezura, H. Genes that influence yield in tomato. Breed. Sci. 2013, 63, 3–13. [Google Scholar] [CrossRef] [PubMed]
- Giovannoni, J.J. Genetic regulation of fruit development and ripening. Plant Cell 2004, 16, S170–S180. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Ding, Y.; Li, C.; Li, M.; Ma, F.; Yuan, Y. Comparative Proteomic Analysis Reveals Key Proteins Linked to the Accumulation of Soluble Sugars and Organic Acids in the Mature Fruits of the Wild Malus Species. Plants 2019, 8, 488. [Google Scholar] [CrossRef]
- Guo, J.; Wang, P.; Cheng, Q.; Sun, L.; Wang, H.; Wang, Y.; Kao, L.; Li, Y.; Qiu, T.; Yang, W.; et al. Proteomic analysis reveals strong mitochondrial involvement in cytoplasmic male sterility of pepper (Capsicum annuum L.). J. Proteom. 2017, 168, 15–27. [Google Scholar] [CrossRef]
- Voelckel, C.; Borevitz, J.O.; Kramer, E.M.; Hodges, S.A. Within and between whorls: Comparative transcriptional profiling of Aquilegia and Arabidopsis. PLoS ONE 2010, 5, e9735. [Google Scholar] [CrossRef]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef]
- Lee, T.A.; Wetering, S.W.V.; Brusslan, J.A. Stromal protein degradation is incomplete in Arabidopsis thaliana autophagy mutants undergoing natural senescence. BMC Res. Notes 2013, 6, 17. [Google Scholar] [CrossRef]
- Launholt, D.; Merkle, T.; Houben, A.; Schulz, A.; Grasser, K.D. Arabidopsis chromatin-associated HMGA and HMGB use different nuclear targeting signals and display highly dynamic localization within the nucleus. Plant Cell 2006, 18, 2904–2918. [Google Scholar] [CrossRef]
- Kobayashi, K.; Suzuki, T.; Iwata, E.; Nakamichi, N.; Suzuki, T.; Chen, P.; Ohtani, M.; Ishida, T.; Hosoya, H.; Müller, S.; et al. Transcriptional repression by MYB3R proteins regulates plant organ growth. EMBO J. 2015, 34, 1992–2007. [Google Scholar] [CrossRef]
- Shin, J.; Jeong, G.; Park, J.Y.; Kim, H.; Lee, I. MUN (MERISTEM UNSTRUCTURED), encoding a SPC24 homolog of NDC80 kinetochore complex, affects development through cell division in arabidopsis thaliana. Plant J. 2018, 93, 977–991. [Google Scholar] [CrossRef]
- Yao, M.; Wakamatsu, Y.; Itoh, T.J.; Shoji, T.; Hashimoto, T. Arabidopsis SPIRAL2 promotes uninterrupted microtubule growth by suppressing the pause state of microtubule dynamics. J. Cell Sci. 2008, 121, 2372–2381. [Google Scholar] [CrossRef] [PubMed]
- Wightman, R.; Chomicki, G.; Kumar, M.; Carr, P.; Turner, S.R. SPIRAL2 determines plant microtubule organization by modulating microtubule severing. Curr. Biol. 2013, 23, 1902–1907. [Google Scholar] [CrossRef] [PubMed]
- Buschmann, H.; Fabri, C.O.; Hauptmann, M.; Hutzler, P.; Laux, T.; Lloyd, C.W.; Schäffner, A.R. Helical growth of the Arabidopsis mutant tortifolia1 reveals a plant-specific microtubule-associated protein. Curr. Biol. 2004, 14, 1515–1521. [Google Scholar] [CrossRef]
- Petrovská, B.; Jeřábková, H.; Kohoutová, L.; Cenklová, V.; Pochylová, Ž.; Gelová, Z.; Kočárová, G.; Váchová, L.; Kurejová, M.; Tomaštíková, E.; et al. Overexpressed TPX2 causes ectopic formation of microtubular arrays in the nuclei of acentrosomal plant cells. J. Exp. Bot. 2013, 64, 4575–4587. [Google Scholar] [CrossRef]
- Vos, J.W.; Pieuchot, L.; Evrard, J.L.; Janski, N.; Bergdoll, M.; de Ronde, D.; Perez, L.H.; Sardon, T.; Vernos, I.; Schmit, A.C. The plant TPX2 protein regulates prospindle assembly before nuclear envelope breakdown. Plant Cell 2008, 20, 2783–2797. [Google Scholar] [CrossRef]
- Nashima, K.; Shimizu, T.; Nishitani, C.; Yamamoto, T.; Takahashi, H.; Nakazono, M.; Itai, A.; Isuzugawa, K.; Hanada, T.; Takashina, T.; et al. Microarray analysis of gene expression patterns during fruit development in European pear (Pyrus communis). Sci. Hortic. 2013, 164, 466–473. [Google Scholar] [CrossRef]
- Steiner, A.; Rybak, K.; Altmann, M.; McFarlane, H.E.; Klaeger, S.; Nguyen, N.; Facher, E.; Ivakov, A.; Wanner, G.; Kuster, B.; et al. Cell cycle-regulated PLEIADE/At MAP 65–3 links membrane and microtubule dynamics during plant cytokinesis. Plant J. 2016, 88, 531–541. [Google Scholar] [CrossRef] [PubMed]
- Pignocchi, C.; Minns, G.E.; Nesi, N.; Koumproglou, R.; Kitsios, G.; Benning, C.; Lloyd, C.W.; Doonan, J.H.; Hills, M.J. ENDOSPERM DEFECTIVE1 is a novel microtubule-associated protein essential for seed development in Arabidopsis. Plant Cell 2009, 21, 90–105. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Marcus, A.; Li, W.; Hu, Y.; Calzada, J.P.V.; Grossniklaus, U.; Cyr, R.J.; Ma, H. The Arabidopsis ATK1 gene is required for spindle morphogenesis in male meiosis. Development 2002, 129, 2401–2409. [Google Scholar] [PubMed]
- Cifuentes, M.; Jolivet, S.; Cromer, L.; Harashima, H.; Bulankova, P.; Renne, C.; Crismani, W.; Nomura, Y.; Nakagami, H.; Sugimoto, K.; et al. TDM1 regulation determines the number of meiotic divisions. PLoS Genet. 2016, 12, e1005856. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, B.E.; Hirota, T.; Kudo, N.R.; Peters, J.M.; Nasmyth, K. Shugoshin prevents dissociation of cohesin from centromeres during mitosis in vertebrate cells. PLoS Biol. 2005, 3, e86. [Google Scholar] [CrossRef] [PubMed]
- Kitajima, T.S.; Kawashima, S.A.; Watanabe, Y. The conserved kinetochore protein shugoshin protects centromeric cohesion during meiosis. Nature 2004, 427, 510–517. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Sun, B.; Cai, W.; Zhou, X.; Mao, Y.; Chen, C.; Wei, J.; Cao, B.; Chen, C.; Chen, G.; et al. Natural variations in the MYB transcription factor MYB31 determine the evolution of extremely pungent peppers. New Phytol. 2019, 223, 922–938. [Google Scholar] [CrossRef]
- Sun, B.; Zhu, Z.; Chen, C.; Chen, G.; Cao, B.; Chen, C.; Lei, J. Jasmonate-inducible R2R3-MYB transcription factor regulates capsaicinoid biosynthesis and stamen development in Capsicum. J. Agric. Food Chem. 2019, 67, 10891–10903. [Google Scholar] [CrossRef]
- Pickersgill, B. Domestication of plants in the Americas: Insights from Mendelian and molecular genetics. Ann. Bot. 2007, 100, 925–940. [Google Scholar] [CrossRef]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2013. [Google Scholar]
- Escoto-Sandoval, C.; Flores-Díaz, A.; Reyes-Valdés, M.H.; Ochoa-Alejo, N.; Martinez, O. An R package for data mining chili pepper fruit transcriptomes. Evaluation 2020. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Martínez, O.; Escoto-Sandoval, C. Salsa: An R Package of Data Mining Facilities for Capsicum Gene Expression Profiles; 2021. [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Accession | Type | Key | Maturity (DAA) |
|---|---|---|---|
| Ancho San Luis | D | AS | 80 |
| Criollo de Morelos 334 (CM334) | D | CM | 60 |
| California Wonder | D | CW | 70 |
| Jalapeño Espinalteco | D | JE | 70 |
| Serrano Tampiqueño 74 | D | ST | 60 |
| Zunla-1 | D | ZU | 60 |
| Piquín Coahuila | W | CO | 60 |
| Piquín Queretaro | W | QU | 60 |
| Piquín Sonora Red | W | SR | 60 |
| Piquín Sonora Yellow | W | SY | 60 |
| Figure 3A Legend | Protein ID | Description | Ortholog |
|---|---|---|---|
| (1)-MAP/ASE1 (1). | XP_016564755.1 | 65-kDa microtubule-associated protein 3 | AT5G51600 |
| (2)-DUF566. | XP_016538322.1 | ENDOSPERM DEFECTIVE 1 (DUF566) | AT2G44190 |
| (3)-Kinesin 3, isoform X3. | XP_016541615.1 | Kinesin 3 isoform X3 | AT4G21270 |
| (4)-MAP/ASE 1 (2). | XP_016575449.1 | 65-kDa microtubule-associated protein 3 isoform X1 | AT5G51600 |
| (5)-POLENLESS 3. | XP_016577799.1 | Protein POLLENLESS 3 | AT4G20900 |
| (6)-Shugoshin. | XP_016548908.1 | Shugoshin-1; chromosome segregation | AT3G44960 |
| (7)-AP2/B3-TF (1). | XP_016568750.1 | AP2/B3-like TF family protein | AT5G42700 |
| (8)-HGM B, 6. | XP_016555757.1 | High-mobility group B protein 6 | AT4G11080 |
| (9)-Cyclin S13-7. | XP_016543946.1 | G2/mitotic-specific cyclin S13-7 | AT3G11520 |
| (10)-SET domain protein 35. | XP_016547461.1 | SET domain; methyltransferase activity. | AT1G26760 |
| (11)-AP2/B3-TF (2). | XP_016575946.1 | AP2/B3-like TF family protein | AT5G58280 |
| (12)-FHA, PS1. | XP_016574880.1 | FHA domain-containing protein PS1 | AT1G34355 |
| (13)-Myb 3R-1.6 | XP_016537977.1 | Myb-related protein 3R-1 | AT4G32730 |
| (14)-Tesmin / TSO1 CX3 3. | XP_016565918.1 | Protein tesmin/TSO1 CXC 3 | AT3G22780 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martínez, O.; Arce-Rodríguez, M.L.; Hernández-Godínez, F.; Escoto-Sandoval, C.; Cervantes-Hernández, F.; Hayano-Kanashiro, C.; Ordaz-Ortiz, J.J.; Reyes-Valdés, M.H.; Razo-Mendivil, F.G.; Garcés-Claver, A.; et al. Transcriptome Analyses Throughout Chili Pepper Fruit Development Reveal Novel Insights into the Domestication Process. Plants 2021, 10, 585. https://doi.org/10.3390/plants10030585
Martínez O, Arce-Rodríguez ML, Hernández-Godínez F, Escoto-Sandoval C, Cervantes-Hernández F, Hayano-Kanashiro C, Ordaz-Ortiz JJ, Reyes-Valdés MH, Razo-Mendivil FG, Garcés-Claver A, et al. Transcriptome Analyses Throughout Chili Pepper Fruit Development Reveal Novel Insights into the Domestication Process. Plants. 2021; 10(3):585. https://doi.org/10.3390/plants10030585
Chicago/Turabian StyleMartínez, Octavio, Magda L. Arce-Rodríguez, Fernando Hernández-Godínez, Christian Escoto-Sandoval, Felipe Cervantes-Hernández, Corina Hayano-Kanashiro, José J. Ordaz-Ortiz, M. Humberto Reyes-Valdés, Fernando G. Razo-Mendivil, Ana Garcés-Claver, and et al. 2021. "Transcriptome Analyses Throughout Chili Pepper Fruit Development Reveal Novel Insights into the Domestication Process" Plants 10, no. 3: 585. https://doi.org/10.3390/plants10030585
APA StyleMartínez, O., Arce-Rodríguez, M. L., Hernández-Godínez, F., Escoto-Sandoval, C., Cervantes-Hernández, F., Hayano-Kanashiro, C., Ordaz-Ortiz, J. J., Reyes-Valdés, M. H., Razo-Mendivil, F. G., Garcés-Claver, A., & Ochoa-Alejo, N. (2021). Transcriptome Analyses Throughout Chili Pepper Fruit Development Reveal Novel Insights into the Domestication Process. Plants, 10(3), 585. https://doi.org/10.3390/plants10030585