
Genome-Wide Identification and Expression Analysis of MADS-Box Family Genes in Litchi (Litchi chinensis Sonn.) and Their Involvement in Floral Sex Determination

 and
and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Results
2.1. Identification and Characterization of MADS-Box Genes in Litchi
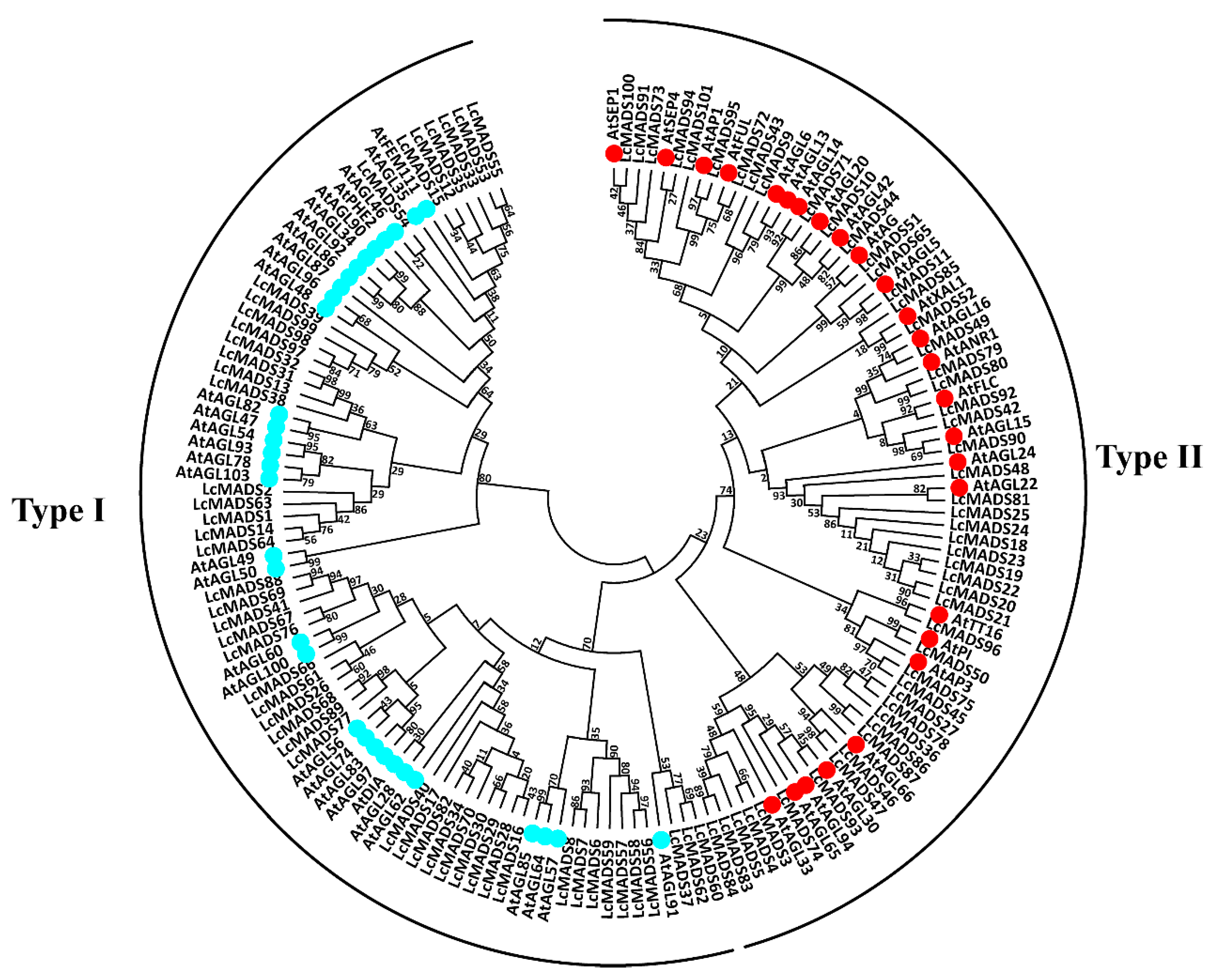
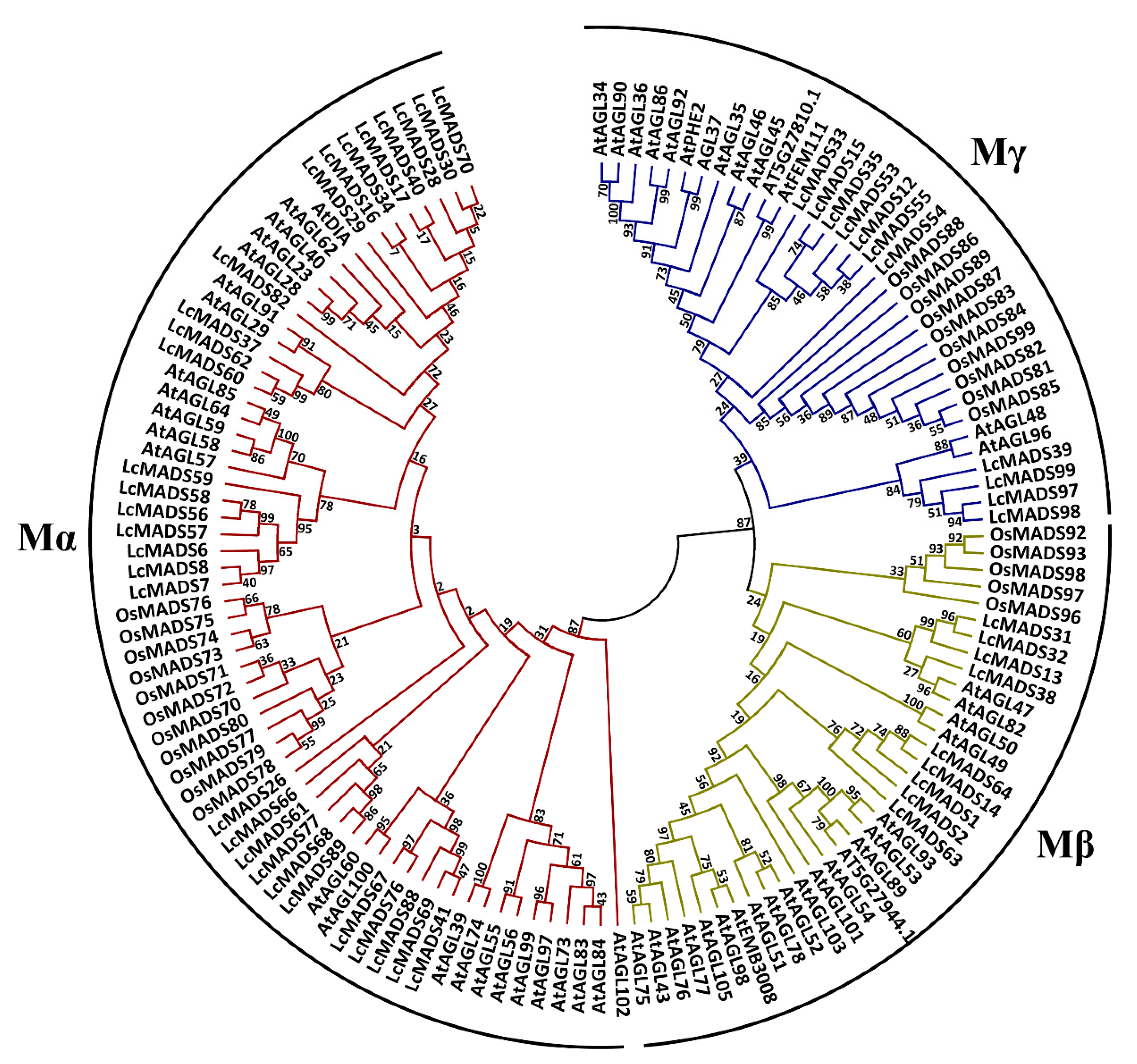
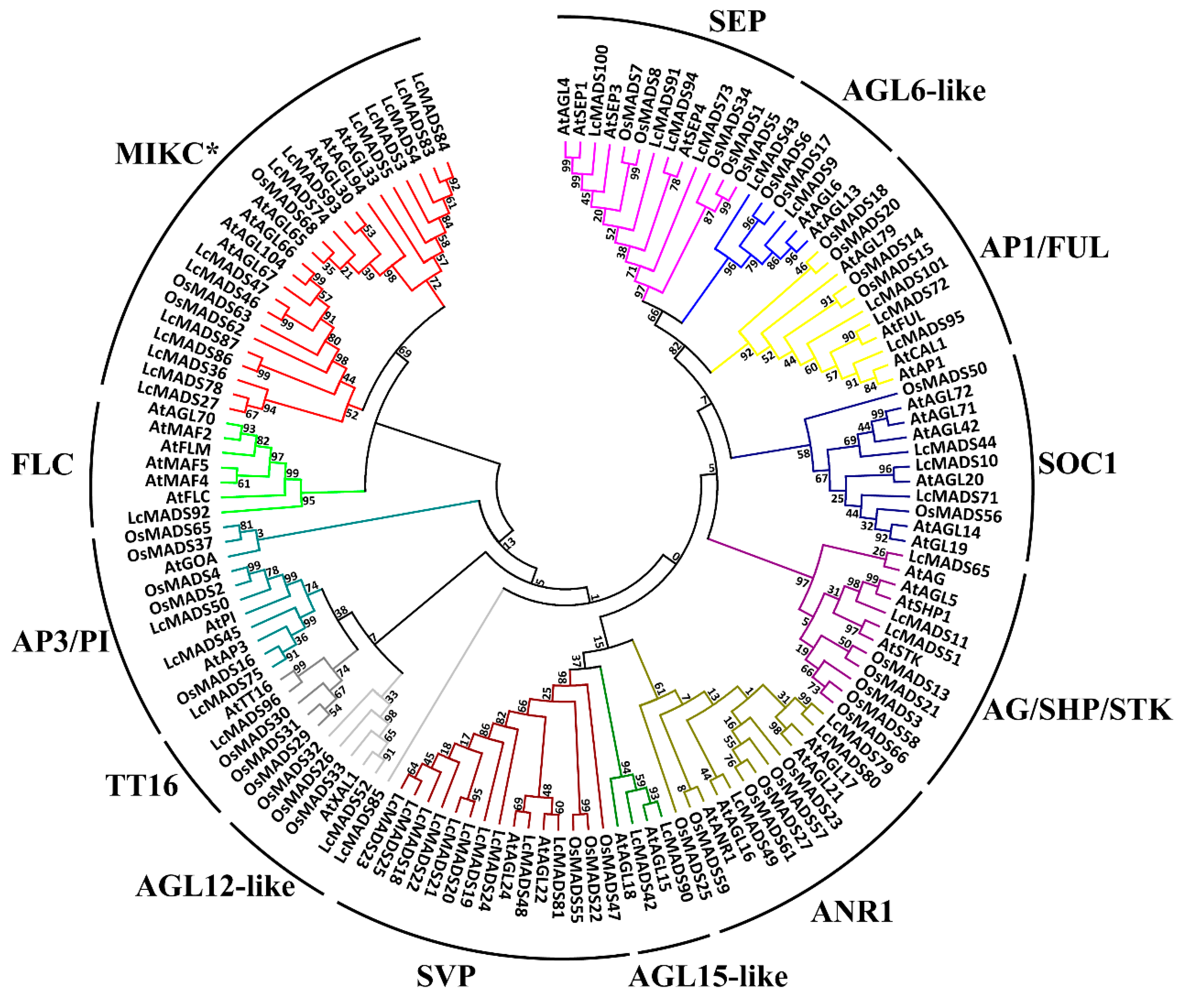
2.2. Phylogenetic Analysis of Litchi MADS-Box Genes
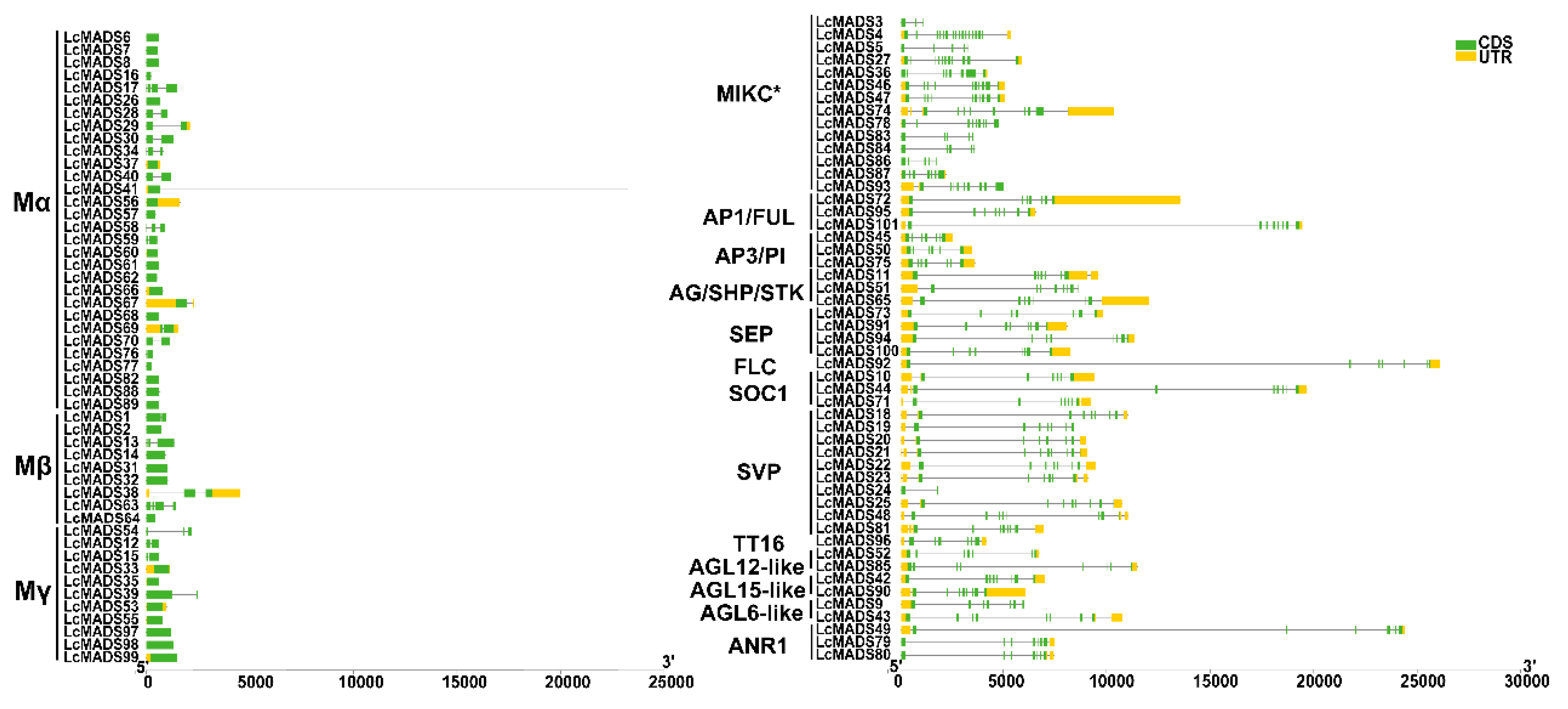
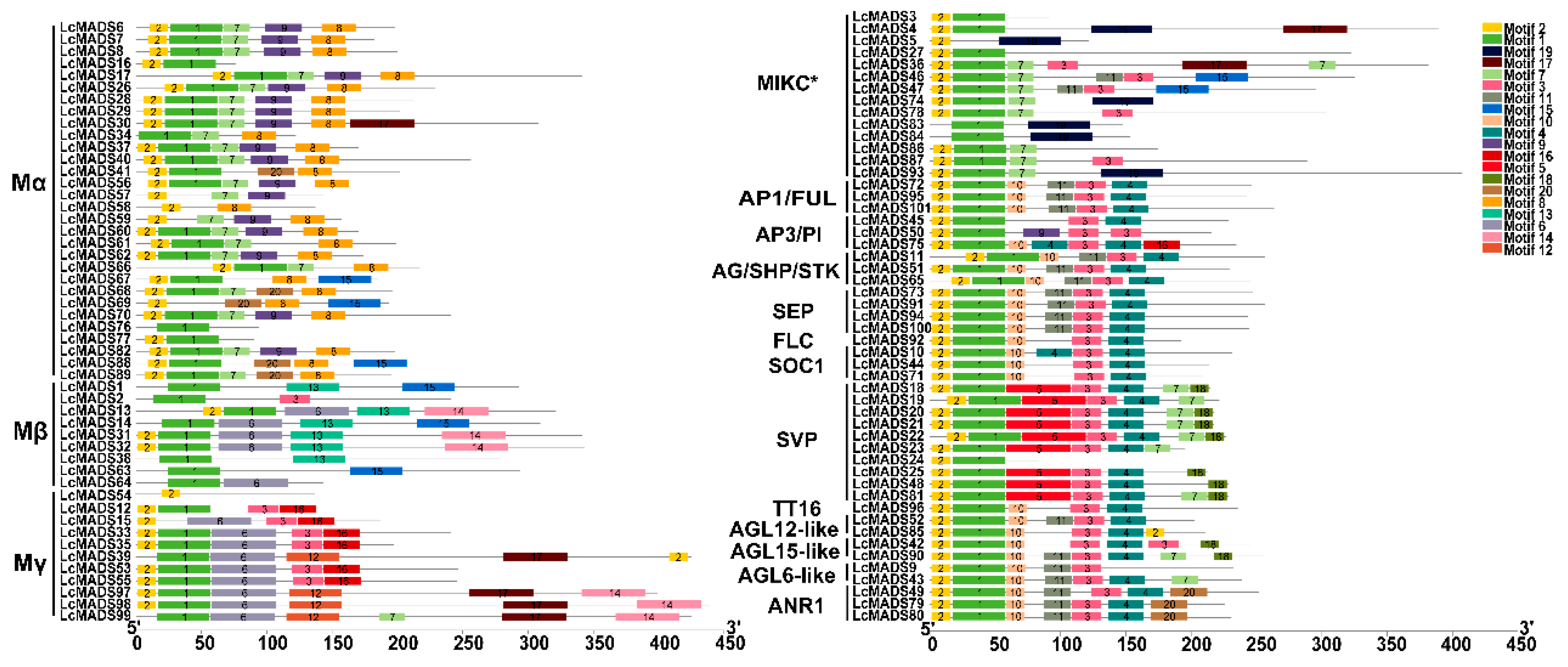
2.3. Identification of Gene Domain, Structure, and Conserved Motif
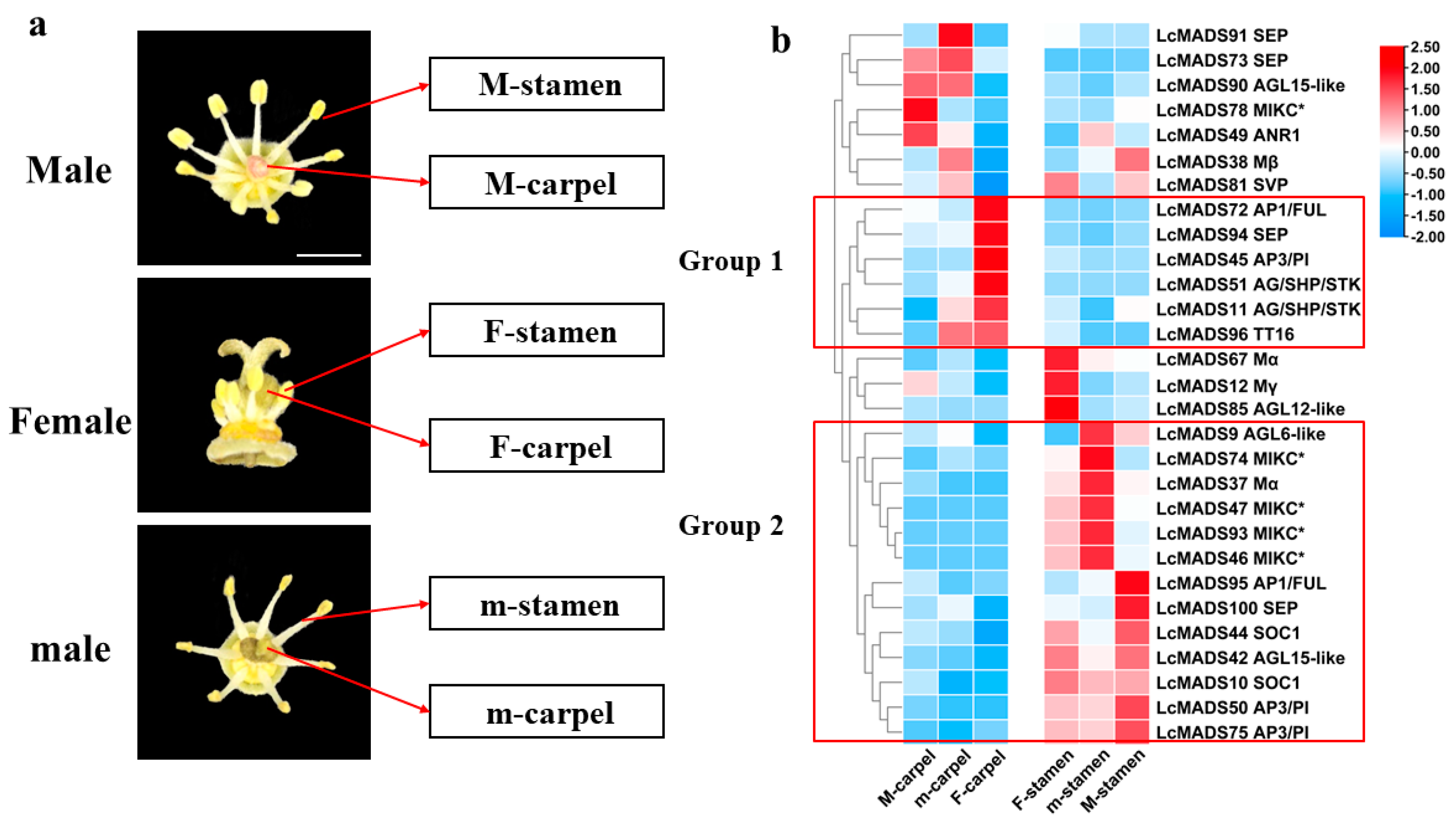
2.4. Expression Profiles of Litchi MADS-Box Genes in Different Floral Organ Tissues
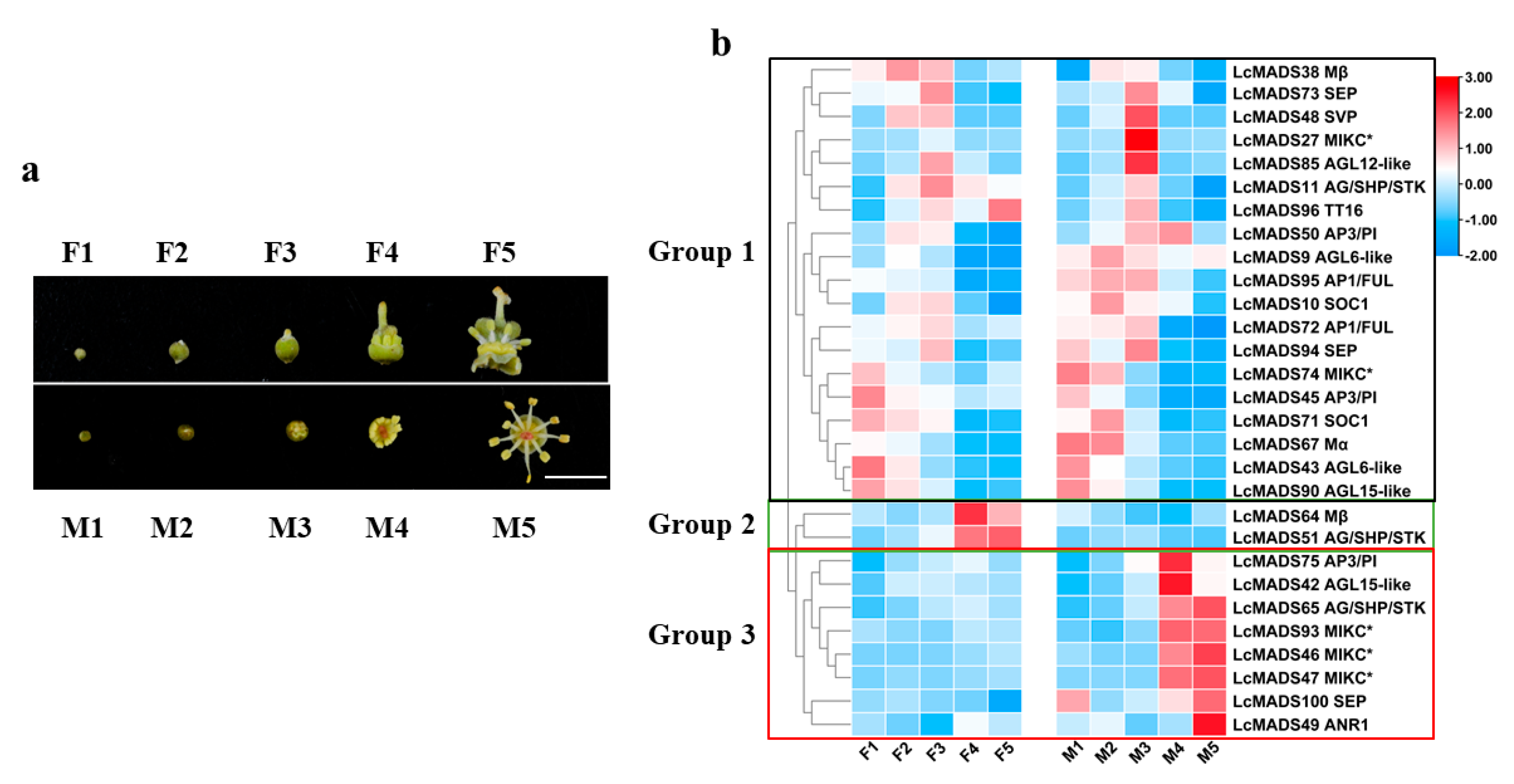
2.5. Gene Expression of Litchi MADS-Box Genes during Flower Development
2.6. Prediction of Promoter Elements in Litchi MADS-Box Genes
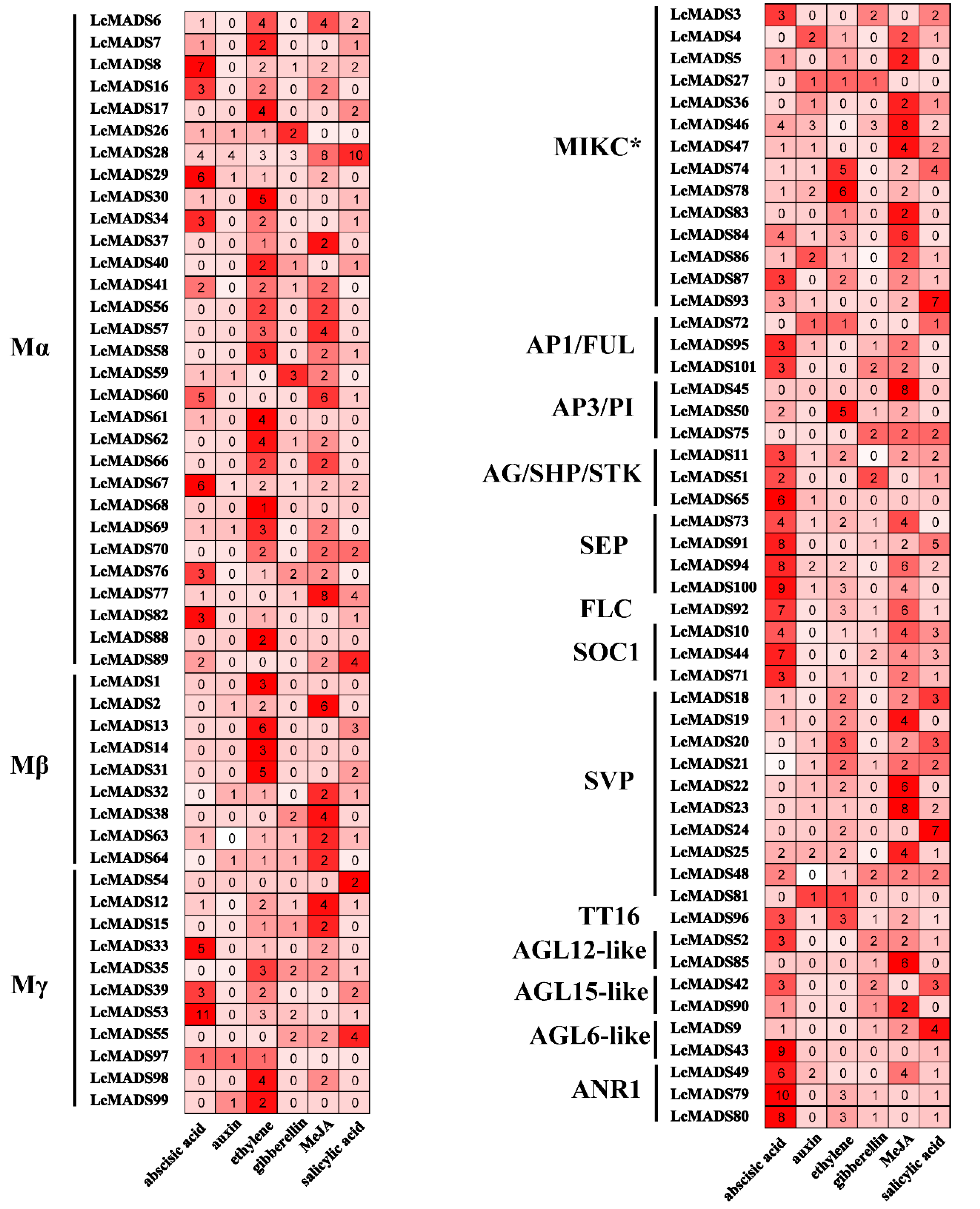
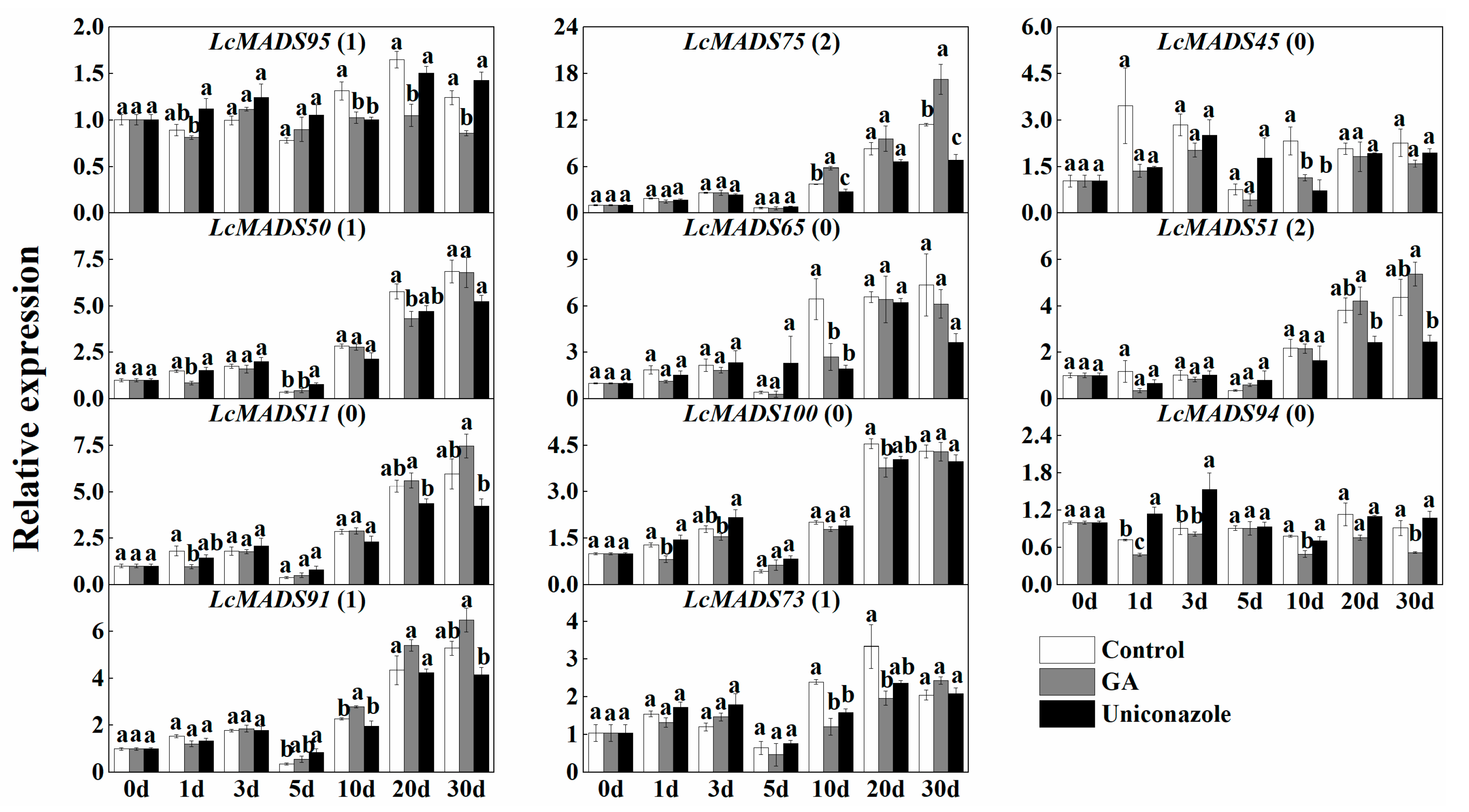
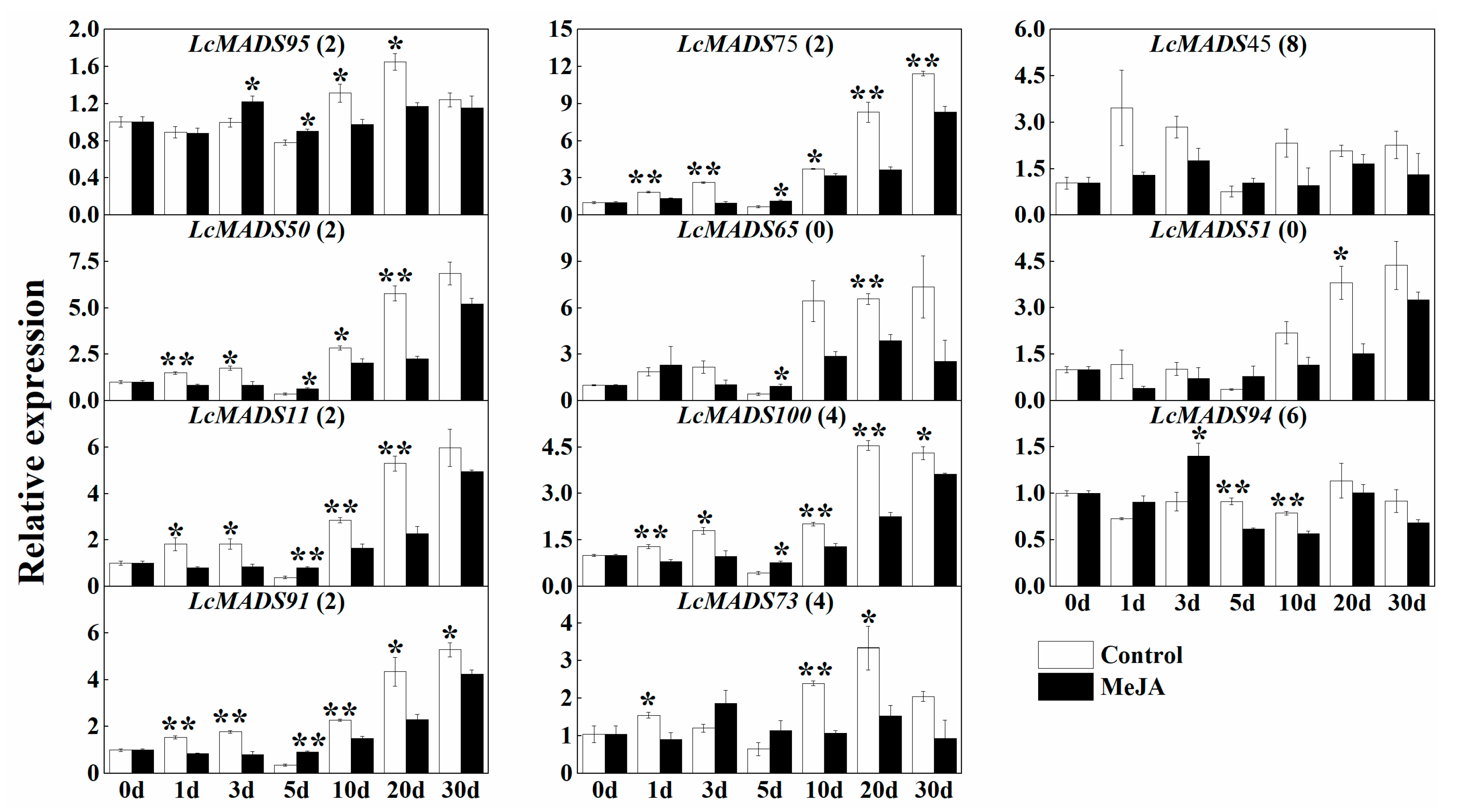
2.7. Expression of MADS-Box Genes in Response to the Treatment of Hormones
3. Discussion
4. Materials and Methods
4.1. Whole-Genome Identification of MADS-Box Genes
4.2. Phylogenetic Analysis of MADS-Box Genes and Mapping on Chromosomes
4.3. Gene Domain, Structure, and Conserved Motif Analysis
4.4. Cis-Element Enrichment Analysis
4.5. Plant Materials
4.6. Analysis of Gene Expression by RNA-Sequencing and Quantitative Real-Time PCR
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Smaczniak, C.; Immink, R.G.H.; Angenent, G.C.; Kaufmann, K. Developmental and evolutionary diversity of plant MADS-domain factors: Insights from recent studies. Development 2012, 139, 3081–3098. [Google Scholar] [CrossRef]
- Passmore, S.; Maine, G.T.; Elble, R.; Christ, C.; Type, B.K. Saccharomyces cerevisiae protein involved in plasmid maintenance is necessary for mating of MATa Cells. J. Mol. Biol. 1988, 204, 593–606. [Google Scholar] [CrossRef]
- Norman, C.; Runswick, M.; Pollock, R.; Treisman, R. Isolation and properties of cDNA clones encoding SRF, a transcription factor that binds to the c-fos serum response element. Cell 1988, 55, 989–1003. [Google Scholar] [CrossRef]
- Yanofsky, M.F.; Ma, H.; Bowman, J.L.; Drews, G.N.; Feldmann, K.A.; Meyerowitz, E.M. The protein encoded by the Arabidopsis homeotic gene agamous resembles transcription factors. Nature 1990, 346, 35–39. [Google Scholar] [CrossRef] [PubMed]
- Sommer, H.; Beltran, J.P.; Huijser, P.; Pape, H.; Lonnig, W.E.; Saedler, H.; Schwarz-Sommer, Z. Deficiens, a homeotic gene involved in the control of flower morphogenesis in Antirrhinum majus: The protein shows homology to transcription factors. EMBO J. 1990, 9, 605–613. [Google Scholar] [CrossRef] [PubMed]
- De Bodt, S.; Raes, J.; Van de Peer, Y.; Theissen, G. And then there were many: MADS goes genomic. Trends Plant Sci. 2003, 8, 475–483. [Google Scholar] [CrossRef] [PubMed]
- Becker, A.; Theissen, G. The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 2003, 29, 464–489. [Google Scholar] [CrossRef]
- Parenicova, L.; De Folter, S.; Kieffer, M.; Horner, D.S.; Favalli, C.; Busscher, J.; Cook, H.E.; Ingram, R.M.; Kater, M.M.; Davies, B.; et al. Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis. Plant Cell 2003, 15, 1538–1551. [Google Scholar] [CrossRef] [PubMed]
- Henschel, K.; Kofuji, R.; Hasebe, M.; Saedler, H.; Munster, T.; Theissen, G. Two ancient classes of MIKC-type MADS-box genes are present in the moss Physcomitrella patens. Mol. Biol. Evol. 2002, 19, 801–814. [Google Scholar] [CrossRef]
- Weigel, D.; Meyerowitz, E.M. The ABCs of floral homeotic genes. Cell 1994, 78, 203–209. [Google Scholar] [CrossRef]
- Meyerowitz, E.M.; Coen, E.S. The war of the whorls: Genetic interactions controlling flower development. Nature 1991, 353, 31–37. [Google Scholar]
- Jack, T. Plant development going MADS. Plant Mol. Biol. 2001, 46, 515–520. [Google Scholar] [CrossRef]
- Ma, H.; DePamphilis, C. The ABCs of floral evolution. Cell 2000, 101, 5–8. [Google Scholar] [CrossRef]
- Pelaz, S.; Ditta, G.S.; Baumann, E.; Yanofsky, M.F. B and C foral organ identity functions require SEPALLATA MADS-box genes. Nature 2000, 405, 200–203. [Google Scholar] [CrossRef]
- Kaufmann, K.; Melzer, R.; Theissen, G. MIKC-type MADS-domain proteins: Structural modularity, protein interactions and network evolution in land plants. Gene 2005, 347, 183–198. [Google Scholar] [CrossRef] [PubMed]
- Robbertse, H.; Fivaz, J.; Menzel, C. A reevaluation of tree model, inflorescence morphology, and sex ratio in lychee (Litchi Chinensis Sonn.). J. Amer. Soc. Hort. Sci. 1995, 120, 914–920. [Google Scholar] [CrossRef]
- Wei, Y.; Dong, C.; Zhang, H.; Zheng, X.; Shu, B.; Shi, S.; Li, W. Transcriptional changes in litchi (Litchi chinensis Sonn.) inflorescences treated with uniconazole. PLoS ONE 2017, 12, e176053. [Google Scholar] [CrossRef]
- Chen, Y.; Tan, B. New insight in the Gibberellin biosynthesis and signal transduction. Plant Signal. Behav. 2015, 10, e1000140. [Google Scholar] [CrossRef][Green Version]
- De Folter, S.; Shchennikova, A.V.; Franken, J.; Busscher, M.; Baskar, R.; Grossniklaus, U.; Angenent, G.C.; Immink, R.G.H. A Bsister MADS-box gene involved in ovule and seed development in petunia and Arabidopsis. Plant J. 2006, 47, 934–946. [Google Scholar] [CrossRef]
- Mondragon-Palomino, M.; Theissen, G. Conserved differential expression of paralogous DEFICIENS- and GLOBOSA-like MADS-box genes in the flowers of Orchidaceae: Refining the ‘orchid code’. Plant J. 2011, 66, 1008–1019. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Ming, M.; Li, J.; Shi, D.; Qiao, X.; Li, L.; Zhang, S.; Wu, J. Genome-wide identification of the MADS-box transcription factor family in pear (Pyrus bretschneideri) reveals evolution and functional divergence. PeerJ 2017, 5, e3776. [Google Scholar] [CrossRef] [PubMed]
- Leseberg, C.H.; Li, A.; Kang, H.; Duvall, M.; Mao, L. Genome-wide analysis of the MADS-box gene family in Populus trichocarpa. Gene 2006, 378, 84–94. [Google Scholar] [CrossRef]
- Velasco, R.; Zharkikh, A.; Affourtit, J.; Dhingra, A.; Cestaro, A.; Kalyanaraman, A.; Fontana, P.; Bhatnagar, S.K.; Troggio, M.; Pruss, D.; et al. The genome of the domesticated apple (Malus × domestica Borkh.). Nat. Genet. 2010, 42, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Arora, R.; Agarwal, P.; Ray, S.; Singh, A.K.; Singh, V.P.; Tyagi, A.K.; Kapoor, S. MADS-box gene family in rice: Genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genom. 2007, 8, 242. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Fatima, M.; Zhou, P.; Ma, Q.; Ming, R. Analysis of MADS-box genes revealed modified flowering gene network and diurnal expression in pineapple. BMC Genom. 2020, 21, 8. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Tang, D.; Lin, X.; Ding, M.; Tong, Z. Genome-wide identification of MADS-box family genes in moso bamboo (Phyllostachys edulis) and a functional analysis of PeMADS5 in flowering. BMC Plant Biol. 2018, 18, 176. [Google Scholar] [CrossRef]
- Li, X.; Shen, X.; Zhang, S.; Jiang, M.; Zhang, Z.; Lin, Y.; Lai, Z. Genome-wide identification and function prediction of MADS-box gene family in Dimocarpus longan Lour. Chin. J. Appl. Environ. Biol. 2021. (In Chinese) [Google Scholar] [CrossRef]
- Ming, R.; VanBuren, R.; Wai, C.M.; Tang, H.; Schatz, M.C.; Bowers, J.E.; Lyons, E.; Wang, M.; Chen, J.; Biggers, E.; et al. The pineapple genome and the evolution of CAM photosynthesis. Nat. Genet. 2015, 47, 1435–1442. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Yoo, S.J.; Park, S.H.; Hwang, I.; Lee, J.S.; Ahn, J.H. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 2007, 21, 397–402. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.; Shen, L.; Wu, Y.; Chen, H.; Robertson, M.; Helliwell, C.A.; Ito, T.; Meyerowitz, E.; Yu, H. A repressor complex governs the integration of flowering signals in Arabidopsis. Dev. Cell 2008, 15, 110–120. [Google Scholar] [CrossRef]
- Xiao, Q.; Su, Z.; Chen, H.; Shen, J. Genome-wide identification and involvement of litchi SPL genes in flowering in response to cold and leaf maturity. J. Hortic. Sci. Biotechnol. 2019, 94, 428–440. [Google Scholar] [CrossRef]
- Ray, P.K.; Ruby, R. Effect of paclobutrazol on flowering in “China” litchi (Litchi chinensis Sonn.). Ind. J. Plant Physiol. 2004, 9, 208–211. [Google Scholar]
- Hu, J.; Kim, H.J.; Chen, H.; Zhou, B. Litchi flowering is regulated by expression of short vegetative phase genes. J. Amer. Soc. Hort. Sci. 2018, 143, 101–109. [Google Scholar] [CrossRef]
- Gregis, V.; Sessa, A.; Dorca-Fornell, C.; Kater, M.M. The Arabidopsis floral meristem identity genes AP1, AGL24 and SVP directly repress class B and C floral homeotic genes. Plant J. 2009, 60, 626–637. [Google Scholar] [CrossRef] [PubMed]
- Trevaskis, B.; Tadege, M.; Hemming, M.N.; Peacock, W.J.; Dennis, E.S.; Sheldon, C. Short Vegetative Phase-Like MADS-box genes inhibit floral meristem identity in barley. Plant Physiol. 2007, 143, 225–235. [Google Scholar] [CrossRef]
- Wu, R.; Walton, E.F.; Richardson, A.C.; Wood, M.; Hellens, R.P.; Varkonyi-Gasic, E. Conservation and divergence of four kiwifruit SVP-like MADS-box genes suggest distinct roles in kiwifruit bud dormancy and flowering. J. Exp. Bot. 2012, 63, 797–807. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Rouse, D.T.; Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. The molecular basis of vernalization: The central role of FLOWERING LOCUS C (FLC). Proc. Natl. Acad. Sci. USA 2000, 97, 3753–3758. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.; Dean, C. Arabidopsis, the rosetta stone of flowering time? Science 2002, 296, 285–289. [Google Scholar] [CrossRef]
- Hepworth, J.; Dean, C. Flowering Locus C’s lessons: Conserved chromatin switches underpinning developmental timing and adaptation. Plant Physiol. 2015, 168, 1237–1245. [Google Scholar] [CrossRef]
- Csorba, T.; Questa, J.; Sun, Q.; Dean, C. Antisense COOLAIR mediates the coordinated switching of chromatin states at FLC during vernalization. Proc. Natl. Acad. Sci. USA 2014, 111, 16160–16165. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Zhu, P.; Hepworth, J.; Bloomer, R.; Antoniou-Kourounioti, R.L.; Doughty, J.; Heckmann, A.; Xu, C.Y.; Yang, H.C.; Dean, C. Natural temperature fluctuations promote COOLAIR regulation of FLC. Genes Dev. 2021, 35, 12. [Google Scholar] [CrossRef]
- Samach, A.; Onouchi, H.; Gold, S.; Ditta, G.; Schwarz-Sommer, Z.; Yanofsky, M.; Coupland, G. Distinct roles of CONSTANS target genes in reproductive development of Arabidopsis. Science 2000, 288, 1613–1616. [Google Scholar] [CrossRef]
- Shen, J.; Xiao, Q.; Qiu, H.; Chen, C.; Chen, H. Integrative effect of drought and low temperature on litchi (Litchi chinensis Sonn.) floral initiation revealed by dynamic genome-wide transcriptome analysis. Sci. Rep. 2016, 6, 32005. [Google Scholar] [CrossRef]
- Li, W.; Mao, J.; Su, J.; Li, X.; Chen, B. Exogenous ABA and its inhibitor regulate flower bud induction of apple cv. ‘Nagafu No. 2′ grafted on different rootstocks. Trees 2021, 35, 609–620. [Google Scholar] [CrossRef]
- Cui, Z.; Zhou, B.; Zhang, Z.; Hu, Z. Abscisic acid promotes flowering and enhances LcAP1 expression in Litchi chinensis Sonn. S. Afr. J. Bot. 2013, 88, 76–79. [Google Scholar] [CrossRef]
- Frankowski, K.; Wilmowicz, E.; Kucko, A.; Kesy, J.; Swiezawska, B.; Kopcewicz, J. Ethylene, auxin, and abscisic acid interactions in the control of photoperiodic flower induction in Pharbitis nil. Biol. Plant. 2014, 58, 305–310. [Google Scholar] [CrossRef]
- Wang, W.; Chen, W.; Chen, W.; Hung, L.; Chang, P. Influence of abscisic acid on flowering in Phalaenopsis hybrida. Plant Physiol. Biochem. 2002, 40, 97–100. [Google Scholar] [CrossRef]
- Berger, B.; Baldwin, I.T. Silencing the hydroxyproline-rich glycopeptide systemin precursor in two accessions of Nicotiana attenuata alters flower morphology and rates of self-pollination. Plant Physiol. 2009, 149, 1690–1700. [Google Scholar] [CrossRef] [PubMed]
- Nagpal, P.; Ellis, C.M.; Weber, H.; Ploense, S.E.; Barkawi, L.S.; Guilfoyle, T.J.; Hagen, G.; Alonso, J.M.; Cohen, J.D.; Farmer, E.E.; et al. Auxin response factors ARF6 and ARF8 promote jasmonic acid production and flower maturation. Development 2005, 132, 4107–4118. [Google Scholar] [CrossRef]
- Stintzi, A.; Browse, J. The Arabidopsis male-sterile mutant, opr3, lacks the 12-oxophytodienoic acid reductase required for jasmonate synthesis. Proc. Natl. Acad. Sci. USA 2000, 97, 10625–10630. [Google Scholar] [CrossRef] [PubMed]
- Song, S.; Qi, T.; Huang, H.; Ren, Q.; Wu, D.; Chang, C.; Peng, W.; Liu, Y.; Peng, J.; Xie, D. The jasmonate-ZIM domain proteins interact with the R2R3-MYB transcription factors MYB21 and MYB24 to affect jasmonate-regulated stamen development in Arabidopsis. Plant Cell 2011, 23, 1000–1013. [Google Scholar] [CrossRef]
- Dreni, L.; Jacchia, S.; Fornara, F.; Fornari, M.; Ouwerkerk, P.; An, G.; Colombo, L.; Kater, M. The D-lineage MADS-box gene OsMADS13 controls ovule identity in rice. Plant J. 2007, 52, 690–699. [Google Scholar] [CrossRef]
- Makwana, V.; Shukla, P.; Robin, P. GA application induces alteration in sex ratio and cell death in Jatropha curcas. Plant Growth Regul. 2010, 61, 121–125. [Google Scholar] [CrossRef]
- Rood, S.; Pharis, R. Changes of endogenous gibberellin-like substances with sex reversal of the apical inflorescence of corn. Plant Physiol. 1980, 66, 793–796. [Google Scholar] [CrossRef]
- Tanimoto, T. Modification of sex expression in Sagittaria latifolia by the application of gibberellic acid and paclobutrazol. J. Japan. Soc. Hort. Sci. 2007, 76, 47–53. [Google Scholar] [CrossRef][Green Version]
- Cheng, H.; Song, S.; Xiao, L.; Soo, H.; Cheng, Z.; Xie, D.; Peng, J. Gibberellin acts through jasmonate to control the expression of MYB21, MYB24, and MYB57 to promote stamen filament growth in Arabidopsis. PLoS Genet. 2009, 5, 13. [Google Scholar] [CrossRef]
- West, N.; Golenberg, E. Gender-specific expression of GIBBERELLIC ACID INSENSITIVE is critical for unisexual organ initiation in dioecious Spinacia oleracea. New Phytol. 2018, 217, 1322–1334. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, X.; Liu, B.; Wang, W.; Liu, X.; Chen, C.; Liu, X.; Yang, S.; Ren, H. A GAMYB homologue CsGAMYB1 regulates sex expression of cucumber via an ethylene-independent pathway. J. Exp. Bot. 2014, 65, 3201–3213. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Letunic, I.; Doerks, T.; Bork, P. SMART 7: Recent updates to the protein domain annotation resource. Nucleic Acids Res. 2011, 40, D302–D305. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef]
- Guindon, S.; Gascuel, O. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.; Geer, R.; Gonzales, N.; Gwadz, M.; Hurwitz, D.; Marchler, G.; Song, J.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2020, 48, D265–D268. [Google Scholar] [CrossRef]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Lescot, M.; Dehais, P.; Thijs, G.; Marchal, K.; Moreau, Y.; Van de Peer, Y.; Rouze, P.; Rombauts, S. PlantCARE, a database of plant cis-acting regulatory elements and a portal to tools for in silico analysis of promoter sequences. Nucleic Acids Res. 2002, 30, 325–327. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Williams, B.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.; Salzberg, S.; Wold, B.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed]
- Zhong, H.; Chen, J.; Li, C.; Chen, L.; Wu, J.; Chen, J.; Lu, W.; Li, J. Selection of reliable reference genes for expression studies by reverse transcription quantitative real-time PCR in litchi under different experimental conditions. Plant Cell Rep. 2011, 30, 641–653. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, H.; Wang, H.; Huang, J.; Liu, M.; Chen, T.; Shan, X.; Chen, H.; Shen, J. Genome-Wide Identification and Expression Analysis of MADS-Box Family Genes in Litchi (Litchi chinensis Sonn.) and Their Involvement in Floral Sex Determination. Plants 2021, 10, 2142. https://doi.org/10.3390/plants10102142
Guan H, Wang H, Huang J, Liu M, Chen T, Shan X, Chen H, Shen J. Genome-Wide Identification and Expression Analysis of MADS-Box Family Genes in Litchi (Litchi chinensis Sonn.) and Their Involvement in Floral Sex Determination. Plants. 2021; 10(10):2142. https://doi.org/10.3390/plants10102142
Chicago/Turabian StyleGuan, Hongling, Han Wang, Jianjun Huang, Mingxin Liu, Ting Chen, Xiaozhen Shan, Houbin Chen, and Jiyuan Shen. 2021. "Genome-Wide Identification and Expression Analysis of MADS-Box Family Genes in Litchi (Litchi chinensis Sonn.) and Their Involvement in Floral Sex Determination" Plants 10, no. 10: 2142. https://doi.org/10.3390/plants10102142
APA StyleGuan, H., Wang, H., Huang, J., Liu, M., Chen, T., Shan, X., Chen, H., & Shen, J. (2021). Genome-Wide Identification and Expression Analysis of MADS-Box Family Genes in Litchi (Litchi chinensis Sonn.) and Their Involvement in Floral Sex Determination. Plants, 10(10), 2142. https://doi.org/10.3390/plants10102142